
Synaptic Plasticity Abnormalities in Fetal Alcohol Spectrum Disorders


Abstract
1. Introduction
Molecular Basis of Neuronal Plasticity
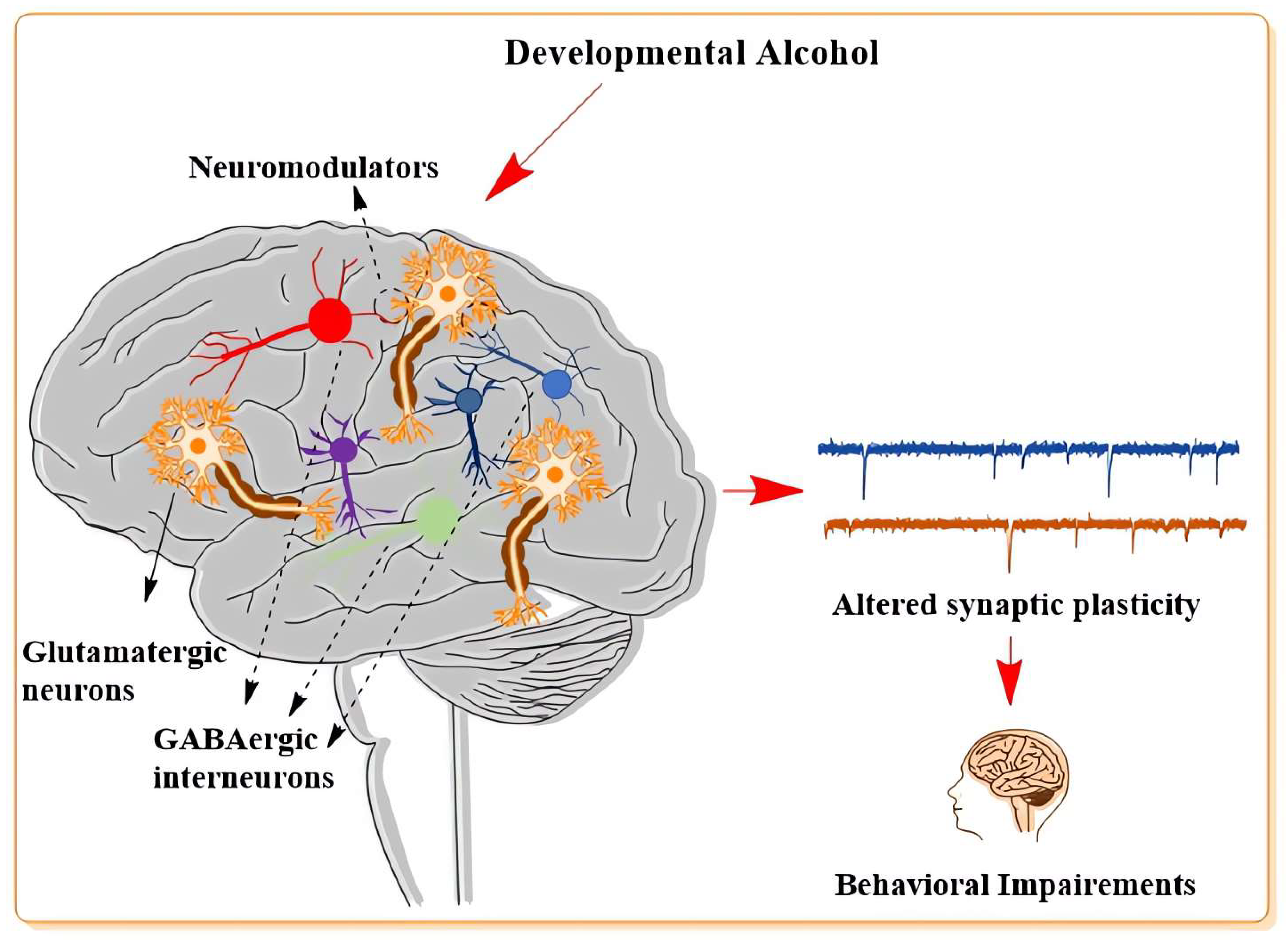
2. Influence of Developmental Alcohol on Neuronal Plasticity
2.1. Developmental Alcohol Influence on the Glutamatergic Neurotransmitter System
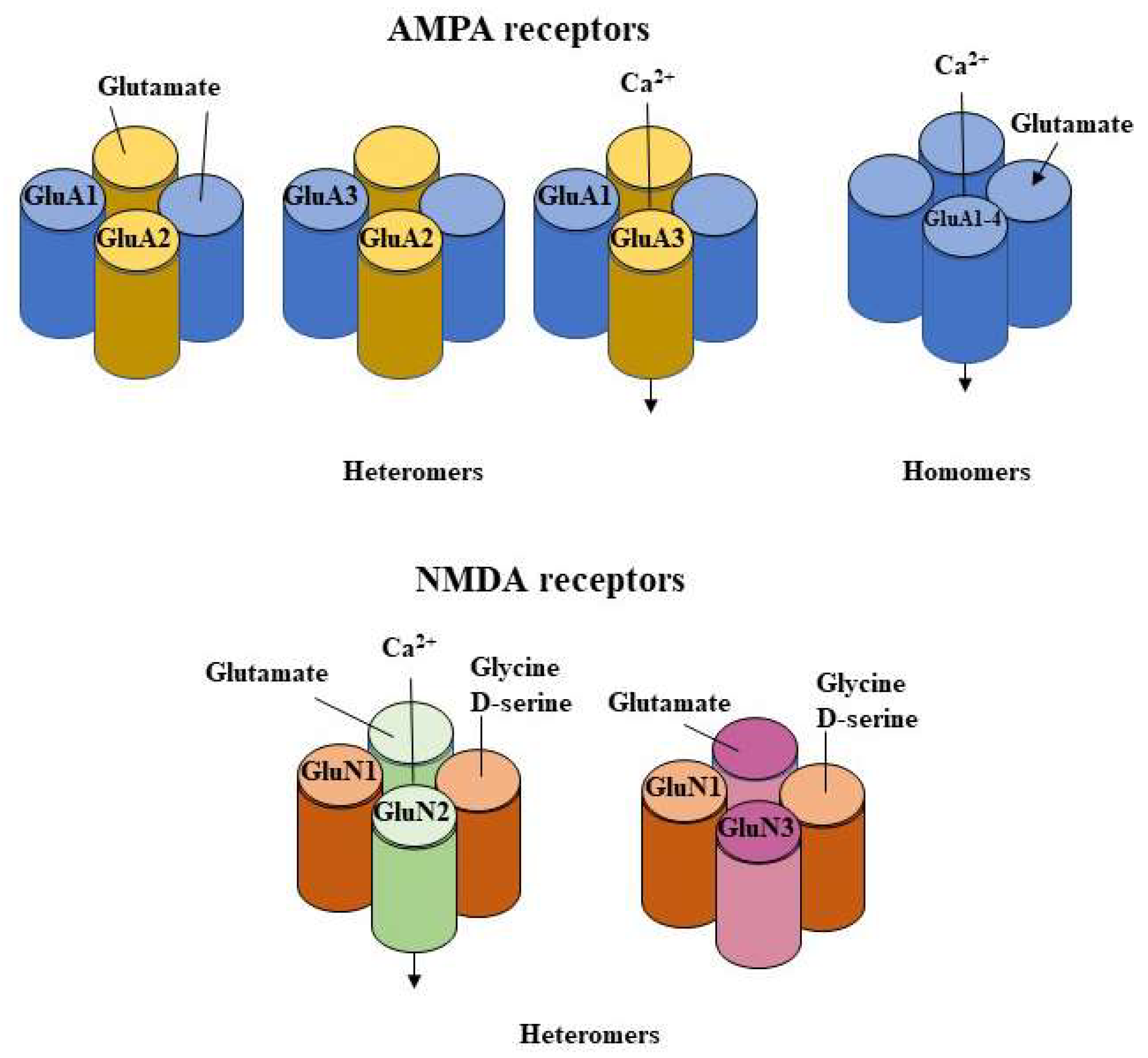
2.1.1. Influence on AMPA Function
2.1.2. Influence on AMPA Subunits Changes
2.1.3. Influence on NMDA Receptors Subunits

{kind=link}
{kind=link}
{kind=link}
| Alcohol Model | BAC | Tissue/Region | Effects |
|---|---|---|---|
| AMPAR function | |||
| PD4–9; (vapor, 3h/day) (W rats) | 330 mg/dL | NC | Reduced GluR1 (PD10) [76]. |
| GD2–67; Oral (4 g/kg) (Pigs) | 327 mg/dL, maternal | Cerebral CE | Reduced GluR3 (PD61) [77]. |
| GD3–21; (4 g/kg/day) (SD Rats) | 184 mg/dL, maternal | HP | Reduced AMPA-mediated mEPSCs [78]. |
| GD8–20; (3 & 4 g/kg/day) (SD Rats) | 281–341 mg/dL, maternal | VTA | Enhanced Depression of AMPAR-EPSCs [78] and increased GluR3 (PD35–60). |
| GD1–21 (Alc liquid diet) (LE Rats) | 60 mg/dL | HP (DG) | Decreased GluR1 (PD90) [106] |
| GD8–20 (3 &4 g/kg/day) (SD Rats) | 281–341 mg/dL, maternal | VTA | Enhanced Ca2+-permeable AMPAR& increased GluR3 (PD14–84) [79]. |
| GD10–11 (2 g/kg/day); GD12–18 (4 g/kg/day); PD4–5 (1.5 g/kg/day); PD6–14 (3 g/kg/day) (C57 mice) | 255 mg/dL on PD10 | mPFC DG | Increased AMPA receptor function (PD74) [80]. Increased GluR1, 2 & 3 (PD74) [81]. |
| GD1–PD21 (10,15 & 20% Alc sol) (C57 mice) | 80 mg/dL, maternal | HP | Reduced GluR1/GluR2 ratio (PD30–58) [82] |
| NMDAR function | |||
| GD1–21 (3.35% Alc liquid diet) (SD Rats) | 35–40 mg/dL, maternal | Dorsal HP | Reduced NMDA sensitive [3H]-glutamate binding (PD45) [95]. |
| GD2–62 (4 g/kg/day) (SD Rats) | 269 mg/dL, maternal | HP | Reduced [3H]MK-801 binding (GD63) [96]. |
| GD12–18 (5 g/kg/day) (SD Rats) | 143 mg/dL, maternal | HP & CE | Reduced [3H]MK-801 binding (PD20–22); Increased high-affinity state [97]. |
| PD4–14 (10.2 % Alc diet) (SD rats) | 429 mg/dL, PD8 | HP & CE | Reduced [3H]MK-801 binding (PD20–22); Increased high-affinity state [97]. |
