
Opportunities and Challenges for the Development of MRCK Kinases Inhibitors as Potential Cancer Chemotherapeutics

 , , , ,
, , , ,
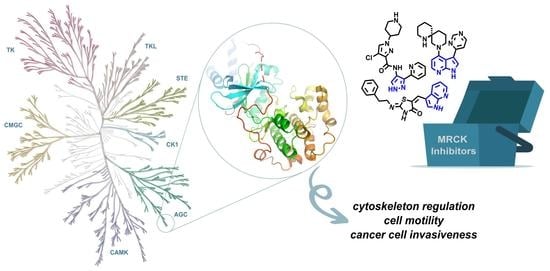
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Cancer and the Cytoskeleton
2. Actin–Myosin Cytoskeleton Regulation
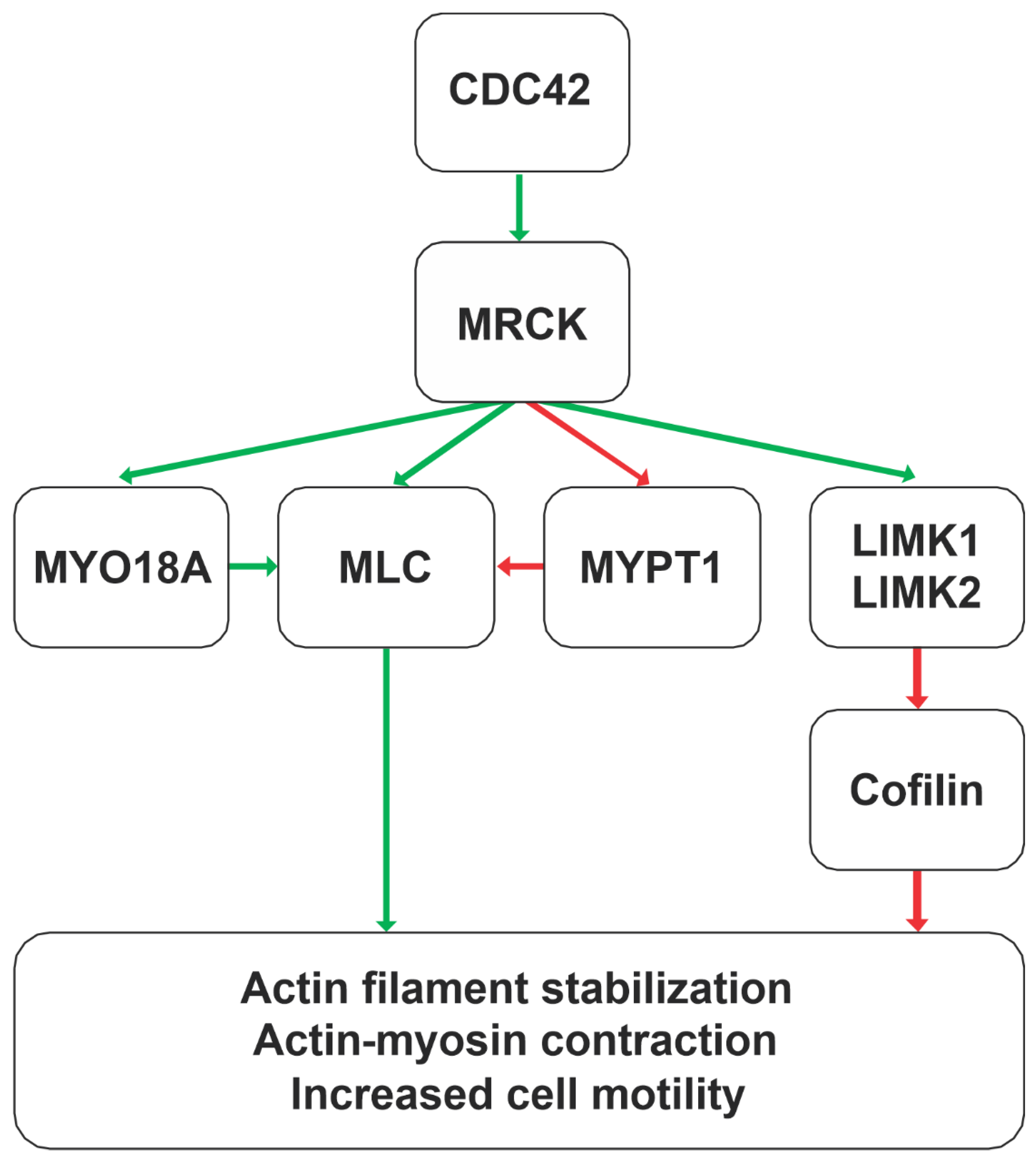
3. MRCK Function
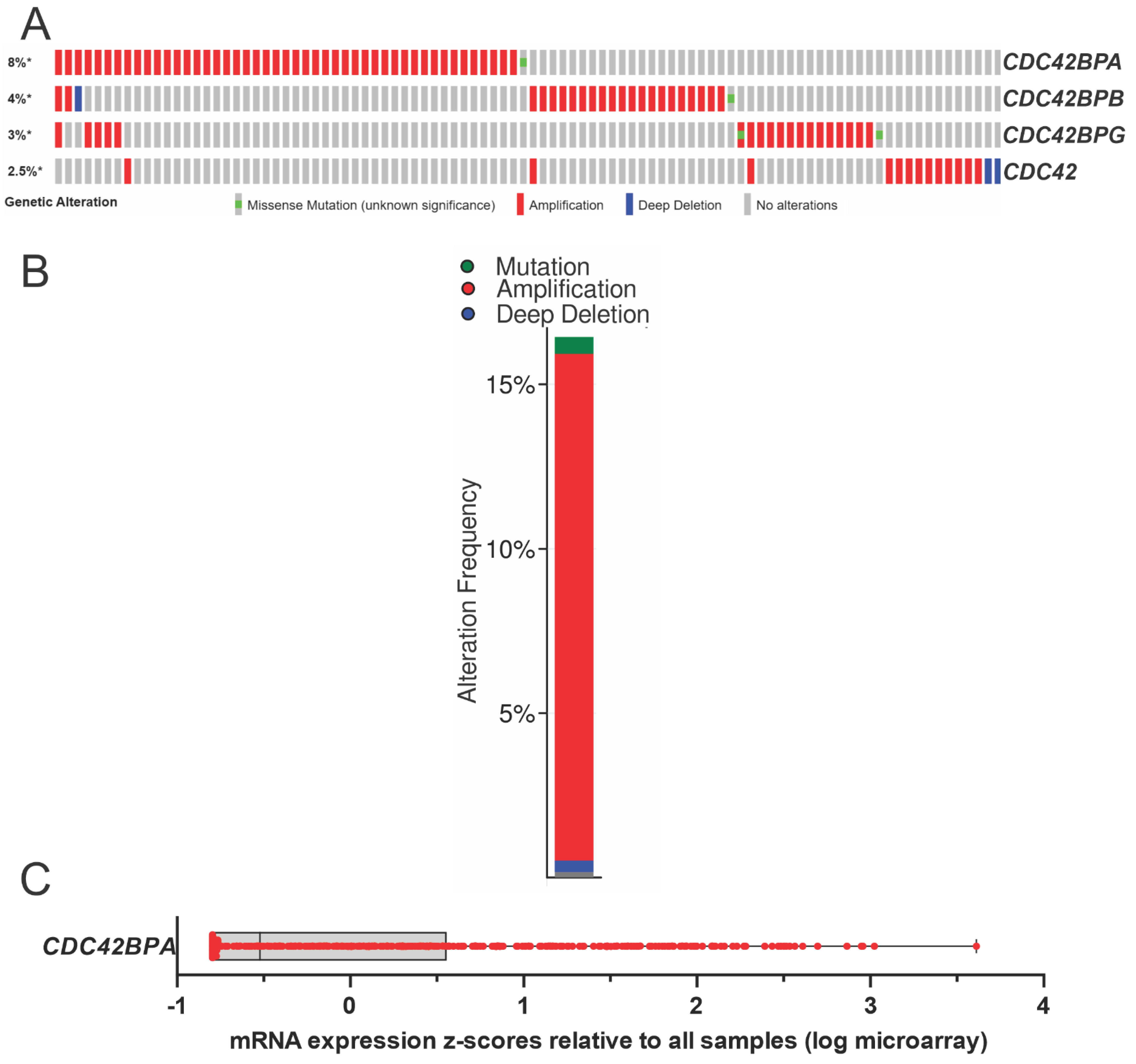
4. MRCK Expression and Cancer Association
5. Small-Molecule MRCK Pharmacological Inhibitors
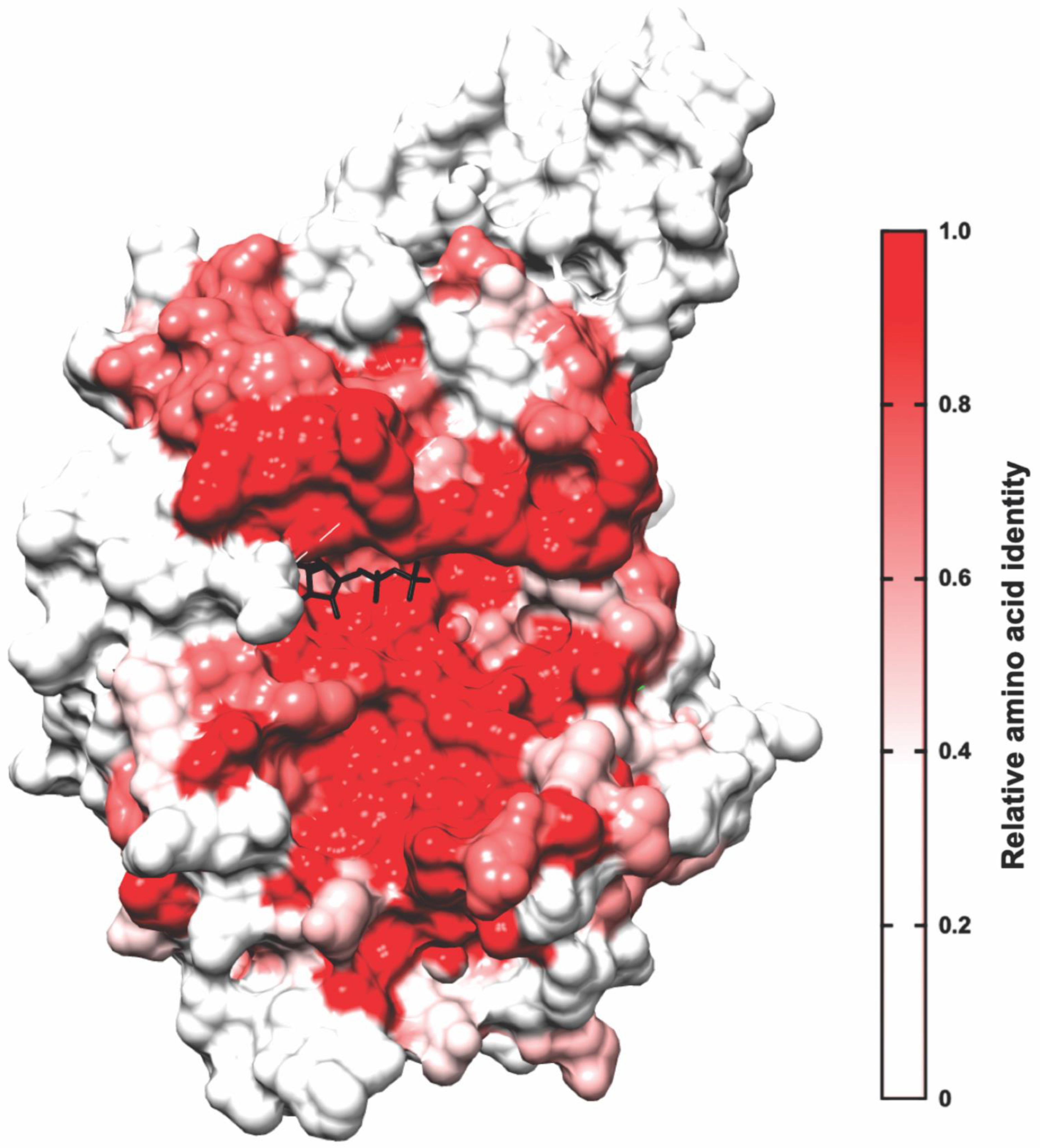
6. Binding Mode of MRCK Inhibitors
7. MRCK Inhibitors in Biological Studies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- East, M.P.; Asquith, C.R.M. Cdc42BPA/MRCKα: A Kinase Target for Brain, Ovarian and Skin Cancers. Nat. Rev. Drug Discov. 2021, 20, 167. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Olson, M.F.; Sahai, E. The Actin Cytoskeleton in Cancer Cell Motility. Clin. Exp. Metastasis 2009, 26, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.J.-Y.; Chen, I.-H.; Kuo, B.Y.-T.; Yu, C.-C.; Lai, M.-T.; Lin, J.-T.; Lin, L.Y.-T.; Chen, C.-M.; Hwang, T.; Sheu, J.J.-C. Alterations of Cytoskeleton Networks in Cell Fate Determination and Cancer Development. Biomolecules 2022, 12, 1862. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Samain, R.; Kohlhammer, L.; Orgaz, J.L.; George, S.L.; Maiques, O.; Barcelo, J.; Sanz-Moreno, V. Rho GTPase Signaling in Cancer Progression and Dissemination. Physiol. Rev. 2022, 102, 455–510. [Google Scholar] [CrossRef]
- Garrido-Casado, M.; Asensio-Juárez, G.; Vicente-Manzanares, M. Nonmuscle Myosin II Regulation Directs Its Multiple Roles in Cell Migration and Division. Annu. Rev. Cell Dev. Biol. 2021, 37, 285–310. [Google Scholar] [CrossRef]
- Debold, E.P. Recent Insights into the Relative Timing of Myosin’s Powerstroke and Release of Phosphate. Cytoskeleton 2021, 78, 448–458. [Google Scholar] [CrossRef]
