
Promotion of Lymphangiogenesis by Targeted Delivery of VEGF-C Improves Diabetic Wound Healing

 , , , , , and
, , , , , and 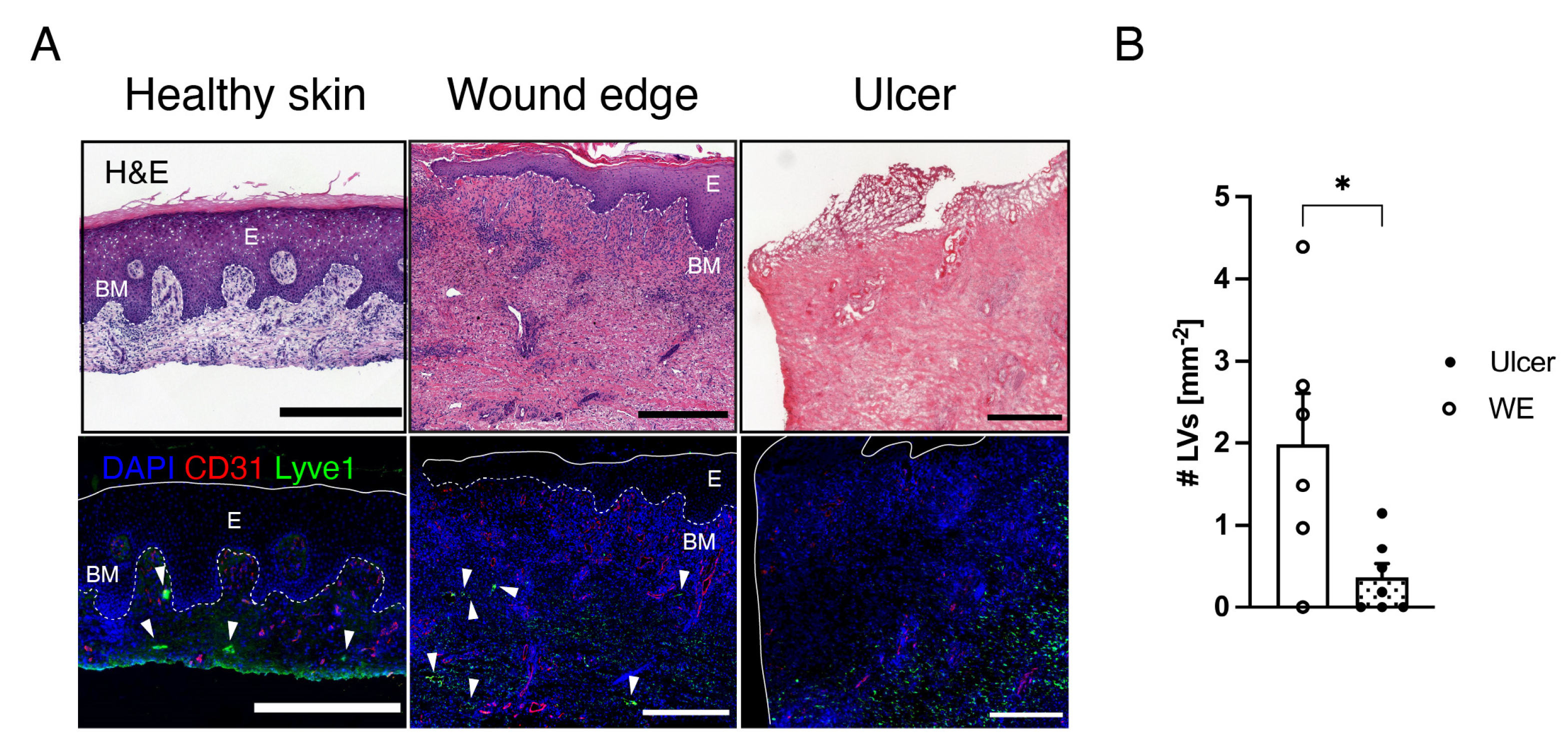{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Wounding Experiments
2.2. Antibody Treatment of Mice
2.3. Histology and Immunostaining
2.4. Image Acquisition and Analysis
2.5. RNA Isolation, Sequencing, and Bioinformatics
2.6. Human Wounds
2.7. Statistics
3. Results
3.1. Lymphatic Vessels Are Largely Absent in Human Chronic Ulcers
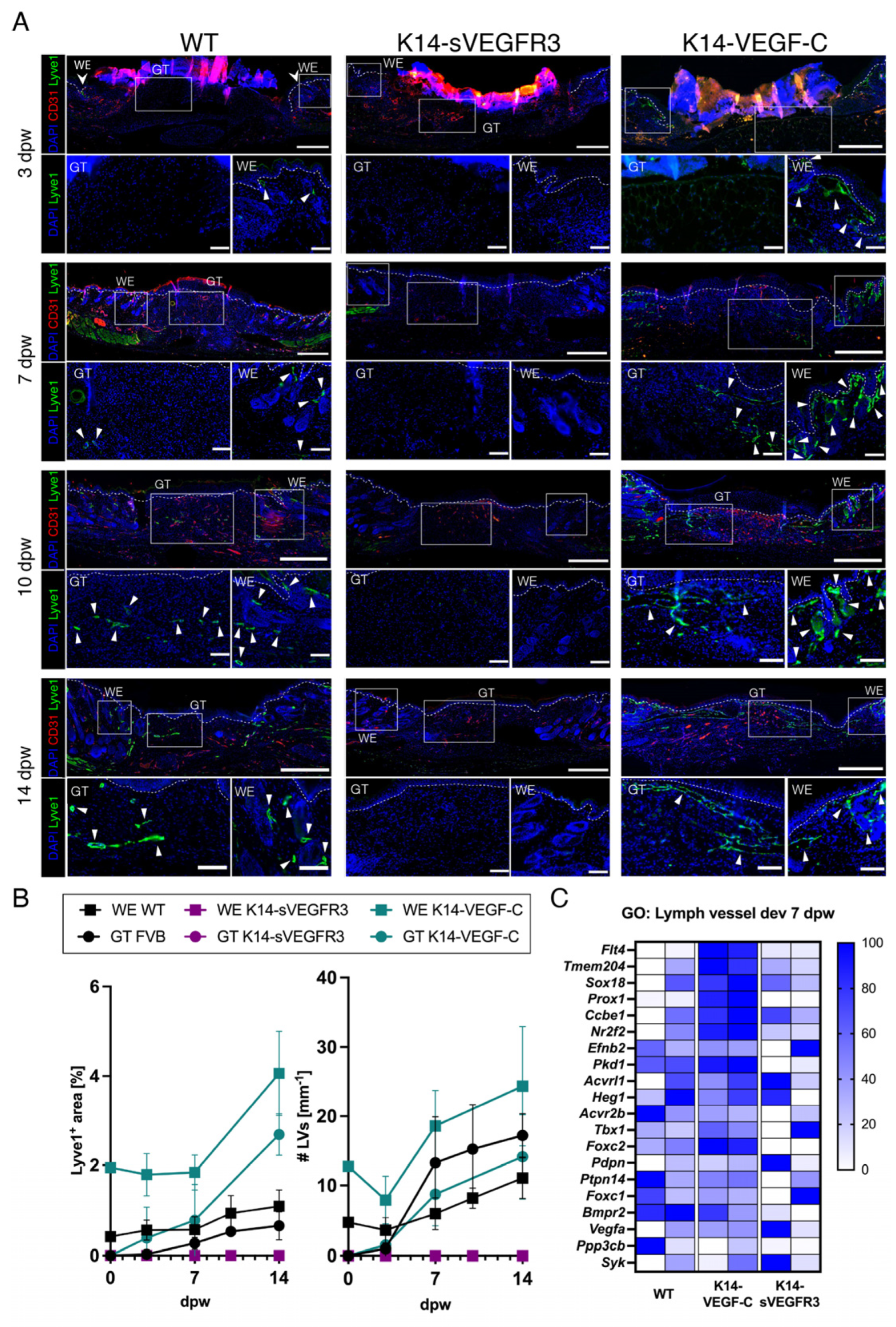
3.2. Lymphangiogenesis in Wounds of Wildtype, K14-VEGF-C, and K14-sVEGFR3 Mice
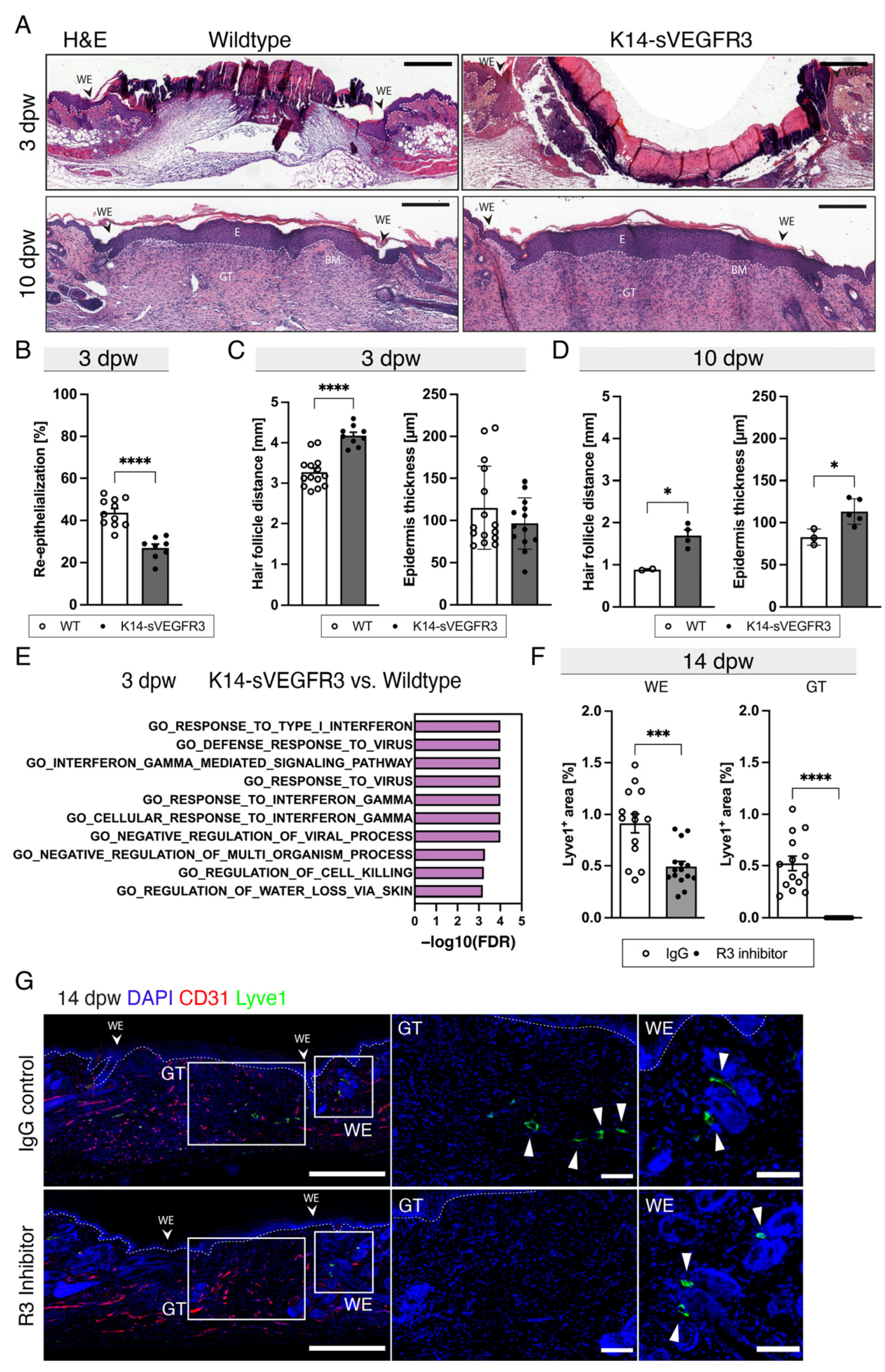
3.3. The Lack of Cutaneous Lymphatic Vessels Delays Wound Closure and Blocking VEGFR3 Completely Inhibits Wound Lymphangiogenesis
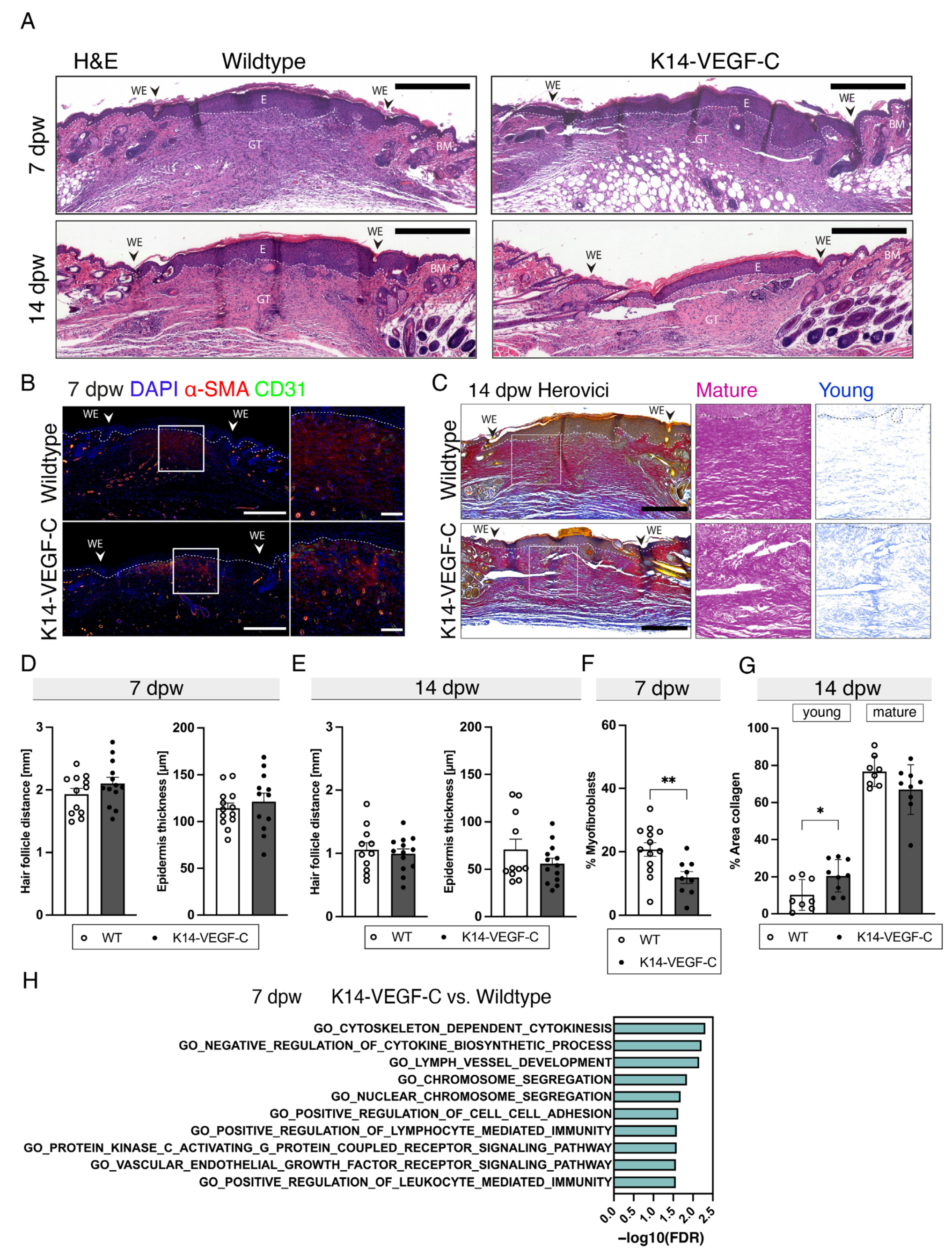
3.4. Reduced Myofibroblast Density and Delayed Collagen Maturation in Wounds of K14-VEGF-C Mice
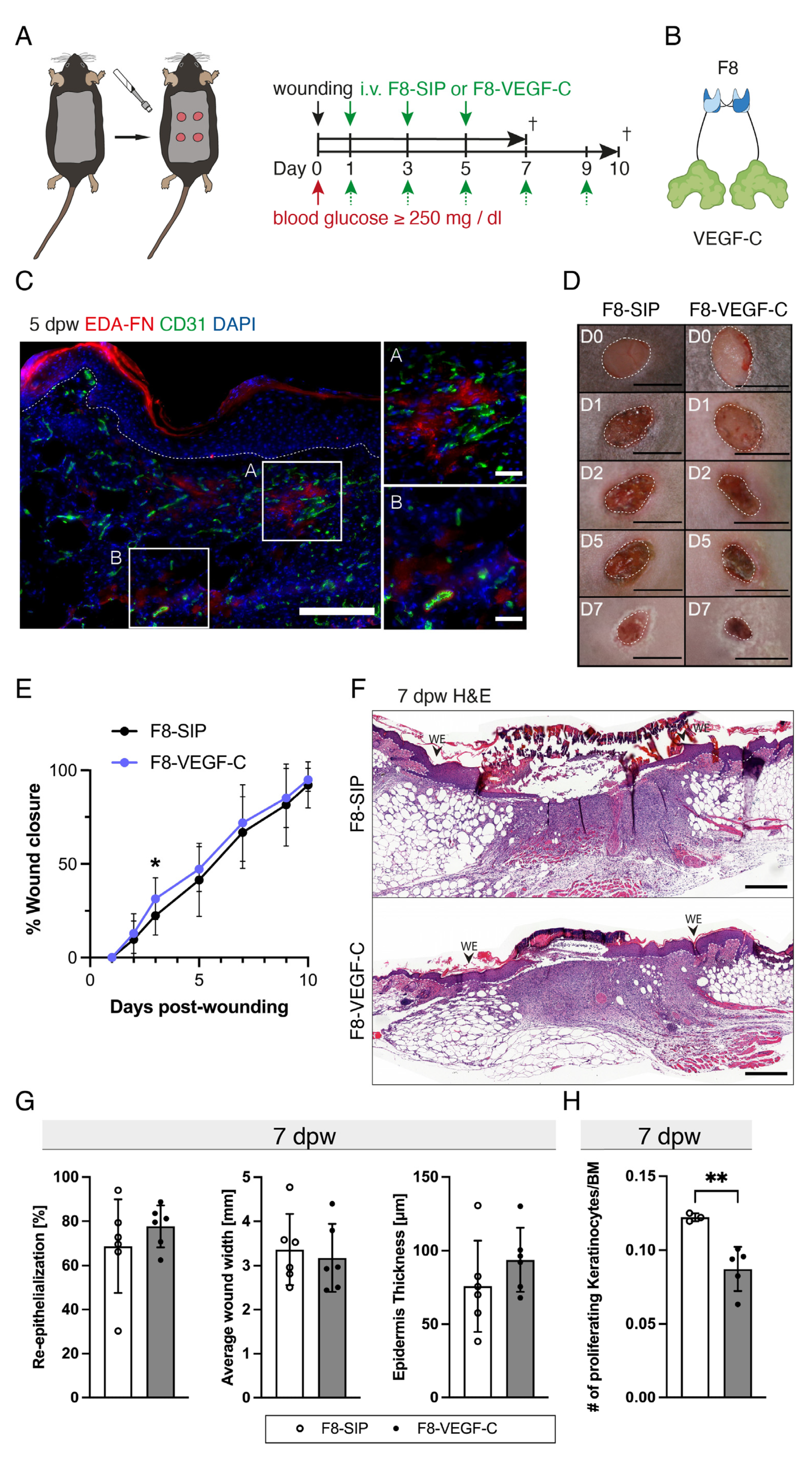
3.5. The F8–VEGF-C Fusion Protein Is Specific for Regenerating Tissue and Improves Diabetic Wound Healing
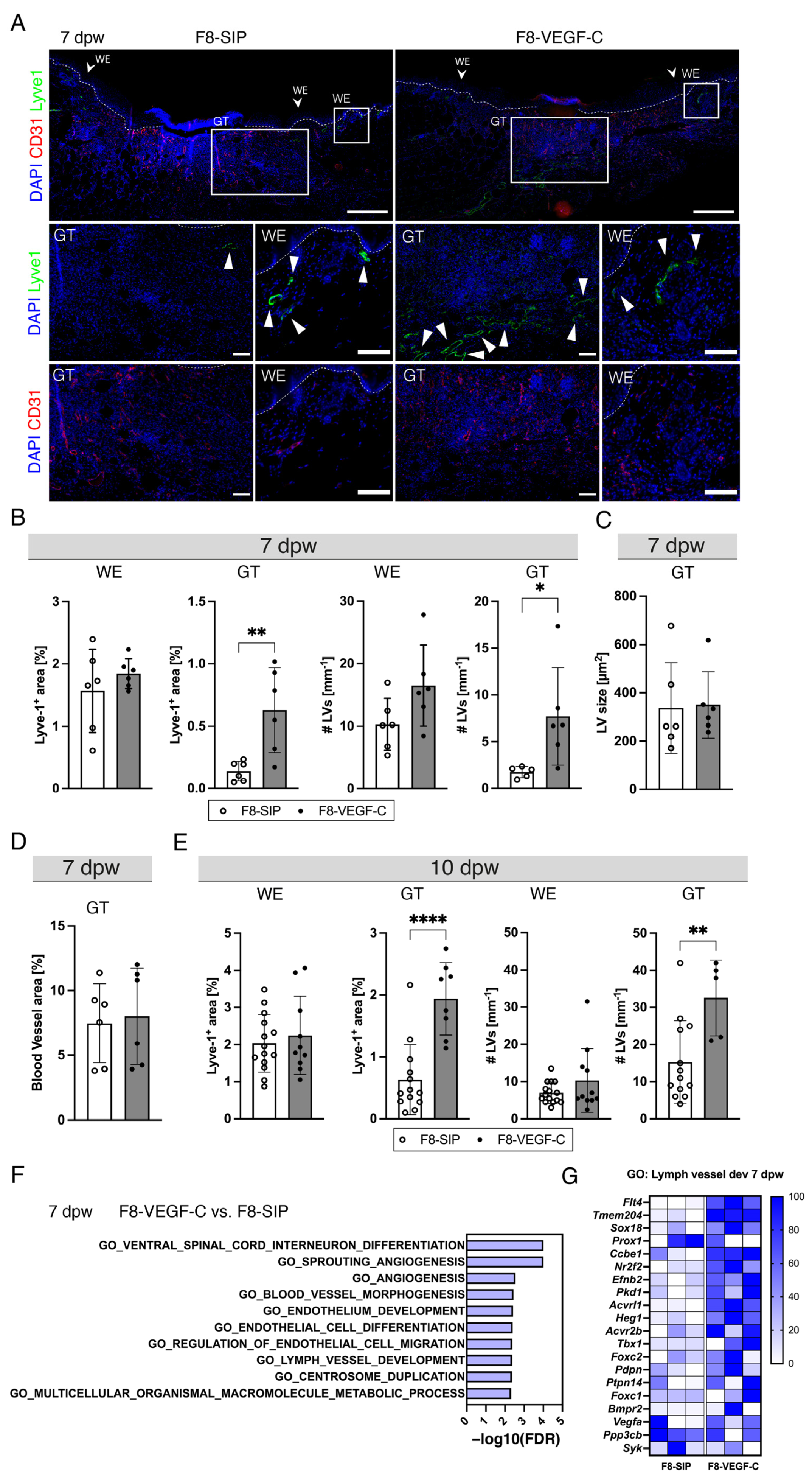
3.6. Targeted Delivery of VEGF-C Potently Induces Lymphangiogenesis in Diabetic Wounds
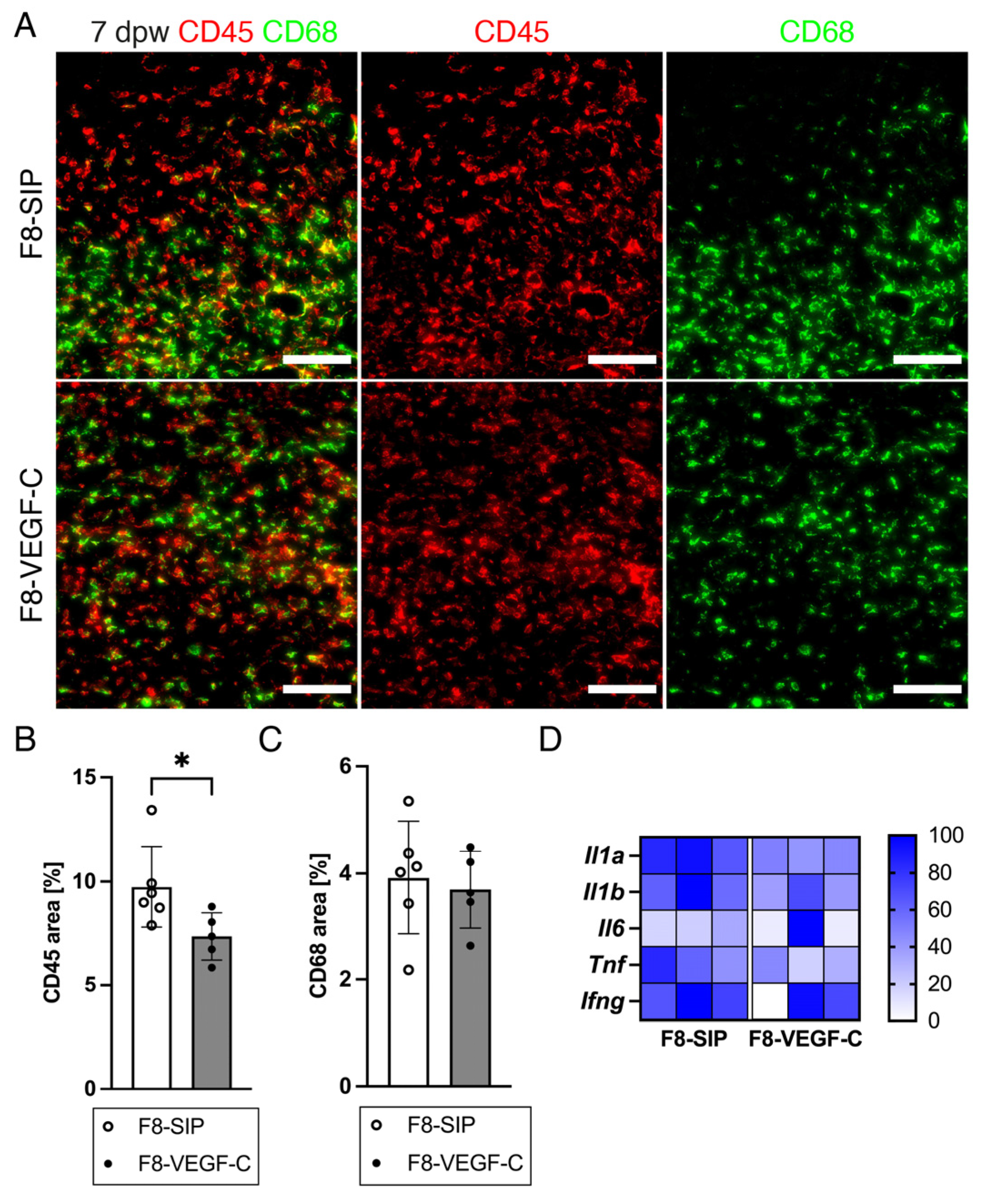
3.7. Targeted Delivery of VEGF-C Reduces Immune Cell Density in Diabetic Wounds
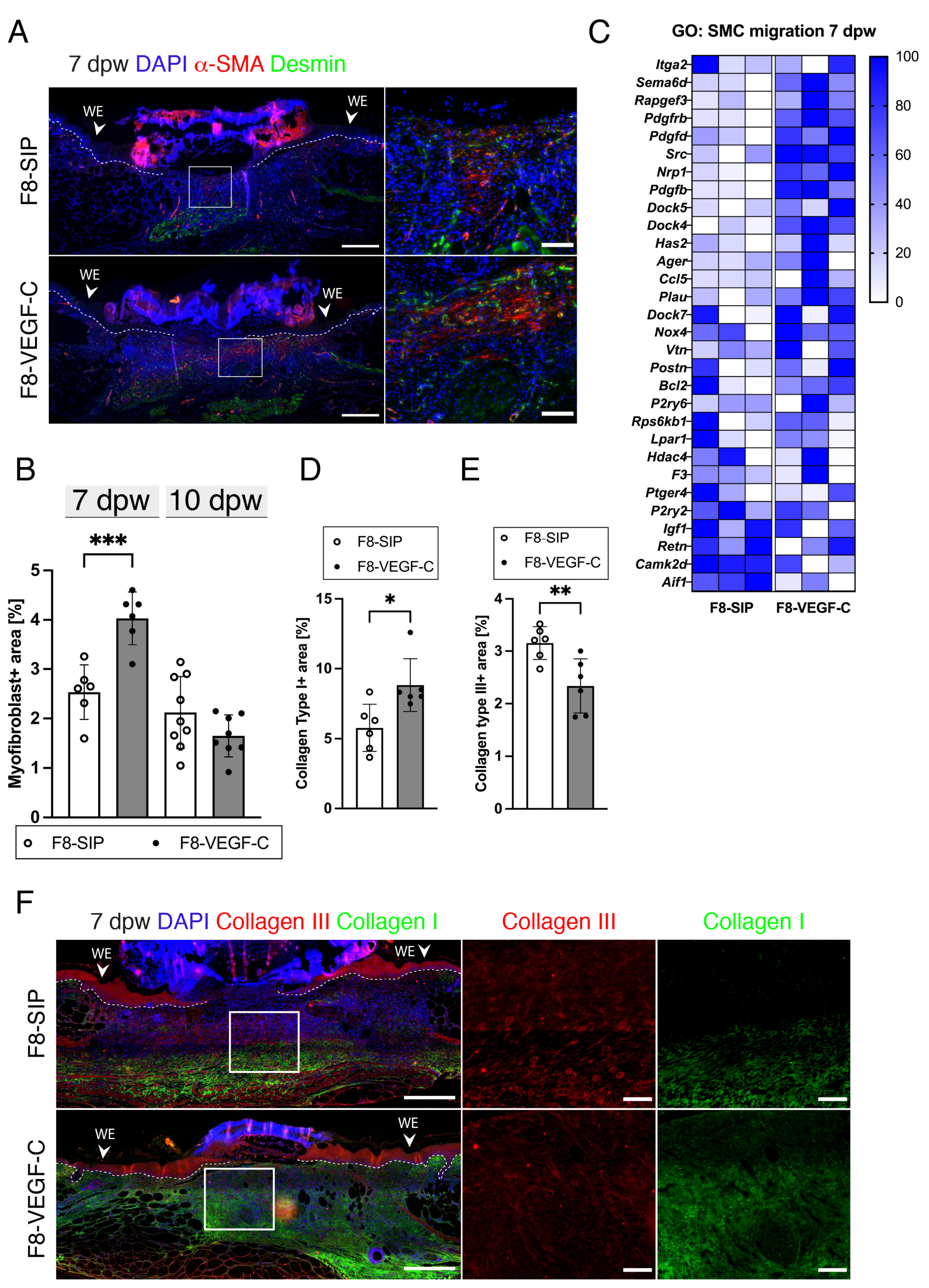
3.8. Targeted Delivery of VEGF-C Increases Myofibroblast Density and Collagen I Deposition in Diabetic Wounds
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef] [PubMed]
- Falanga, V. Wound healing and its impairment in the diabetic foot. Lancet 2005, 366, 1736–1743. [Google Scholar] [CrossRef]
- Cueni, L.N.; Detmar, M. Lymphatic Vascular System and Lymphangiogenesis. In Angiogenesis; Figg, W.D., Folkman, J., Eds.; Springer: Boston, MA, USA, 2008; Chapter 43; pp. 505–516. [Google Scholar]
- Dieterich, L.C.; Tacconi, C.; Ducoli, L.; Detmar, M. Lymphatic vessels in cancer. Physiol. Rev. 2022, 102, 1837–1879. [Google Scholar] [CrossRef] [PubMed]
- Petrova, T.V.; Koh, G.Y. Biological functions of lymphatic vessels. Science 2020, 369, eaax4063. [Google Scholar] [CrossRef] [PubMed]