| GD1–21 (6.5% Alc liquid diet (LE Rats) | 133 mg/dL, maternal | Barrel field CE | Reduced GluN1, GluN2A & B (PD90) [98]. |
| GD1–PD9 (10% Alc sol (SD Rats) | 86–112 mg/dL, maternal | HP | Increased GluN1 & GluN2D mRNA (PD7 & 14) [99]. Reduced GluN2C (PD1) [99]. Increased [3H]MK-801 binding [99]. |
| GD3–21 (20–36% Alc liquid diet (SD Rats) | 119–138 mg/dL, maternal | HP and FB | Reduced GluN2A and GluN2B (PD14) in FB; Reduced GluN2B (PD7) in HP [107]. |
| PD4–9 (6.2 g/kg/day) | 307 mg/dL (PD4–9) | CE | Increased GluN2A (PD21) [100]. |
| GD1–21 (20–36% Alc liquid diet (SD Rats) | 120–145 mg/dL, maternal | Cerebral CE | Reduced cell surface c2-terminal variant postsynaptic GluN2A (PD21) [101]. |
| GD (5 g/kg) & PD4–9 (6.2 g/kg) (SD Rats) | 95 mg/dL, maternal; 35 mg/dL, pup | HP | Increased GluN2A (PD10) [102]. |
| GD2–67; Oral (4 g/kg) (Pigs) | 327 mg/dL, maternal | WB | Reduced GluN2B (PD61) [77]. |
| GD8; 25% alc, i.p. (C57 mice) | ND | WB | Reduced GluN2B mRNA; increased GluN2A mRNA (P90) [103]. |
| GD2–63; Oral (4 g/kg) (Pigs) | 283 mg/dL, maternal | HP | Increased GluN1 mRNA (GD63) [104]. |
| GD1–PD14; 10% Alc sol (SD Rats) | 86–112 mg/dL, maternal | HP | Reduced GluN1 mRNA (PD60&90) [105]. |
2.1.4. Influence on NMDA Receptor Functions
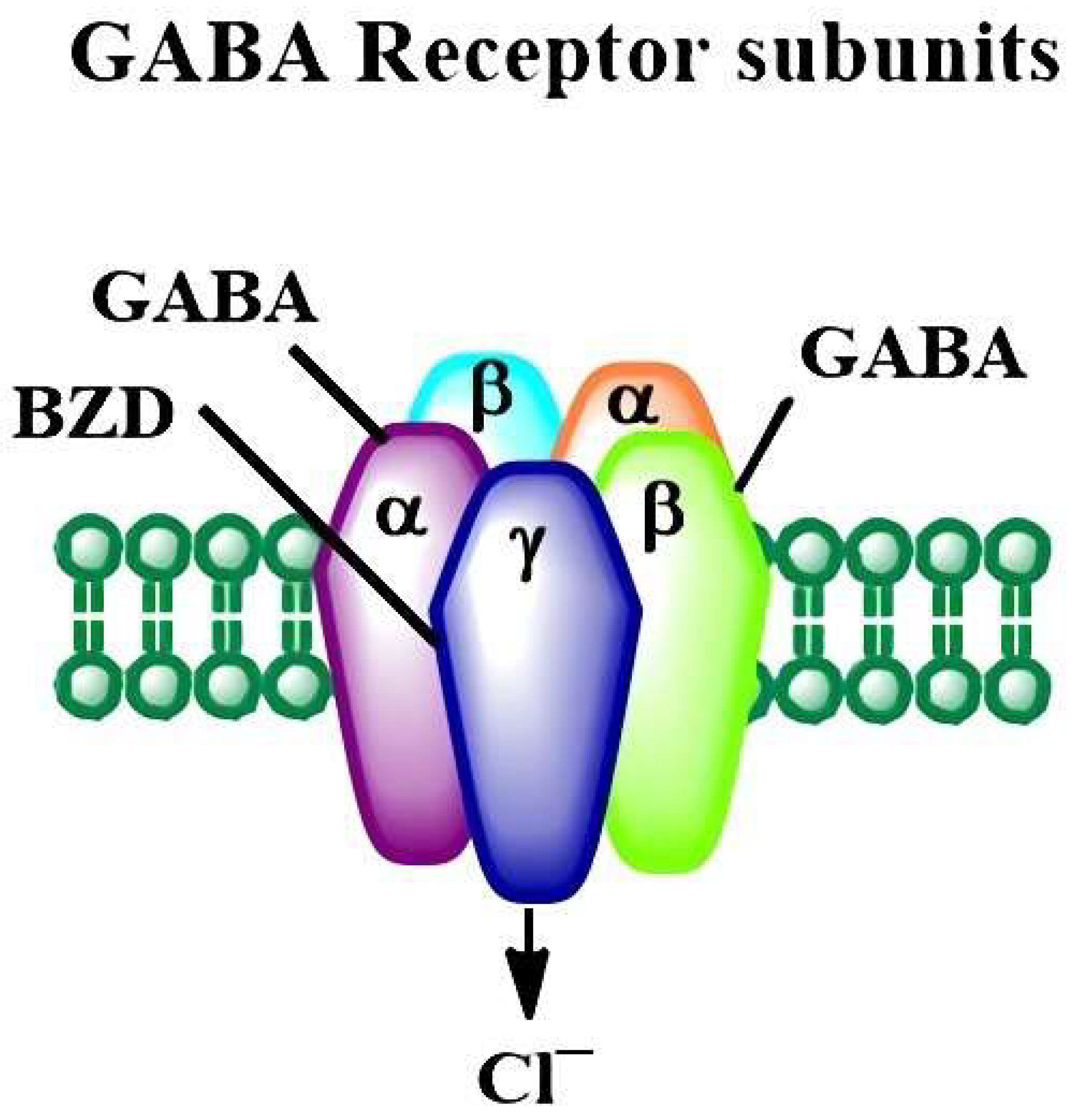
2.2. The Developmental Alcohol Effects on the GABAergic Neurotransmitter System
2.3. The Developmental Effects of Alcohol on Long-Term Synaptic Plasticity
2.4. The Influence of Developmental Alcohol on Modulators of Glutamatergic and GABAergic Neurotransmitter System
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- SAMHSA. Center for Behavioral Health Statistics and Quality. 2019 National Survey on Drug Use and Health. Table 2.18B—Alcohol Use in Past Year among Persons Aged 12 or Older, by Age Group and Demographic Characteristics: Percentages, 2018 and 2019; SAMHSA: Washington, DC, USA, 2019. [Google Scholar]