- Olson, M.F. Rho GTPases, Their Post-Translational Modifications, Disease-Associated Mutations and Pharmacological Inhibitors. Small GTPases 2016, 9, 1–13. [Google Scholar] [CrossRef]
- Mosaddeghzadeh, N.; Ahmadian, M.R. The RHO Family GTPases: Mechanisms of Regulation and Signaling. Cells 2021, 10, 1831. [Google Scholar] [CrossRef]
- Julian, L.; Olson, M.F. Rho-Associated Coiled-Coil Containing Kinases (ROCK). Small GTPases 2014, 5, e29846. [Google Scholar] [CrossRef]
- Unbekandt, M.; Olson, M.F. The Actin-Myosin Regulatory MRCK Kinases: Regulation, Biological Functions and Associations with Human Cancer. J. Mol. Med. 2014, 92, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Manser, E. Myotonic Dystrophy Kinase-Related Cdc42-Binding Kinases (MRCK), the ROCK-like Effectors of Cdc42 and Rac1. Small GTPases 2015, 6, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Uehata, M.; Ishizaki, T.; Satoh, H.; Ono, T.; Kawahara, T.; Morishita, T.; Tamakawa, H.; Yamagami, K.; Inui, J.; Maekawa, M.; et al. Calcium Sensitization of Smooth Muscle Mediated by a Rho-Associated Protein Kinase in Hypertension. Nature 1997, 389, 990–994. [Google Scholar] [CrossRef]
- Rath, N.; Olson, M.F. Rho-Associated Kinases in Tumorigenesis: Re-Considering ROCK Inhibition for Cancer Therapy. EMBO Rep. 2012, 13, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Barcelo, J.; Samain, R.; Sanz-Moreno, V. Preclinical to Clinical Utility of ROCK Inhibitors in Cancer. Trends Cancer, 2023; in press. [Google Scholar] [CrossRef]
- UniProt: A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Pearson, W.R. Selecting the Right Similarity-Scoring Matrix. Curr. Protoc. Bioinform. 2013, 43, 3.5.1–3.5.9. [Google Scholar] [CrossRef]