- Oliver, G.; Kipnis, J.; Randolph, G.J.; Harvey, N.L. The Lymphatic Vasculature in the 21st Century: Novel Functional Roles in Homeostasis and Disease. Cell 2020, 182, 270–296. [Google Scholar] [CrossRef]
- Bull, R.H.; Gane, J.N.; Evans, J.E.; Joseph, A.E.; Mortimer, P.S. Abnormal lymph drainage in patients with chronic venous leg ulcers. J. Am. Acad. Dermatol. 1993, 28, 585–590. [Google Scholar] [CrossRef]
- Paavonen, K.; Puolakkainen, P.; Jussila, L.; Jahkola, T.; Alitalo, K. Vascular endothelial growth factor receptor-3 in lymphangiogenesis in wound healing. Am. J. Pathol. 2000, 156, 1499–1504. [Google Scholar] [CrossRef]
- Lymboussaki, A.; Partanen, T.A.; Olofsson, B.; Thomas-Crusells, J.; Fletcher, C.D.; de Waal, R.M.; Kaipainen, A.; Alitalo, K. Expression of the vascular endothelial growth factor C receptor VEGFR-3 in lymphatic endothelium of the skin and in vascular tumors. Am. J. Pathol. 1998, 153, 395–403. [Google Scholar] [CrossRef]
- Scallan, J.P.; Hill, M.A.; Davis, M.J. Lymphatic vascular integrity is disrupted in type 2 diabetes due to impaired nitric oxide signalling. Cardiovasc. Res. 2015, 107, 89–97. [Google Scholar] [CrossRef]
- Zhang, P.; Lu, J.; Jing, Y.; Tang, S.; Zhu, D.; Bi, Y. Global epidemiology of diabetic foot ulceration: A systematic review and meta-analysis. Ann. Med. 2017, 49, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.G.; Boulton, A.J.M.; Bus, S.A. Diabetic Foot Ulcers and Their Recurrence. N. Engl. J. Med. 2017, 376, 2367–2375. [Google Scholar] [CrossRef] [PubMed]
- Falanga, V. Classifications for wound bed preparation and stimulation of chronic wounds. Wound Repair Regen. 2000, 8, 347–352. [Google Scholar] [CrossRef]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: Molecular and cellular mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef]
- Grinnell, F.; Billingham, R.E.; Burgess, L. Distribution of fibronectin during wound healing in vivo. J. Investig. Dermatol. 1981, 76, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Beyeler, J.; Katsaros, C.; Chiquet, M. Impaired Contracture of 3D Collagen Constructs by Fibronectin-Deficient Murine Fibroblasts. Front. Physiol. 2019, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Bielefeld, K.A.; Amini-Nik, S.; Whetstone, H.; Poon, R.; Youn, A.; Wang, J.; Alman, B.A. Fibronectin and beta-catenin act in a regulatory loop in dermal fibroblasts to modulate cutaneous healing. J. Biol. Chem. 2011, 286, 27687–27697. [Google Scholar] [CrossRef]
