
Modeling Blast Crisis Using Mutagenized Chronic Myeloid Leukemia-Derived Induced Pluripotent Stem Cells (iPSCs)
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. iPSC Cultures
2.3. Teratoma Assays
2.4. N-ethyl-N-Nitrosourea-(ENU) Induced Mutagenesis
2.5. Karyotyping
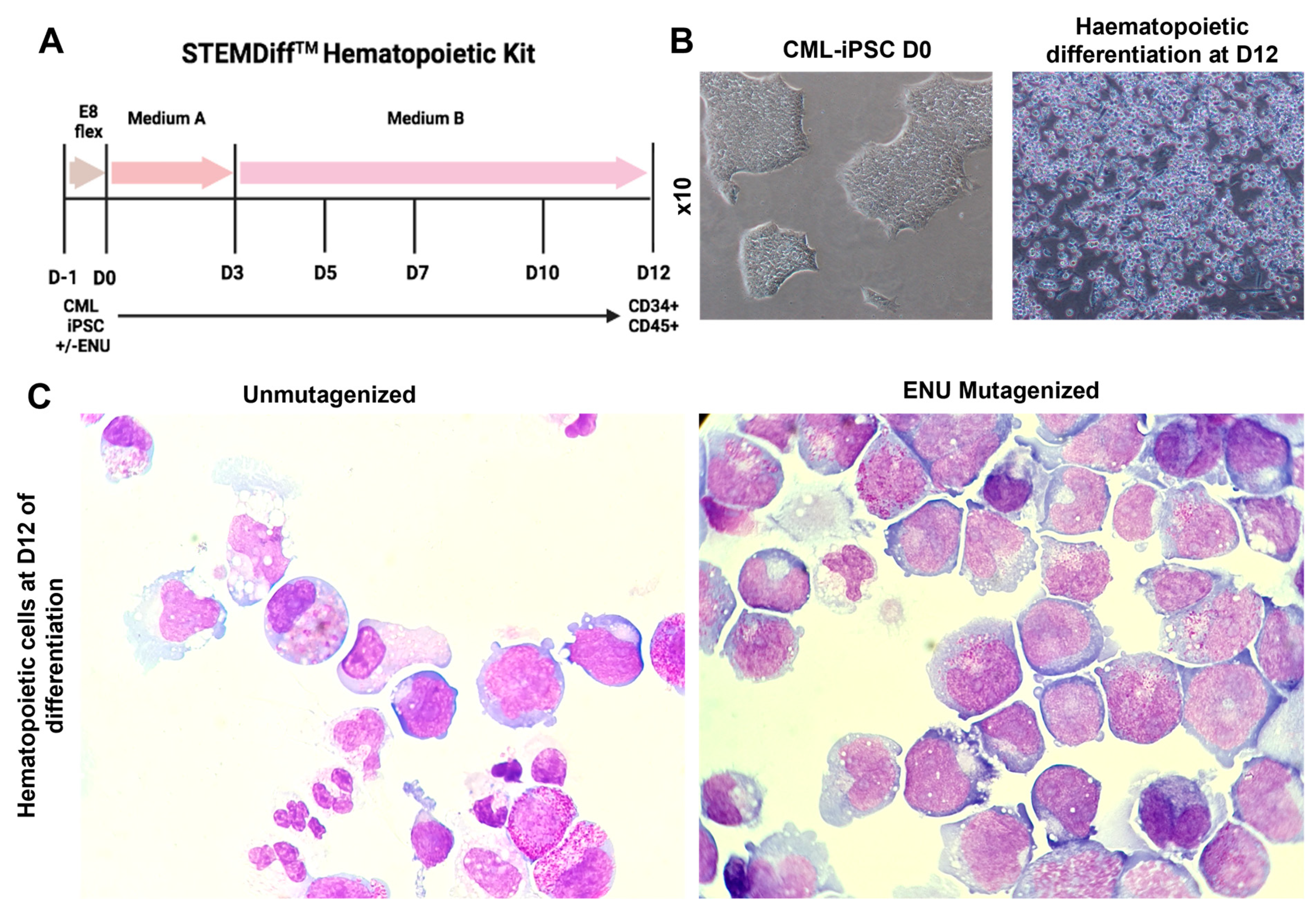
2.6. Hematopoietic Differentiation from iPSCs before and after Mutagenesis
2.7. Evaluation of Cytological Characteristics of Cells before and after Mutagenesis
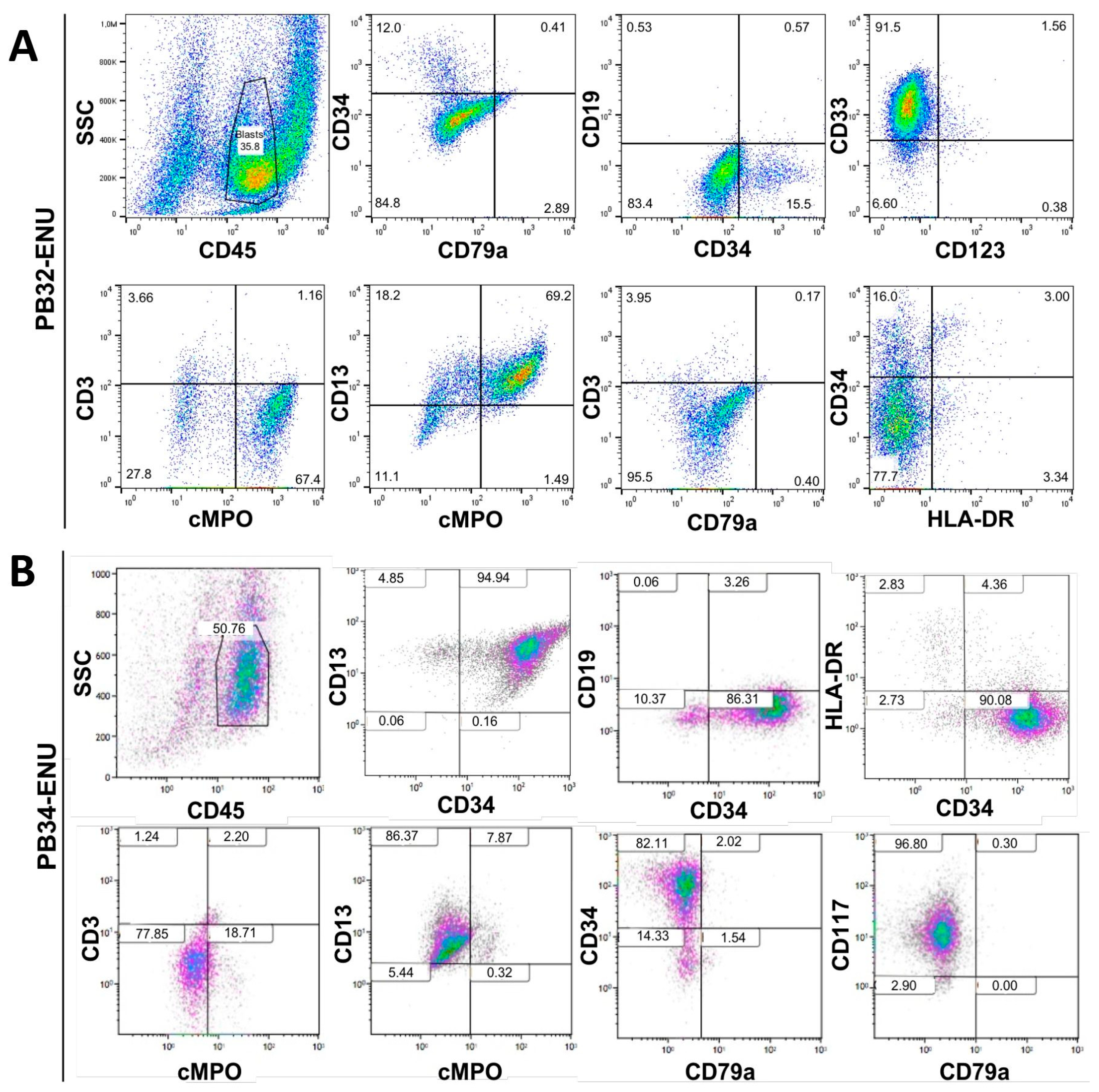
2.8. Flow Cytometry
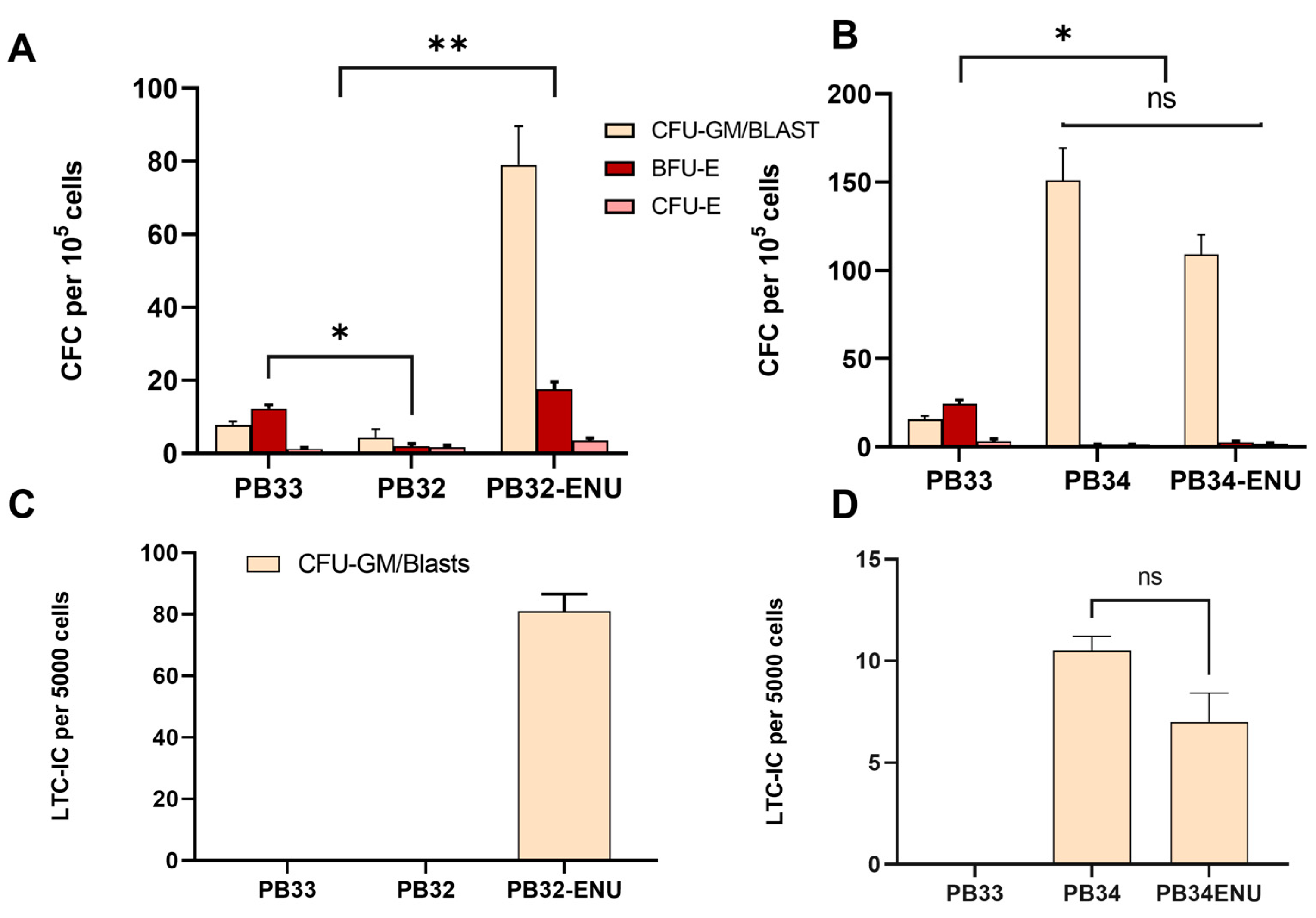
2.9. Clonogenic Assays
2.10. Long-Term Culture Initiating Cell (LTC-IC) Assays
2.11. Evaluation of Ruxolitinib and Imatinib Sensitivity of PB34 and PB34-ENU Hematopoietic Cells
2.12. Patients and Healthy Donors
2.13. Quantitative RT-PCR Assays
2.14. Single-Cell RNA-Sequencing of Mutagenized and Unmutagenized CML-iPSC-Derived Hematopoietic Cells
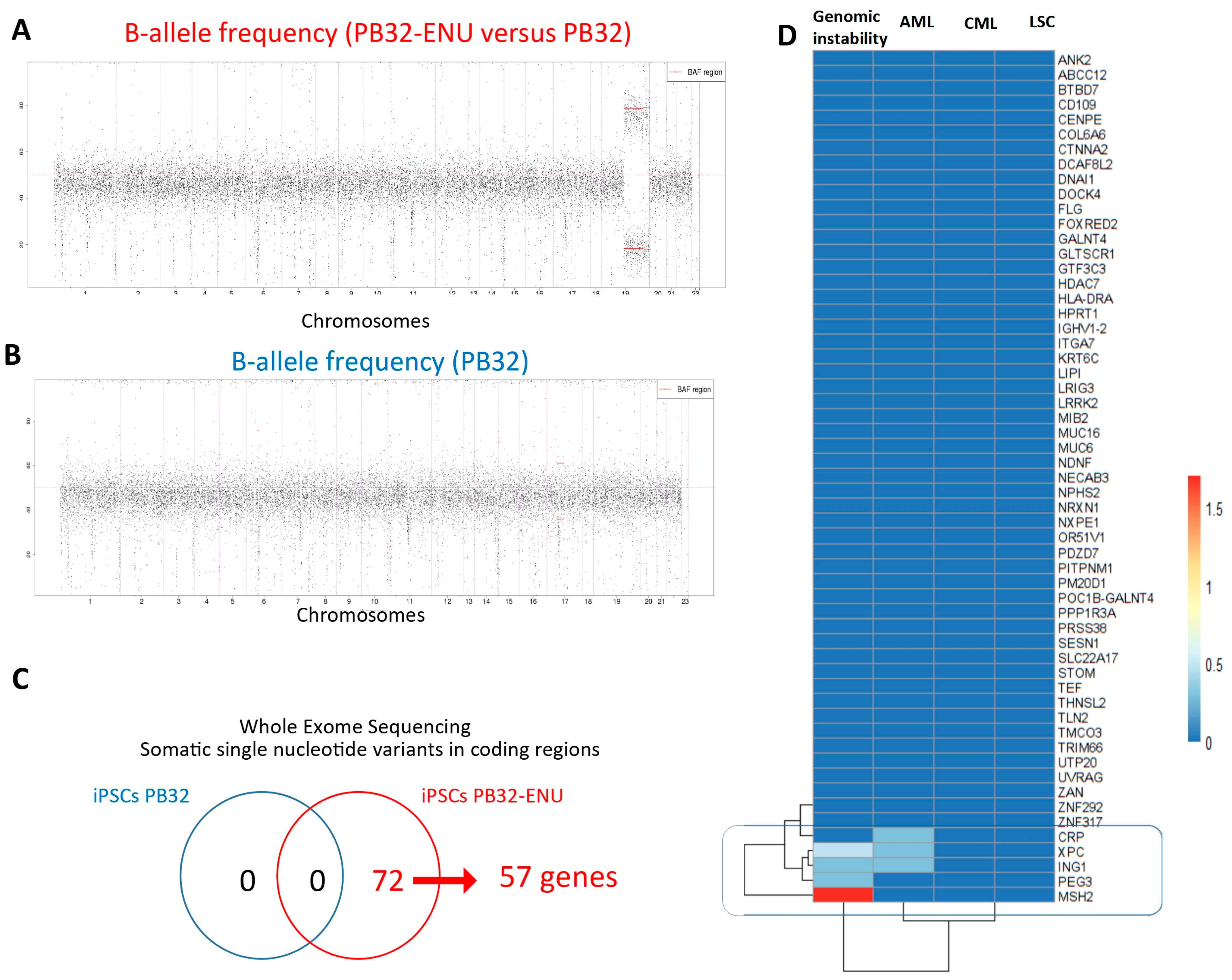
2.15. Exome Analysis of Mutagenized CML-iPSCs
2.16. Bulk RNA Transcriptomics of CML-iPSC-Derived Hematopoietic Cells before and after Mutagenesis
3. Results
3.1. Generation and Characterization of Mutagenized CML-iPS Cells
3.2. Induction of Hematopoietic Differentiation from ENU-Mutagenized and Unmutagenized iPSCs
3.3. Characterization of Mutagenized Hematopoietic Cells Using Flow Cytometry
3.4. Long-Term ENU Exposure Induces an Enhancement of CML-iPSC Hematopoiesis as Compared to Normal iPSC-Derived Clonogenic Activity
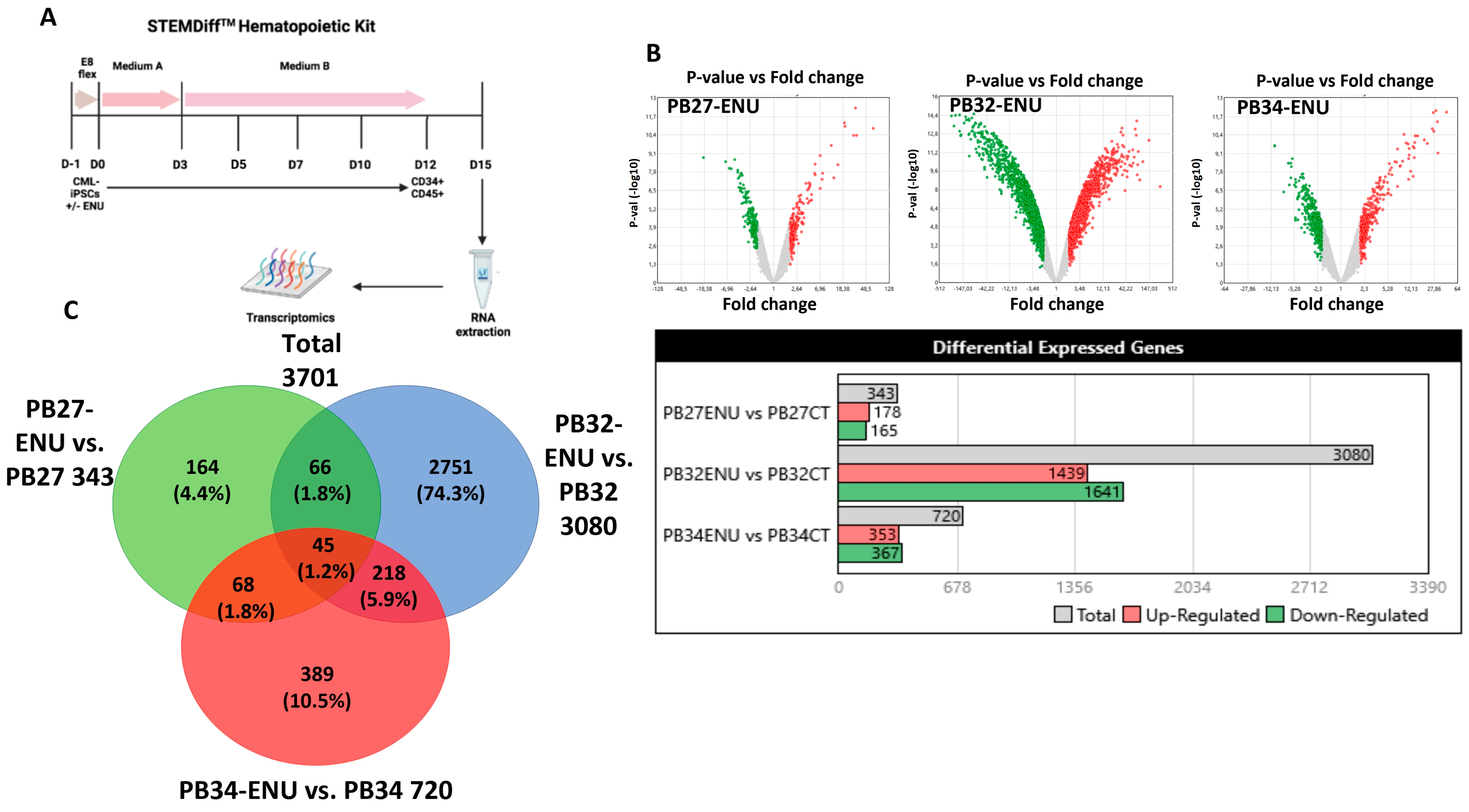
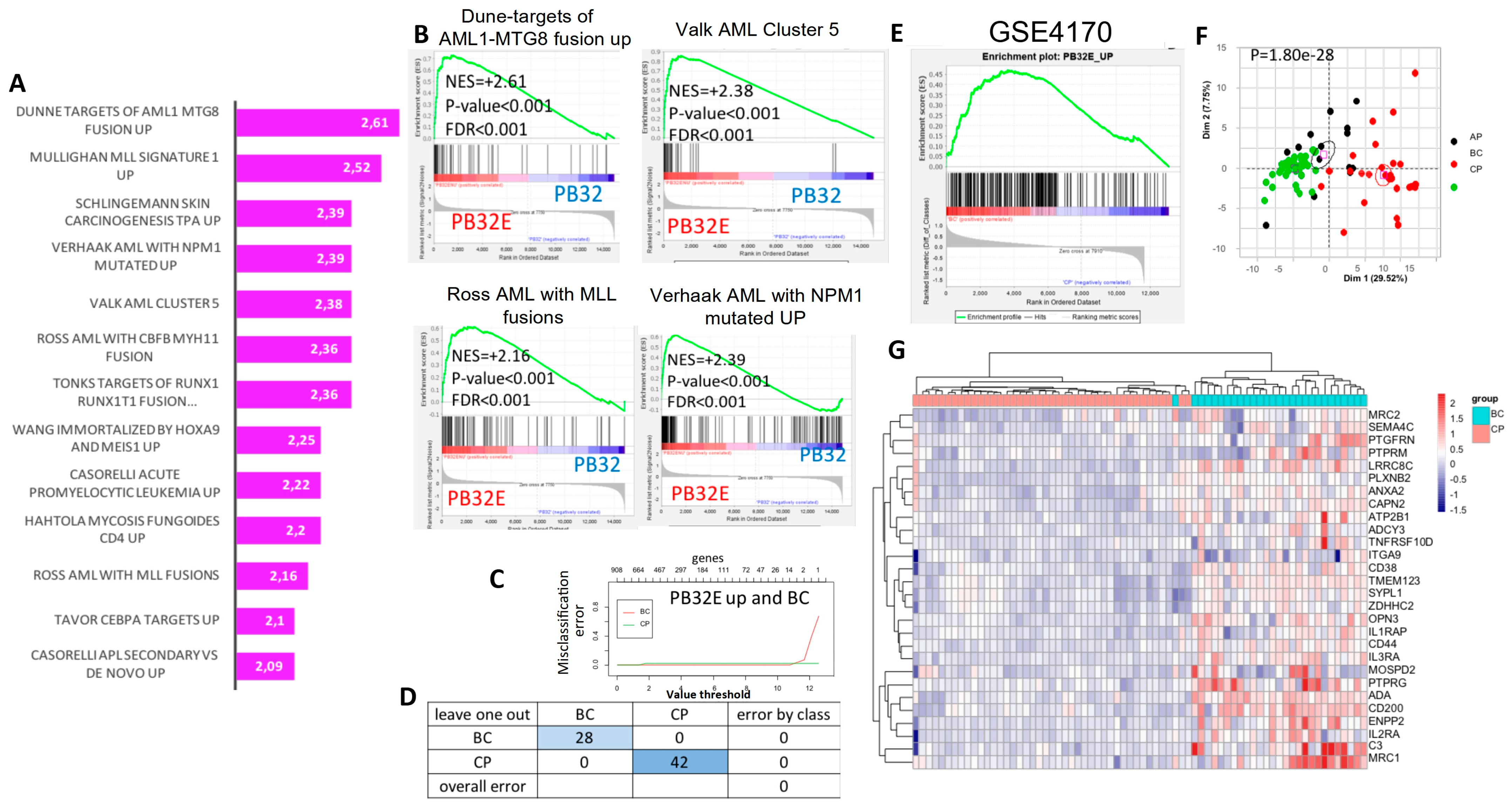
3.5. Transcriptomic Analysis of Hematopoietic Cells Derived from Mutagenized and Non-Mutagenized CML-iPSCs
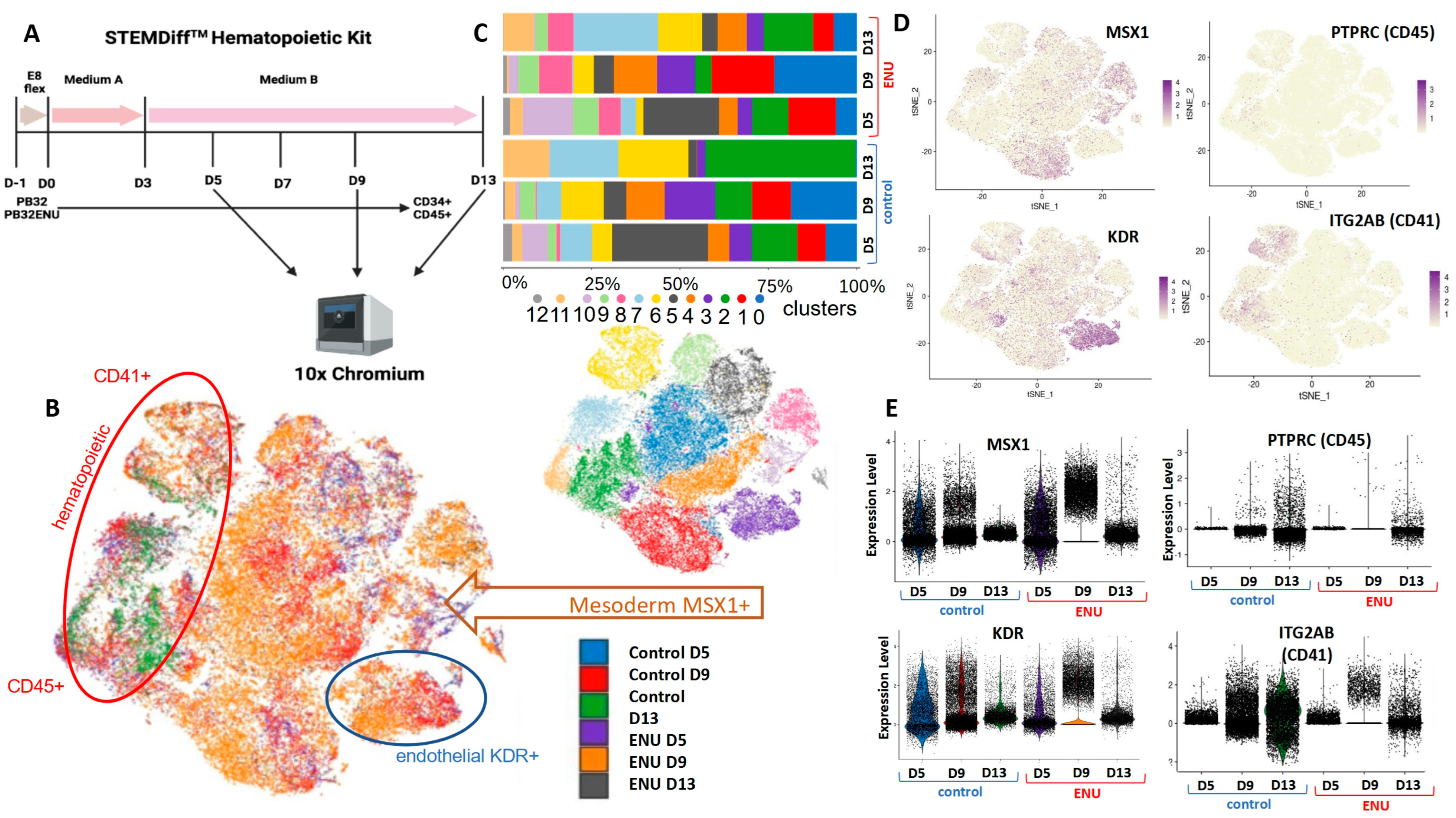
3.6. Single-Cell Transcriptomics during Hematopoietic Differentiation of ENU-Mutagenized iPSCs as Compared to Their Non-Mutagenized Counterparts
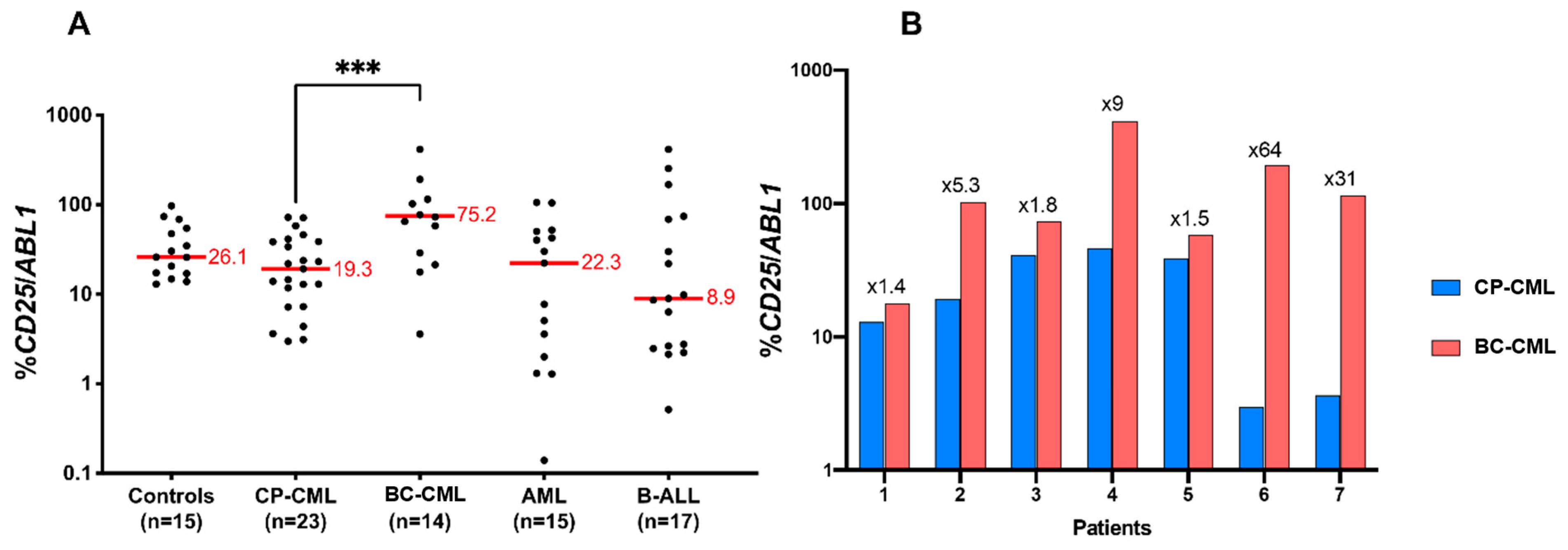