- Heikkila, T.; Wheatley, E.; Crighton, D.; Schroder, E.; Boakes, A.; Kaye, S.J.; Mezna, M.; Pang, L.; Rushbrooke, M.; Turnbull, A.; et al. Co-Crystal Structures of Inhibitors with MRCKβ, a Key Regulator of Tumor Cell Invasion. PLoS ONE 2011, 6, e24825. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Sumi, T.; Matsumoto, K.; Shibuya, A.; Nakamura, T. Activation of LIM Kinases by Myotonic Dystrophy Kinase-Related Cdc42-Binding Kinase Alpha. J. Biol. Chem. 2001, 276, 23092–23096. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.M.; Leung, T.; Manser, E.; Lim, L. Cdc42 Antagonizes Inductive Action of CAMP on Cell Shape, via Effects of the Myotonic Dystrophy Kinase-Related Cdc42-Binding Kinase (MRCK) on Myosin Light Chain Phosphorylation. Eur. J. Cell Biol. 2002, 81, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.; Paterson, H.F.; Marshall, C.J. Cdc42-MRCK and Rho-ROCK Signalling Cooperate in Myosin Phosphorylation and Cell Invasion. Nat. Cell Biol. 2005, 7, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Tan, I.; Ng, C.H.; Lim, L.; Leung, T. Phosphorylation of a Novel Myosin Binding Subunit of Protein Phosphatase 1 Reveals a Conserved Mechanism in the Regulation of Actin Cytoskeleton. J. Biol. Chem. 2001, 276, 21209–21216. [Google Scholar] [CrossRef]
- Tan, I.; Yong, J.; Dong, J.M.; Lim, L.; Leung, T. A Tripartite Complex Containing MRCK Modulates Lamellar Actomyosin Retrograde Flow. Cell 2008, 135, 123–136. [Google Scholar] [CrossRef]
- Lee, I.C.J.; Leung, T.; Tan, I. Adaptor Protein LRAP25 Mediates Myotonic Dystrophy Kinase-Related Cdc42-Binding Kinase (MRCK) Regulation of LIMK1 Protein in Lamellipodial F-Actin Dynamics. J. Biol. Chem. 2014, 289, 26989–27003. [Google Scholar] [CrossRef]
- Tan, I.; Seow, K.T.; Lim, L.; Leung, T. Intermolecular and Intramolecular Interactions Regulate Catalytic Activity of Myotonic Dystrophy Kinase-Related Cdc42-Binding Kinase Alpha. Mol. Cell Biol. 2001, 21, 2767–2778. [Google Scholar] [CrossRef]
- Elkins, J.M.; Amos, A.; Niesen, F.H.; Pike, A.C.; Fedorov, O.; Knapp, S. Structure of Dystrophia Myotonica Protein Kinase. Protein Sci. 2009, 18, 782–791. [Google Scholar] [CrossRef]