- Dejong, K.; Olyaei, A.; Lo, J. Alcohol Use in Pregnancy. Clin. Obstet. Gynecol. 2019, 62, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Denny, C.H.; Acero, C.; Naimi, T.; Kim, S. Consumption of Alcohol Beverages and Binge Drinking Among Pregnant Women Aged 18–44 Years—United States, 2015–2017. MMWR Morb. Mortal Wkly. Rep. 2019, 68, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Muggli, E.; Hearps, S.; Halliday, J.; Elliott, E.; Penington, A.; Thompson, D.; Spittle, A.; Forster, D.; Lewis, S.; Anderson, P. A data driven approach to identify trajectories of prenatal alcohol consumption in an Australian population-based cohort of pregnant women. Sci. Rep. 2022, 12, 4353. [Google Scholar] [CrossRef]
- Muggli, E.; O’Leary, C.; Donath, S.; Orsini, F.; Forster, D.; Anderson, P.; Lewis, S.; Nagle, C.; Craig, J.; Elliott, E.; et al. “Did you ever drink more?” A detailed description of pregnant women’s drinking patterns. BMC Public Health 2016, 16, 683. [Google Scholar] [CrossRef] [PubMed]
- Nykjaer, C.; Alwan, N.; Greenwood, D.; Simpson, N.; Hay, A.; White, K.; Cade, J. Maternal alcohol intake prior to and during pregnancy and risk of adverse birth outcomes: Evidence from a British cohort. J. Epidemiol. Community Health 2014, 68, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Denny, C.; Cheal, N.; Sniezek, J.; Kanny, D. Alcohol use and binge drinking among women of childbearing age—United States, 2011–2013. MMWR Morb. Mortal Wkly. Rep. 2015, 64, 1042–1046. [Google Scholar] [CrossRef]
- Jones, K.L.; Smith, D.; Ulleland, C.; Streissguth, P. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet 1973, 1, 1267–1271. [Google Scholar] [CrossRef]
- Jones, K.L.; Smith, D.W. Recognition of the fetal alcohol syndrome in early infancy. Lancet 1973, 2, 999–1001. [Google Scholar] [CrossRef]
- May, P.A.; Keaster, C.; Bozeman, R.; Goodover, J.; Blankenship, J.; Kalberg, W.; Buckley, D.; Brooks, M.; Hasken, J.; Gossage, J.; et al. Prevalence and characteristics of fetal alcohol syndrome and partial fetal alcohol syndrome in a Rocky Mountain Region City. Drug Alcohol Depend. 2015, 155, 118–127. [Google Scholar] [CrossRef]