3.7. CD25 Is Overexpressed in BC-CML as Compared to CP-CML
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldman, J.M.; Melo, J.V. Chronic Myeloid Leukemia—Advances in Biology and New Approaches to Treatment. N. Engl. J. Med. 2003, 349, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Kantarjian, H. Chronic Myeloid Leukemia: 2014 Update on Diagnosis, Monitoring, and Management. Am. J. Hematol. 2014, 89, 547–556. [Google Scholar] [CrossRef]
- Sloma, I.; Jiang, X.; Eaves, A.C.; Eaves, C.J. Insights into the Stem Cells of Chronic Myeloid Leukemia. Leukemia 2010, 24, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Talpaz, M.; Gutterman, J. Biologic Therapy of Chronic Myelogenous Leukemia. Oncology 1987, 1, 35–40, 48, 49, 52. [Google Scholar] [PubMed]
- Shah, N.P.; Nicoll, J.M.; Nagar, B.; Gorre, M.E.; Paquette, R.L.; Kuriyan, J.; Sawyers, C.L. Multiple BCR-ABL Kinase Domain Mutations Confer Polyclonal Resistance to the Tyrosine Kinase Inhibitor Imatinib (STI571) in Chronic Phase and Blast Crisis Chronic Myeloid Leukemia. Cancer Cell 2002, 2, 117–125. [Google Scholar] [CrossRef]
- Soverini, S.; Martinelli, G.; Colarossi, S.; Gnani, A.; Castagnetti, F.; Rosti, G.; Bosi, C.; Paolini, S.; Rondoni, M.; Piccaluga, P.P.; et al. Presence or the Emergence of a F317L BCR-ABL Mutation May Be Associated with Resistance to Dasatinib in Philadelphia Chromosome-Positive Leukemia. J. Clin. Oncol. 2006, 24, e51–e52. [Google Scholar] [CrossRef]
- Crisan, A.M.; Coriu, D.; Arion, C.; Colita, A.; Jardan, C. The Impact of Additional Cytogenetic Abnormalities at Diagnosis and during Therapy with Tyrosine Kinase Inhibitors in Chronic Myeloid Leukaemia. J. Med. Life 2015, 8, 502–508. [Google Scholar]
- Mitelman, F. The Cytogenetic Scenario of Chronic Myeloid Leukemia. Leuk. Lymphoma 1993, 11, 11–15. [Google Scholar] [CrossRef]