- Jacobs, M.; Hayakawa, K.; Swenson, L.; Bellon, S.; Fleming, M.; Taslimi, P.; Doran, J. The Structure of Dimeric ROCK I Reveals the Mechanism for Ligand Selectivity. J. Biol. Chem. 2006, 281, 260–268. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Kasa, M.; Amano, M.; Kaibuchi, K.; Hakoshima, T. Molecular Mechanism for the Regulation of Rho-Kinase by Dimerization and Its Inhibition by Fasudil. Structure 2006, 14, 589–600. [Google Scholar] [CrossRef]
- Unbekandt, M.; Belshaw, S.; Bower, J.; Clarke, M.; Cordes, J.; Crighton, D.; Croft, D.R.; Drysdale, M.J.; Garnett, M.J.; Gill, K.; et al. Discovery of Potent and Selective MRCK Inhibitors with Therapeutic Effect on Skin Cancer. Cancer Res. 2018, 78, 2096–2114. [Google Scholar] [CrossRef] [PubMed]
- Unbekandt, M.; Lilla, S.; Zanivan, S.; Olson, M.F. The Cdc42 Effector Protein MRCKβ Autophosphorylates on Threonine 1108. Small GTPases 2020, 11, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Van’t Veer, L.J.; Dai, H.; Van De Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; Van Der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene Expression Profiling Predicts Clinical Outcome of Breast Cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Roepman, P.; Van’t Veer, L.J.; Bernards, R.; de Snoo, F.; Glas, A.M. Biological Functions of the Genes in the Mammaprint Breast Cancer Profile Reflect the Hallmarks of Cancer. Biomark. Insights 2010, 5, 129–138. [Google Scholar] [CrossRef]
- Buyse, M.; Loi, S.; van’t Veer, L.; Viale, G.; Delorenzi, M.; Glas, A.M.; Saghatchian d’Assignies, M.; Bergh, J.; Lidereau, R.; Ellis, P.; et al. Validation and Clinical Utility of a 70-Gene Prognostic Signature for Women With Node-Negative Breast Cancer. J. Natl. Cancer Inst. 2006, 98, 1183–1192. [Google Scholar] [CrossRef]