- Alati, R.; Al Mamun, A.; Williams, G.; O’Callaghan, M.; Najman, J.; Bor, W. In utero alcohol exposure and prediction of alcohol disorders in early adulthood: A birth cohort study. Arch. Gen. Psychiatry 2006, 63, 1009–1016. [Google Scholar] [CrossRef]
- Autti-Ramo, I.; Fagerlund, A.; Ervalahti, N.; Loimu, L.; Korkman, M.; Hoyme, H. Fetal alcohol spectrum disorders in Finland: Clinical delineation of 77 older children and adolescents. Am. J. Med. Genet. A 2006, 140, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Ceccanti, M.; Spagnolo, P.A.; Tarani, L.; Attilia, M.L.; Chessa, L.; Mancinelli, R.; Stegagno, M.; Sasso, G.F.; Romeo, M.; Jones, K.; et al. Clinical delineation of fetal alcohol spectrum disorders (FASD) in Italian children: Comparison and contrast with other racial/ethnic groups and implications for diagnosis and prevention. Neurosci. Biobehav. Rev. 2007, 31, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Spohr, H.L.; Willms, J.; Steinhausen, H. Fetal alcohol spectrum disorders in young adulthood. J. Pediatr. 2007, 150, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Streissguth, A.P.; Aase, J.; Clarren, S.; Randels, S.; LaDue, R.; Smith, D. Fetal alcohol syndrome in adolescents and adults. JAMA 1991, 265, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; Delaney-Black, V.; Nordstrom, B. Fetal alcohol spectrum disorder. JAMA 2003, 290, 2996–2999. [Google Scholar] [CrossRef]
- May, P.A.; Baete, A.; Russo, J.; Elliott, A.; Blankenship, J.; Kalberg, W.; Buckley, D.; Brooks, M.; Hasken, J.; Abdul-Rahman, O.; et al. Prevalence and characteristics of fetal alcohol spectrum disorders. Pediatrics 2014, 134, 855–866. [Google Scholar] [CrossRef]
- May, P.A.; Chambers, C.; Kalberg, W.; Zellner, J.; Feldman, H.; Buckley, D.; Kopald, D.; Hasken, J.; Xu, R.; Honerkamp-Smith, G.; et al. Prevalence of Fetal Alcohol Spectrum Disorders in 4 US Communities. JAMA 2018, 319, 474–482. [Google Scholar] [CrossRef]