- Perrotti, D.; Jamieson, C.; Goldman, J.; Skorski, T. Chronic Myeloid Leukemia: Mechanisms of Blastic Transformation. J. Clin. Investig. 2010, 120, 2254–2264. [Google Scholar] [CrossRef]
- Canitrot, Y.; Lautier, D.; Laurent, G.; Fréchet, M.; Ahmed, A.; Turhan, A.G.; Salles, B.; Cazaux, C.; Hoffmann, J.S. Mutator Phenotype of BCR—ABL Transfected Ba/F3 Cell Lines and Its Association with Enhanced Expression of DNA Polymerase Beta. Oncogene 1999, 18, 2676–2680. [Google Scholar] [CrossRef]
- Koptyra, M.; Cramer, K.; Slupianek, A.; Richardson, C.; Skorski, T. BCR/ABL Promotes Accumulation of Chromosomal Aberrations Induced by Oxidative and Genotoxic Stress. Leukemia 2008, 22, 1969–1972. [Google Scholar] [CrossRef] [Green Version]
- Bolton-Gillespie, E.; Schemionek, M.; Klein, H.-U.; Flis, S.; Hoser, G.; Lange, T.; Nieborowska-Skorska, M.; Maier, J.; Kerstiens, L.; Koptyra, M.; et al. Genomic Instability May Originate from Imatinib-Refractory Chronic Myeloid Leukemia Stem Cells. Blood 2013, 121, 4175–4183. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Dugray, A.; AbdulKarim, B.; Marangoni, E.; Maggiorella, L.; Vaganay, S.; M’Kacher, R.; Rasy, S.D.; Eschwege, F.; Vainchenker, W.; et al. BCR-ABL down-Regulates the DNA Repair Protein DNA-PKcs. Blood 2001, 97, 2084–2090. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Jarrousse, S.; Buet, D.; Dugray, A.; Bonnet, M.-L.; Vozenin-Brotons, M.-C.; Guilhot, F.; Turhan, A.G.; Feunteun, J.; Bourhis, J. Down-Regulation of BRCA1 in BCR-ABL-Expressing Hematopoietic Cells. Blood 2003, 101, 4583–4588. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Gurdek, E.; Koptyra, M.; Nowicki, M.O.; Siddiqui, K.M.; Groden, J.; Skorski, T. BLM Helicase Is Activated in BCR/ABL Leukemia Cells to Modulate Responses to Cisplatin. Oncogene 2005, 24, 3914–3922. [Google Scholar] [CrossRef] [PubMed]