- Mook, S.; Schmidt, M.K.; Weigelt, B.; Kreike, B.; Eekhout, I.; van de Vijver, M.J.; Glas, A.M.; Floore, A.; Rutgers, E.J.T.; van ’t Veer, L.J. The 70-Gene Prognosis Signature Predicts Early Metastasis in Breast Cancer Patients between 55 and 70 Years of Age. Ann. Oncol. 2010, 21, 717–722. [Google Scholar] [CrossRef]
- Knauer, M.; Mook, S.; Rutgers, E.J.T.; Bender, R.A.; Hauptmann, M.; van de Vijver, M.J.; Koornstra, R.H.T.; Bueno-de-Mesquita, J.M.; Linn, S.C.; van ’t Veer, L.J. The Predictive Value of the 70-Gene Signature for Adjuvant Chemotherapy in Early Breast Cancer. Breast Cancer Res. Treat. 2010, 120, 655–661. [Google Scholar] [CrossRef]
- Cardoso, F.; van’t Veer, L.J.; Bogaerts, J.; Slaets, L.; Viale, G.; Delaloge, S.; Pierga, J.-Y.; Brain, E.; Causeret, S.; DeLorenzi, M.; et al. 70-Gene Signature as an Aid to Treatment Decisions in Early-Stage Breast Cancer. N. Engl. J. Med. 2016, 375, 717–729. [Google Scholar] [CrossRef]
- Birch, J.L.; Strathdee, K.; Gilmour, L.; Vallatos, A.; McDonald, L.; Kouzeli, A.; Vasan, R.; Qaisi, A.H.; Croft, D.R.; Crighton, D.; et al. A Novel Small-Molecule Inhibitor of MRCK Prevents Radiation-Driven Invasion in Glioblastoma. Cancer Res. 2018, 78, 6509–6522. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic Reprogramming of the Kinome in Response to Targeted MEK Inhibition in Triple-Negative Breast Cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Kurimchak, A.M.; Herrera-Montávez, C.; Brown, J.; Johnson, K.J.; Sodi, V.; Srivastava, N.; Kumar, V.; Deihimi, S.; O’Brien, S.; Peri, S.; et al. Functional Proteomics Interrogation of the Kinome Identifies MRCKA as a Therapeutic Target in High-Grade Serous Ovarian Carcinoma. Sci. Signal. 2020, 13, eaax8238. [Google Scholar] [CrossRef] [PubMed]