- Aragon, A.S.; Kalberg, W.; Buckley, D.; Barela-Scott, L.; Tabachnick, B.; May, P. Neuropsychological study of FASD in a sample of American Indian children: Processing simple versus complex information. Alcohol Clin. Exp. Res. 2008, 32, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Green, C.R.; Mihic, A.; Nikkel, S.; Stade, B.; Rasmussen, C.; Munoz, D.; Reynolds, J. Executive function deficits in children with fetal alcohol spectrum disorders (FASD) measured using the Cambridge Neuropsychological Tests Automated Battery (CANTAB). J. Child Psychol. Psychiatry 2009, 50, 688–697. [Google Scholar] [CrossRef]
- Mattson, S.N.; Crocker, N.; Nguyen, T. Fetal alcohol spectrum disorders: Neuropsychological and behavioral features. Neuropsychol. Rev. 2011, 21, 81–101. [Google Scholar] [CrossRef]
- Mattson, S.N.; Roesch, S.; Fagerlund, A.; Autti-Ramo, I.; Jones, K.; May, P.; Adnams, C.; Konovalova, V.; Riley, E. Toward a neurobehavioral profile of fetal alcohol spectrum disorders. Alcohol Clin. Exp. Res. 2010, 34, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- May, P.A.; Blankenship, J.; Marais, A.; Gossage, J.; Kalberg, W.; Joubert, B.; Cloete, M.; Barnard, R.; De Vries, M.; Hasken, J.; et al. Maternal alcohol consumption producing fetal alcohol spectrum disorders (FASD): Quantity, frequency, and timing of drinking. Drug Alcohol Depend. 2013, 133, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Lebel, C.; Mattson, S.; Riley, E.; Jones, K.; Adnams, C.; May, P.; Bookheimer, S.; O’Connor, M.J.; Narr, K.; Kan, E.; et al. A longitudinal study of the long-term consequences of drinking during pregnancy: Heavy in utero alcohol exposure disrupts the normal processes of brain development. J. Neurosci. 2012, 32, 15243–15251. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.L.; O’Brien, J.W.; Spadoni, A.; Tapert, S.; Jones, K.; Riley, E.; Mattson, S. A functional magnetic resonance imaging study of spatial working memory in children with prenatal alcohol exposure: Contribution of familial history of alcohol use disorders. Alcohol Clin. Exp. Res. 2013, 37, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Sood, B.; Delaney-Black, V.; Covington, C.; Nordstrom-Klee, B.; Ager, J.; Templin, T.; Janisse, J.; Martier, S.; Sokol, R. Prenatal alcohol exposure and childhood behavior at age 6 to 7 years: I. dose-response effect. Pediatrics 2001, 108, E34. [Google Scholar] [CrossRef]