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef]
- Midgley, A.C.; Rogers, M.; Hallett, M.B.; Clayton, A.; Bowen, T.; Phillips, A.O.; Steadman, R. Transforming growth factor-beta1 (TGF-beta1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J. Biol. Chem. 2013, 288, 14824–14838. [Google Scholar] [CrossRef]
- Wall, I.B.; Moseley, R.; Baird, D.M.; Kipling, D.; Giles, P.; Laffafian, I.; Price, P.E.; Thomas, D.W.; Stephens, P. Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. J. Investig. Dermatol. 2008, 128, 2526–2540. [Google Scholar] [CrossRef] [PubMed]
- Loots, M.A.; Lamme, E.N.; Mekkes, J.R.; Bos, J.D.; Middelkoop, E. Cultured fibroblasts from chronic diabetic wounds on the lower extremity (non-insulin-dependent diabetes mellitus) show disturbed proliferation. Arch. Dermatol. Res. 1999, 291, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Lerman, O.Z.; Galiano, R.D.; Armour, M.; Levine, J.P.; Gurtner, G.C. Cellular dysfunction in the diabetic fibroblast: Impairment in migration, vascular endothelial growth factor production, and response to hypoxia. Am. J. Pathol. 2003, 162, 303–312. [Google Scholar] [CrossRef]
- Wan, R.; Weissman, J.P.; Grundman, K.; Lang, L.; Grybowski, D.J.; Galiano, R.D. Diabetic wound healing: The impact of diabetes on myofibroblast activity and its potential therapeutic treatments. Wound Repair Regen. 2021, 29, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Junker, J.P.; Philip, J.; Kiwanuka, E.; Hackl, F.; Caterson, E.J.; Eriksson, E. Assessing quality of healing in skin: Review of available methods and devices. Wound Repair Regen. 2014, 22 (Suppl. 1), 2–10. [Google Scholar] [CrossRef]
- Corr, D.T.; Hart, D.A. Biomechanics of Scar Tissue and Uninjured Skin. Adv. Wound Care 2013, 2, 37–43. [Google Scholar] [CrossRef]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68–69, 81–93. [Google Scholar] [CrossRef]
- Schwager, S.; Detmar, M. Inflammation and Lymphatic Function. Front. Immunol. 2019, 10, 308. [Google Scholar] [CrossRef]