- Elkin, R.; Oh, J.H.; Liu, Y.L.; Selenica, P.; Weigelt, B.; Reis-Filho, J.S.; Zamarin, D.; Deasy, J.O.; Norton, L.; Levine, A.J.; et al. Geometric Network Analysis Provides Prognostic Information in Patients with High Grade Serous Carcinoma of the Ovary Treated with Immune Checkpoint Inhibitors. NPJ Genom. Med. 2021, 6, 99. [Google Scholar] [CrossRef] [PubMed]
- Manning-Geist, B.; Gordhandas, S.; Liu, Y.L.; Zhou, Q.; Iasonos, A.; Da Cruz Paula, A.; Mandelker, D.; Roche, K.L.; Zivanovic, O.; Maio, A.; et al. MAPK Pathway Genetic Alterations Are Associated with Prolonged Overall Survival in Low-Grade Serous Ovarian Carcinoma. Clin. Cancer Res. 2022, 28, 4456–4465. [Google Scholar] [CrossRef] [PubMed]
- Tan, I.; Lai, J.; Yong, J.; Li, S.F.; Leung, T. Chelerythrine Perturbs Lamellar Actomyosin Filaments by Selective Inhibition of Myotonic Dystrophy Kinase-Related Cdc42-Binding Kinase. FEBS Lett. 2011, 585, 1260–1268. [Google Scholar] [CrossRef]
- Lin, W.; Huang, J.; Yuan, Z.; Feng, S.; Xie, Y.; Ma, W. Protein Kinase C Inhibitor Chelerythrine Selectively Inhibits Proliferation of Triple-Negative Breast Cancer Cells. Sci. Rep. 2017, 7, 2022. [Google Scholar] [CrossRef]
- Herbert, J.M.; Augereau, J.M.; Gleye, J.; Maffrand, J.P. Chelerythrine Is a Potent and Specific Inhibitor of Protein Kinase C. Biochem. Biophys. Res. Commun. 1990, 172, 993–999. [Google Scholar] [CrossRef]
- Nan, J.; Du, Y.; Chen, X.; Bai, Q.; Wang, Y.; Zhang, X.; Zhu, N.; Zhang, J.; Hou, J.; Wang, Q.; et al. TPCA-1 Is a Direct Dual Inhibitor of STAT3 and NF-ΚB and Regresses Mutant EGFR-Associated Human Non–Small Cell Lung Cancers. Mol. Cancer Ther. 2014, 13, 617–629. [Google Scholar] [CrossRef]
- Amano, M.; Chihara, K.; Nakamura, N.; Kaneko, T.; Matsuura, Y.; Kaibuchi, K. The COOH Terminus of Rho-Kinase Negatively Regulates Rho-Kinase Activity. J. Biol. Chem. 1999, 274, 32418–32424. [Google Scholar] [CrossRef]