- Streissguth, A.P.; Bookstein, F.; Sampson, P.; Barr, H. Neurobehavioral effects of prenatal alcohol: Part III. PLS analyses of neuropsychologic tests. Neurotoxicol. Teratol. 1989, 11, 493–507. [Google Scholar] [CrossRef]
- Guerri, C.; Bazinet, A.; Riley, E. Foetal Alcohol Spectrum Disorders and alterations in brain and behaviour. Alcohol Alcohol 2009, 44, 108–114. [Google Scholar] [CrossRef]
- Basavarajappa, B.S. Fetal Alcohol Spectrum Disorder: Potential Role of Endocannabinoids Signaling. Brain Sci. 2015, 5, 456–493. [Google Scholar] [CrossRef]
- Brady, M.L.; Diaz, M.; Iuso, A.; Everett, J.; Valenzuela, C.; Caldwell, K. Moderate prenatal alcohol exposure reduces plasticity and alters NMDA receptor subunit composition in the dentate gyrus. J. Neurosci. 2013, 33, 1062–1067. [Google Scholar] [CrossRef]
- Brolese, G.; Lunardi, P.; Broetto, N.; Engelke, D.; Lirio, F.; Batassini, C.; Tramontina, A.; Goncalves, C. Moderate prenatal alcohol exposure alters behavior and neuroglial parameters in adolescent rats. Behav. Brain Res. 2014, 269, 175–184. [Google Scholar] [CrossRef]
- Cui, Z.J.; Zhao, K.; Zhao, H.; Yu, D.; Niu, Y.; Zhang, J.; Deng, J. Prenatal alcohol exposure induces long-term changes in dendritic spines and synapses in the mouse visual cortex. Alcohol Alcohol 2010, 45, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Downing, C.; Johnson, T.; Larson, C.; Leakey, T.; Siegfried, R.; Rafferty, T.; Cooney, C. Subtle decreases in DNA methylation and gene expression at the mouse Igf2 locus following prenatal alcohol exposure: Effects of a methyl-supplemented diet. Alcohol 2011, 45, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Goggin, S.L.; Caldwell, K.; Cunningham, L.; Allan, A. Prenatal alcohol exposure alters p35, CDK5 and GSK3beta in the medial frontal cortex and hippocampus of adolescent mice. Toxicol. Rep. 2014, 1, 544–553. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Horn, K.H. Mechanisms of alcohol-induced damage to the developing nervous system. Alcohol Res. Health 2001, 25, 175–184. [Google Scholar]