- Jeltsch, M.; Kaipainen, A.; Joukov, V.; Meng, X.; Lakso, M.; Rauvala, H.; Swartz, M.; Fukumura, D.; Jain, R.K.; Alitalo, K. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science 1997, 276, 1423–1425. [Google Scholar] [CrossRef]
- Skobe, M.; Hawighorst, T.; Jackson, D.G.; Prevo, R.; Janes, L.; Velasco, P.; Riccardi, L.; Alitalo, K.; Claffey, K.; Detmar, M. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat. Med. 2001, 7, 192–198. [Google Scholar] [CrossRef]
- Trompezinski, S.; Berthier-Vergnes, O.; Denis, A.; Schmitt, D.; Viac, J. Comparative expression of vascular endothelial growth factor family members, VEGF-B, -C and -D, by normal human keratinocytes and fibroblasts. Exp. Dermatol. 2004, 13, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhao, W.; Meng, W.; Liu, C.; Chen, Y.; Sun, Y. Vascular endothelial growth factor-C: Its unrevealed role in fibrogenesis. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H789–H796. [Google Scholar] [CrossRef] [PubMed]
- Huggenberger, R.; Ullmann, S.; Proulx, S.T.; Pytowski, B.; Alitalo, K.; Detmar, M. Stimulation of lymphangiogenesis via VEGFR-3 inhibits chronic skin inflammation. J. Exp. Med. 2010, 207, 2255–2269. [Google Scholar] [CrossRef]
- Huggenberger, R.; Siddiqui, S.S.; Brander, D.; Ullmann, S.; Zimmermann, K.; Antsiferova, M.; Werner, S.; Alitalo, K.; Detmar, M. An important role of lymphatic vessel activation in limiting acute inflammation. Blood 2011, 117, 4667–4678. [Google Scholar] [CrossRef]
- Guc, E.; Briquez, P.S.; Foretay, D.; Fankhauser, M.A.; Hubbell, J.A.; Kilarski, W.W.; Swartz, M.A. Local induction of lymphangiogenesis with engineered fibrin-binding VEGF-C promotes wound healing by increasing immune cell trafficking and matrix remodeling. Biomaterials 2017, 131, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Saaristo, A.; Tammela, T.; Timonen, J.; Yla-Herttuala, S.; Tukiainen, E.; Asko-Seljavaara, S.; Alitalo, K. Vascular endothelial growth factor-C gene therapy restores lymphatic flow across incision wounds. FASEB J. 2004, 18, 1707–1709. [Google Scholar] [CrossRef]
- Saaristo, A.; Tammela, T.; Farkkila, A.; Karkkainen, M.; Suominen, E.; Yla-Herttuala, S.; Alitalo, K. Vascular endothelial growth factor-C accelerates diabetic wound healing. Am. J. Pathol. 2006, 169, 1080–1087. [Google Scholar] [CrossRef]