- Canitrot, Y.; Falinski, R.; Louat, T.; Laurent, G.; Cazaux, C.; Hoffmann, J.-S.; Lautier, D.; Skorski, T. P210 BCR/ABL Kinase Regulates Nucleotide Excision Repair (NER) and Resistance to UV Radiation. Blood 2003, 102, 2632–2637. [Google Scholar] [CrossRef]
- Stoklosa, T.; Poplawski, T.; Koptyra, M.; Nieborowska-Skorska, M.; Basak, G.; Slupianek, A.; Rayevskaya, M.; Seferynska, I.; Herrera, L.; Blasiak, J.; et al. BCR/ABL Inhibits Mismatch Repair to Protect from Apoptosis and Induce Point Mutations. Cancer Res. 2008, 68, 2576–2580. [Google Scholar] [CrossRef]
- Graham, S.M.; Jørgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, Quiescent, Philadelphia-Positive Stem Cells from Patients with Chronic Myeloid Leukemia Are Insensitive to STI571 in Vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef]
- Chomel, J.-C.; Bonnet, M.-L.; Sorel, N.; Bertrand, A.; Meunier, M.-C.; Fichelson, S.; Melkus, M.; Bennaceur-Griscelli, A.; Guilhot, F.; Turhan, A.G. Leukemic Stem Cell Persistence in Chronic Myeloid Leukemia Patients with Sustained Undetectable Molecular Residual Disease. Blood 2011, 118, 3657–3660. [Google Scholar] [CrossRef]
- Chu, S.; McDonald, T.; Lin, A.; Chakraborty, S.; Huang, Q.; Snyder, D.S.; Bhatia, R. Persistence of Leukemia Stem Cells in Chronic Myelogenous Leukemia Patients in Prolonged Remission with Imatinib Treatment. Blood 2011, 118, 5565–5572. [Google Scholar] [CrossRef]
- Herrmann, H.; Sadovnik, I.; Cerny-Reiterer, S.; Rülicke, T.; Stefanzl, G.; Willmann, M.; Hoermann, G.; Bilban, M.; Blatt, K.; Herndlhofer, S.; et al. Dipeptidylpeptidase IV (CD26) Defines Leukemic Stem Cells (LSC) in Chronic Myeloid Leukemia. Blood 2014, 123, 3951–3962. [Google Scholar] [CrossRef] [Green Version]
- Chomel, J.-C.; Turhan, A.G. Chronic Myeloid Leukemia Stem Cells in the Era of Targeted Therapies: Resistance, Persistence and Long-Term Dormancy. Oncotarget 2011, 2, 713–727. [Google Scholar] [CrossRef]