- Kale, V.P.; Hengst, J.A.; Desai, D.H.; Dick, T.E.; Choe, K.N.; Colledge, A.L.; Takahashi, Y.; Sung, S.-S.; Amin, S.G.; Yun, J.K. A Novel Selective Multikinase Inhibitor of ROCK and MRCK Effectively Blocks Cancer Cell Migration and Invasion. Cancer Lett. 2014, 354, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Unbekandt, M.; Croft, D.R.; Crighton, D.; Mezna, M.; McArthur, D.; McConnell, P.; Schüttelkopf, A.W.; Belshaw, S.; Pannifer, A.; Sime, M.; et al. A Novel Small-Molecule MRCK Inhibitor Blocks Cancer Cell Invasion. Cell Commun. Signal. 2014, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Seta, K.; Morisco, C.; Vatner, S.F.; Sadoshima, J. Chelerythrine Rapidly Induces Apoptosis through Generation of Reactive Oxygen Species in Cardiac Myocytes. J. Mol. Cell. Cardiol. 2001, 33, 1829–1848. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Bhowmik, D.; Suresh Kumar, G. The Benzophenanthridine Alkaloid Chelerythrine Binds to DNA by Intercalation: Photophysical Aspects and Thermodynamic Results of Iminium versus Alkanolamine Interaction. J. Photochem. Photobiol. B Biol. 2013, 129, 57–68. [Google Scholar] [CrossRef]
- Brunhofer, G.; Fallarero, A.; Karlsson, D.; Batista-Gonzalez, A.; Shinde, P.; Gopi Mohan, C.; Vuorela, P. Exploration of Natural Compounds as Sources of New Bifunctional Scaffolds Targeting Cholinesterases and Beta Amyloid Aggregation: The Case of Chelerythrine. Bioorg. Med. Chem. 2012, 20, 6669–6679. [Google Scholar] [CrossRef]
- Chan, S.L.; Lee, M.C.; Tan, K.O.; Yang, L.K.; Lee, A.S.; Flotow, H.; Fu, N.Y.; Butler, M.S.; Soejarto, D.D.; Buss, A.D.; et al. Identification of Chelerythrine as an Inhibitor of BclXL Function. J. Biol. Chem. 2003, 278, 20453–20456. [Google Scholar] [CrossRef]
- Bruneau, A.; Delaunay, J.-L.; Durand-Schneider, A.-M.; Vauthier, V.; Ben Saad, A.; Aoudjehane, L.; El Mourabit, H.; Morichon, R.; Falguières, T.; Gautheron, J.; et al. MRCK-Alpha and Its Effector Myosin II Regulatory Light Chain Bind ABCB4 and Regulate Its Membrane Expression. Cells 2022, 11, 617. [Google Scholar] [CrossRef]