- Maruyama, K.; Asai, J.; Ii, M.; Thorne, T.; Losordo, D.W.; D’Amore, P.A. Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am. J. Pathol. 2007, 170, 1178–1191. [Google Scholar] [CrossRef]
- Glinton, K.E.; Ma, W.; Lantz, C.; Grigoryeva, L.S.; DeBerge, M.; Liu, X.; Febbraio, M.; Kahn, M.; Oliver, G.; Thorp, E.B. Macrophage-produced VEGFC is induced by efferocytosis to ameliorate cardiac injury and inflammation. J. Clin. Investig. 2022, 132, e140685. [Google Scholar] [CrossRef]
- Schoppmann, S.F.; Birner, P.; Stockl, J.; Kalt, R.; Ullrich, R.; Caucig, C.; Kriehuber, E.; Nagy, K.; Alitalo, K.; Kerjaschki, D. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am. J. Pathol. 2002, 161, 947–956. [Google Scholar] [CrossRef]
- Villa, A.; Trachsel, E.; Kaspar, M.; Schliemann, C.; Sommavilla, R.; Rybak, J.N.; Rosli, C.; Borsi, L.; Neri, D. A high-affinity human monoclonal antibody specific to the alternatively spliced EDA domain of fibronectin efficiently targets tumor neo-vasculature in vivo. Int. J. Cancer 2008, 122, 2405–2413. [Google Scholar] [CrossRef] [PubMed]
- Schwager, S.; Renner, S.; Hemmerle, T.; Karaman, S.; Proulx, S.T.; Fetz, R.; Golding-Ochsenbein, A.M.; Probst, P.; Halin, C.; Neri, D.; et al. Antibody-mediated delivery of VEGF-C potently reduces chronic skin inflammation. JCI Insight 2018, 3, e124850. [Google Scholar] [CrossRef] [PubMed]
- Tacconi, C.; Schwager, S.; Cousin, N.; Bajic, D.; Sesartic, M.; Sundberg, J.P.; Neri, D.; Detmar, M. Antibody-Mediated Delivery of VEGFC Ameliorates Experimental Chronic Colitis. ACS Pharmacol. Transl. Sci. 2019, 2, 342–352. [Google Scholar] [CrossRef]
- Makinen, T.; Jussila, L.; Veikkola, T.; Karpanen, T.; Kettunen, M.I.; Pulkkanen, K.J.; Kauppinen, R.; Jackson, D.G.; Kubo, H.; Nishikawa, S.; et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat. Med. 2001, 7, 199–205. [Google Scholar] [CrossRef]
- Villa, A.; Lovato, V.; Bujak, E.; Wulhfard, S.; Pasche, N.; Neri, D. A novel synthetic naive human antibody library allows the isolation of antibodies against a new epitope of oncofetal fibronectin. MAbs 2011, 3, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Pytowski, B.; Goldman, J.; Persaud, K.; Wu, Y.; Witte, L.; Hicklin, D.J.; Skobe, M.; Boardman, K.C.; Swartz, M.A. Complete and specific