- Herrmann, H.; Sadovnik, I.; Eisenwort, G.; Rülicke, T.; Blatt, K.; Herndlhofer, S.; Willmann, M.; Stefanzl, G.; Baumgartner, S.; Greiner, G.; et al. Delineation of Target Expression Profiles in CD34+/CD38− and CD34+/CD38+ Stem and Progenitor Cells in AML and CML. Blood Adv. 2020, 4, 5118–5132. [Google Scholar] [CrossRef]
- Melkus, M.; Bennaceur-Griscelli, A.; Valogne, Y.; Flamant, S.; Chomel, J.-C.; Sorel, N.; Bonnet, M.-L.; Deininger, M.W.; Mitjavila-Garcia, M.-T.; Turhan, A.G. Biological Effects of T315I-Mutated BCR-ABL in an Embryonic Stem Cell-Derived Hematopoiesis Model. Exp. Hematol. 2013, 41, 335–345.e3. [Google Scholar] [CrossRef] [PubMed]
- Kumano, K.; Arai, S.; Hosoi, M.; Taoka, K.; Takayama, N.; Otsu, M.; Nagae, G.; Ueda, K.; Nakazaki, K.; Kamikubo, Y.; et al. Generation of Induced Pluripotent Stem Cells from Primary Chronic Myelogenous Leukemia Patient Samples. Blood 2012, 119, 6234–6242. [Google Scholar] [CrossRef]
- Telliam, G.; Desterke, C.; Imeri, J.; M’Kacher, R.; Oudrhiri, N.; Balducci, E.; Fontaine-Arnoux, M.; Acloque, H.; Bennaceur-Griscelli, A.; Turhan, A.G. Modeling Global Genomic Instability in Chronic Myeloid Leukemia (CML) Using Patient-Derived Induced Pluripotent Stem Cells (IPSC). bioRxiv 2022. [Google Scholar] [CrossRef]
- Telliam, G.; Féraud, O.; Griscelli, F.; Opolon, P.; Divers, D.; Bennaceur-Griscelli, A.; Turhan, A.G. Generation of an Induced Pluripotent Stem Cell Line from a Patient with Chronic Myeloid Leukemia (CML) Resistant to Targeted Therapies. Stem Cell Res. 2016, 17, 235–237. [Google Scholar] [CrossRef]
- Sloma, I.; Mitjavila-Garcia, M.T.; Feraud, O.; Griscelli, F.; Oudrhiri, N.; El Marsafy, S.; Gobbo, E.; Divers, D.; Proust, A.; Smadja, D.M.; et al. Whole-Genome Analysis Reveals Unexpected Dynamics of Mutant Subclone Development in a Patient with JAK2-V617F-Positive Chronic Myeloid Leukemia. Exp. Hematol. 2017, 53, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; Amir, E.D.