- Zihni, C.; Vlassaks, E.; Terry, S.; Carlton, J.; Leung, T.K.C.; Olson, M.; Pichaud, F.; Balda, M.S.; Matter, K. An Apical MRCK-Driven Morphogenetic Pathway Controls Epithelial Polarity. Nat. Cell Biol. 2017, 19, 1049–1060. [Google Scholar] [CrossRef]
- Zihni, C.; Georgiadis, A.; Ramsden, C.M.; Sanchez-Heras, E.; Haas, A.J.; Nommiste, B.; Semenyuk, O.; Bainbridge, J.W.B.; Coffey, P.J.; Smith, A.J.; et al. Spatiotemporal Control of Actomyosin Contractility by MRCKβ Signaling Drives Phagocytosis. J. Cell Biol. 2022, 221, e202012042. [Google Scholar] [CrossRef]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and Mechanism of Action of Some Commonly Used Protein Kinase Inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruscetta, V.M.; Seaton, T.J.; Shakeel, A.; Vasconcelos, S.N.S.; Viirre, R.D.; Adler, M.J.; Olson, M.F. Opportunities and Challenges for the Development of MRCK Kinases Inhibitors as Potential Cancer Chemotherapeutics. Cells 2023, 12, 534. https://doi.org/10.3390/cells12040534
Ruscetta VM, Seaton TJ, Shakeel A, Vasconcelos SNS, Viirre RD, Adler MJ, Olson MF. Opportunities and Challenges for the Development of MRCK Kinases Inhibitors as Potential Cancer Chemotherapeutics. Cells. 2023; 12(4):534. https://doi.org/10.3390/cells12040534
Chicago/Turabian StyleRuscetta, Vanessa M., Taj J. Seaton, Aleen Shakeel, Stanley N. S. Vasconcelos, Russell D. Viirre, Marc J. Adler, and Michael F. Olson. 2023. "Opportunities and Challenges for the Development of MRCK Kinases Inhibitors as Potential Cancer Chemotherapeutics" Cells 12, no. 4: 534. https://doi.org/10.3390/cells12040534
APA StyleRuscetta, V. M., Seaton, T. J., Shakeel, A., Vasconcelos, S. N. S., Viirre, R. D., Adler, M. J., & Olson, M. F. (2023). Opportunities and Challenges for the Development of MRCK Kinases Inhibitors as Potential Cancer Chemotherapeutics. Cells, 12(4), 534. https://doi.org/10.3390/cells12040534