inhibition of adult lymphatic regeneration by a novel VEGFR-3 neutralizing antibody. J. Natl. Cancer Inst. 2005, 97, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Jurisic, G.; Sundberg, J.P.; Detmar, M. Blockade of VEGF receptor-3 aggravates inflammatory bowel disease and lymphatic vessel enlargement. Inflamm. Bowel Dis. 2013, 19, 1983–1989. [Google Scholar] [CrossRef]
- Liu, Y.H.; Brunner, L.M.; Rebling, J.; Ben-Yehuda Greenwald, M.; Werner, S.; Detmar, M.; Razansky, D. Non-invasive longitudinal imaging of VEGF-induced microvascular alterations in skin wounds. Theranostics 2022, 12, 558–573. [Google Scholar] [CrossRef]
- Wietecha, M.S.; Pensalfini, M.; Cangkrama, M.; Muller, B.; Jin, J.; Brinckmann, J.; Mazza, E.; Werner, S. Activin-mediated alterations of the fibroblast transcriptome and matrisome control the biomechanical properties of skin wounds. Nat. Commun. 2020, 11, 2604. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Pensalfini, M.; Haertel, E.; Hopf, R.; Wietecha, M.; Werner, S.; Mazza, E. The mechanical fingerprint of murine excisional wounds. Acta Biomater. 2018, 65, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. Targeting the myofibroblast to improve wound healing. In Wound Healing Biomaterials; Woodhead Publishing: Cambridge, UK, 2016; pp. 69–100. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Falanga, V.; Isseroff, R.R.; Soulika, A.M.; Romanelli, M.; Margolis, D.; Kapp, S.; Granick, M.; Harding, K. Chronic wounds. Nat. Rev. Dis. Prim. 2022, 8, 50. [Google Scholar] [CrossRef]
- Cornelsen, H. Die chronische Wunde und das lymphologische Therapiekonzept. Phlebologie 2020, 49, 163–170. [Google Scholar] [CrossRef]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in Chronic Wounds. Int. J. Mol. Sci. 2016, 17, 2085. [Google Scholar] [CrossRef]
- D’Alessio, S.; Correale, C.; Tacconi, C.; Gandelli, A.; Pietrogrande, G.; Vetrano, S.; Genua, M.; Arena, V.; Spinelli, A.; Peyrin-Biroulet, L.; et al. VEGF-C-dependent stimulation of lymphatic function ameliorates experimental inflammatory bowel disease. J. Clin. Investig. 2014, 124, 3863–3878. [Google Scholar] [CrossRef] [PubMed]