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R.; et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells That Correlate with Prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip Probe Level Data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Radich, J.P.; Dai, H.; Mao, M.; Oehler, V.; Schelter, J.; Druker, B.; Sawyers, C.; Shah, N.; Stock, W.; Willman, C.L.; et al. Gene Expression Changes Associated with Progression and Response in Chronic Myeloid Leukemia. Proc. Natl. Acad. Sci. USA 2006, 103, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R.; Hastie, T.; Narasimhan, B.; Chu, G. Diagnosis of Multiple Cancer Types by Shrunken Centroids of Gene Expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6567–6572. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for Gene List Enrichment Analysis and Candidate Gene Prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of Biological Networks and Gene Expression Data Using Cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef] [PubMed]
- Guillem, V.M.; Cervantes, F.; Martínez, J.; Alvarez-Larrán, A.; Collado, M.; Camós, M.; Sureda, A.; Maffioli, M.; Marugán, I.; Hernández-Boluda, J.-C. XPC Genetic Polymorphisms Correlate with the Response to Imatinib Treatment in Patients with Chronic Phase Chronic Myeloid Leukemia. Am. J. Hematol. 2010, 85, 482–486. [Google Scholar] [CrossRef]
- Lakkireddy, S.; Aula, S.; Kapley, A.; Gundeti, S.; Kutala, V.K.; Jamil, K. Association of DNA Repair Gene XPC Ala499Val (Rs2228000 C>T) and Lys939Gln (Rs2228001 A>C) Polymorphisms with the Risk of Chronic Myeloid Leukemia: A Case-Control Study in a South Indian Population. J. Gene Med. 2021, 23, e3339. [Google Scholar] [CrossRef]