- Kajiya, K.; Sawane, M.; Huggenberger, R.; Detmar, M. Activation of the VEGFR-3 pathway by VEGF-C attenuates UVB-induced edema formation and skin inflammation by promoting lymphangiogenesis. J. Investig. Dermatol. 2009, 129, 1292–1298. [Google Scholar] [CrossRef]
- Christiansen, A.J.; Dieterich, L.C.; Ohs, I.; Bachmann, S.B.; Bianchi, R.; Proulx, S.T.; Hollmen, M.; Aebischer, D.; Detmar, M. Lymphatic endothelial cells attenuate inflammation via suppression of dendritic cell maturation. Oncotarget 2016, 7, 39421–39435. [Google Scholar] [CrossRef]
- Thomas, S.N.; Rutkowski, J.M.; Pasquier, M.; Kuan, E.L.; Alitalo, K.; Randolph, G.J.; Swartz, M.A. Impaired humoral immunity and tolerance in K14-VEGFR-3-Ig mice that lack dermal lymphatic drainage. J. Immunol. 2012, 189, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Huggenberger, R.; Detmar, M. The cutaneous vascular system in chronic skin inflammation. J. Investig. Dermatol. Symp. Proc. 2011, 15, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Michaels, J.T.; Churgin, S.S.; Blechman, K.M.; Greives, M.R.; Aarabi, S.; Galiano, R.D.; Gurtner, G.C. db/db mice exhibit severe wound-healing impairments compared with other murine diabetic strains in a silicone-splinted excisional wound model. Wound Repair Regen. 2007, 15, 665–670. [Google Scholar] [CrossRef]
- Ffrench-Constant, C.; Van de Water, L.; Dvorak, H.F.; Hynes, R.O. Reappearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J. Cell Biol. 1989, 109, 903–914. [Google Scholar] [CrossRef]
- Patten, J.; Wang, K. Fibronectin in development and wound healing. Adv. Drug Deliv. Rev. 2021, 170, 353–368. [Google Scholar] [CrossRef]
- McDonald, J.A.; Kelley, D.G.; Broekelmann, T.J. Role of fibronectin in collagen deposition: Fab’ to the gelatin-binding domain of fibronectin inhibits both fibronectin and collagen organization in fibroblast extracellular matrix. J. Cell Biol. 1982, 92, 485–492. [Google Scholar] [CrossRef]
- Kurkinen, M.; Vaheri, A.; Roberts, P.J.; Stenman, S. Sequential appearance of fibronectin and collagen in experimental granulation tissue. Lab. Investig. 1980, 43, 47–51. [Google Scholar]