- Takeda, N.; Shibuya, M.; Maru, Y. The BCR-ABL Oncoprotein Potentially Interacts with the Xeroderma Pigmentosum Group B Protein. Proc. Natl. Acad. Sci. USA 1999, 96, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Quek, L.; Otto, G.W.; Garnett, C.; Lhermitte, L.; Karamitros, D.; Stoilova, B.; Lau, I.-J.; Doondeea, J.; Usukhbayar, B.; Kennedy, A.; et al. Genetically Distinct Leukemic Stem Cells in Human CD34− Acute Myeloid Leukemia Are Arrested at a Hemopoietic Precursor-like Stage. J. Exp. Med. 2016, 213, 1513–1535. [Google Scholar] [CrossRef] [PubMed]
- Plesa, A.; Ciuperca, G.; Louvet, V.; Pujo-Menjouet, L.; Génieys, S.; Dumontet, C.; Thomas, X.; Volpert, V. Diagnostics of the AML with Immunophenotypical Data. Math. Model. Nat. Phenom. 2006, 1, 104–123. [Google Scholar] [CrossRef]
- Sadovnik, I.; Hoelbl-Kovacic, A.; Herrmann, H.; Eisenwort, G.; Cerny-Reiterer, S.; Warsch, W.; Hoermann, G.; Greiner, G.; Blatt, K.; Peter, B.; et al. Identification of CD25 as STAT5-Dependent Growth Regulator of Leukemic Stem Cells in Ph+ CML. Clin. Cancer Res. 2016, 22, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, C.I.; Takubo, K.; Kobayashi, H.; Nakamura-Ishizu, A.; Honda, H.; Kataoka, K.; Kumano, K.; Akiyama, H.; Sudo, T.; Kurokawa, M.; et al. The IL-2/CD25 Axis Maintains Distinct Subsets of Chronic Myeloid Leukemia-Initiating Cells. Blood 2014, 123, 2540–2549. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient # | Age/Sex | Status at Diagnosis | Reprogrammed Cells/Origin |
|---|---|---|---|
| UPN27 (PB27) | 56/M | CP-CML | CD34+ / Blood |
| UPN32 (PB32) | 17/M | CP-CML | CD34+ / Blood |
| UPN34 (PB34) | 70/M | CP-CML | CD34+ AP-CML/Blood |
| Antibody | Fluorophore | Reference |
|---|---|---|
| CD45 | PC7 | IM3548 Beckman Coulter |
| CD13 | PE | A07762 Beckman Coulter |
| CD33 | APC | IM2471 Beckman Coulter |
| CD117 | PC5.5 | B96754 Beckman Coulter |
| MPO | FITC | IM1874 Beckman Coulter |
| CD19 | ECD | 6604551 Beckman Coulter |
| CD43 | PE | A32560 Beckman Coulter |
| CD3 | APC-A750 | B10823 Beckman Coulter |
| CD22 | PC5.5 | A80712 Beckman Coulter |
| CD79a | APC | B36287 Beckman Coulter |