- Hemmerle, T.; Zgraggen, S.; Matasci, M.; Halin, C.; Detmar, M.; Neri, D. Antibody-mediated delivery of interleukin 4 to the neo-vasculature reduces chronic skin inflammation. J. Dermatol. Sci. 2014, 76, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Lemnitzer, P.; Gall, J.; Schwager, S.; Toska, A.; Yvan-Charvet, L.; Detmar, M.; Soehnlein, O. Arterial Delivery of VEGF-C Stabilizes Atherosclerotic Lesions. Circ. Res. 2021, 128, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Mathew-Steiner, S.S.; Roy, S.; Sen, C.K. Collagen in Wound Healing. Bioengineering 2021, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Tobalem, M.; Levigne, D.; Modarressi, A.; Atashi, F.; Villard, F.; Hinz, B.; Pittet-Cuenod, B. Hyperglycemia Interacts with Ischemia in a Synergistic Way on Wound Repair and Myofibroblast Differentiation. Plast. Reconstr. Surg. Glob. Open 2015, 3, e471. [Google Scholar] [CrossRef]
- Alizadeh, N.; Pepper, M.S.; Modarressi, A.; Alfo, K.; Schlaudraff, K.; Montandon, D.; Gabbiani, G.; Bochaton-Piallat, M.L.; Pittet, B. Persistent ischemia impairs myofibroblast development in wound granulation tissue: A new model of delayed wound healing. Wound Repair Regen. 2007, 15, 809–816. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunner, L.M.; He, Y.; Cousin, N.; Scholl, J.; Albin, L.K.; Schmucki, B.; Supersaxo, S.; Restivo, G.; Hafner, J.; Neri, D.; et al. Promotion of Lymphangiogenesis by Targeted Delivery of VEGF-C Improves Diabetic Wound Healing. Cells 2023, 12, 472. https://doi.org/10.3390/cells12030472
Brunner LM, He Y, Cousin N, Scholl J, Albin LK, Schmucki B, Supersaxo S, Restivo G, Hafner J, Neri D, et al. Promotion of Lymphangiogenesis by Targeted Delivery of VEGF-C Improves Diabetic Wound Healing. Cells. 2023; 12(3):472. https://doi.org/10.3390/cells12030472
Chicago/Turabian StyleBrunner, Lorenz M., Yuliang He, Nikola Cousin, Jeannette Scholl, Livia K. Albin, Bianca Schmucki, Sandrin Supersaxo, Gaetana Restivo, Jürg Hafner, Dario Neri, and et al. 2023. "Promotion of Lymphangiogenesis by Targeted Delivery of VEGF-C Improves Diabetic Wound Healing" Cells 12, no. 3: 472. https://doi.org/10.3390/cells12030472
APA StyleBrunner, L. M., He, Y., Cousin, N., Scholl, J., Albin, L. K., Schmucki, B., Supersaxo, S., Restivo, G., Hafner, J., Neri, D., Werner, S., & Detmar, M. (2023). Promotion of Lymphangiogenesis by Targeted Delivery of VEGF-C Improves Diabetic Wound Healing. Cells, 12(3), 472. https://doi.org/10.3390/cells12030472