| HLA-DR | ECD | B92438 Beckman Coulter |
| CD34 | APC-A70 | B92417 Beckman Coulter |
| Controls | CP-CML | BC-CML | AML | B-ALL | |
|---|---|---|---|---|---|
| n | 15 | 23 | 14 | 15 | 17 |
| Gender | |||||
| Male | N/A | 13 | 11 | 9 | 5 |
| Female | N/A | 10 | 3 | 6 | 12 |
| Age, range (years) | N/A | 58.1 [22.7–81.1] | 47.4 [22.9–75.2] | 67.6 [26.0–83.6] | 37.8 [4.6–82.5] |
| Molecular Rearrangements | |||||
| BCR::ABL1 M-BCR (e13a2 or e14a2) | 23 | 14 | |||
| BCR::ABL1 m-BCR (e1a2) | 5 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imeri, J.; Desterke, C.; Marcoux, P.; Telliam, G.; Sanekli, S.; Barreau, S.; Erbilgin, Y.; Latsis, T.; Hugues, P.; Sorel, N.; et al. Modeling Blast Crisis Using Mutagenized Chronic Myeloid Leukemia-Derived Induced Pluripotent Stem Cells (iPSCs). Cells 2023, 12, 598. https://doi.org/10.3390/cells12040598
Imeri J, Desterke C, Marcoux P, Telliam G, Sanekli S, Barreau S, Erbilgin Y, Latsis T, Hugues P, Sorel N, et al. Modeling Blast Crisis Using Mutagenized Chronic Myeloid Leukemia-Derived Induced Pluripotent Stem Cells (iPSCs). Cells. 2023; 12(4):598. https://doi.org/10.3390/cells12040598
Chicago/Turabian StyleImeri, Jusuf, Christophe Desterke, Paul Marcoux, Gladys Telliam, Safa Sanekli, Sylvain Barreau, Yucel Erbilgin, Theodoros Latsis, Patricia Hugues, Nathalie Sorel, and et al. 2023. "Modeling Blast Crisis Using Mutagenized Chronic Myeloid Leukemia-Derived Induced Pluripotent Stem Cells (iPSCs)" Cells 12, no. 4: 598. https://doi.org/10.3390/cells12040598
APA StyleImeri, J., Desterke, C., Marcoux, P., Telliam, G., Sanekli, S., Barreau, S., Erbilgin, Y., Latsis, T., Hugues, P., Sorel, N., Cayssials, E., Chomel, J.-C., Bennaceur-Griscelli, A., & Turhan, A. G. (2023). Modeling Blast Crisis Using Mutagenized Chronic Myeloid Leukemia-Derived Induced Pluripotent Stem Cells (iPSCs). Cells, 12(4), 598. https://doi.org/10.3390/cells12040598