- Lunde-Young, R.; Ramirez, J.; Naik, V.; Orzabal, M.; Lee, J.; Konganti, K.; Hillhouse, A.; Threadgill, D.; Ramadoss, J. Hippocampal transcriptome reveals novel targets of FASD pathogenesis. Brain Behav. 2019, 9, e01334. [Google Scholar] [CrossRef] [PubMed]
- Margret, C.P.; Li, C.; Chappell, T.; Elberger, A.; Matta, S.; Waters, R. Prenatal alcohol exposure delays the development of the cortical barrel field in neonatal rats. Exp. Brain Res. 2006, 172, 1–13. [Google Scholar] [CrossRef]
- Nagre, N.N.; Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Basavarajappa, B. CB1-receptor knockout neonatal mice are protected against ethanol-induced impairments of DNMT1, DNMT3A, and DNA methylation. J. Neurochem. 2015, 132, 429–442. [Google Scholar] [CrossRef]
- Ozer, E.; Sarioglu, S.; Gure, A. Effects of prenatal ethanol exposure on neuronal migration, neuronogenesis and brain myelination in the mice brain. Clin. Neuropathol. 2000, 19, 21–25. [Google Scholar]
- Sari, Y. Activity-dependent neuroprotective protein-derived peptide, NAP, preventing alcohol-induced apoptosis in fetal brain of C57BL/6 mouse. Neuroscience 2009, 158, 1426–1435. [Google Scholar] [CrossRef]
- Schaffner, S.L.; Lussier, A.; Baker, J.; Goldowitz, D.; Hamre, K.; Kobor, M. Neonatal Alcohol Exposure in Mice Induces Select Differentiation- and Apoptosis-Related Chromatin Changes Both Independent of and Dependent on Sex. Front. Genet. 2020, 11, 35. [Google Scholar] [CrossRef]
- Siler-Marsiglio, K.I.; Madorsky, I.; Pan, Q.; Paiva, M.; Neeley, A.; Shaw, G.; Heaton, M. Effects of acute ethanol exposure on regulatory mechanisms of Bcl-2-associated apoptosis promoter, bad, in neonatal rat cerebellum: Differential effects during vulnerable and resistant developmental periods. Alcohol Clin. Exp. Res. 2006, 30, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Nagre, N.; Shivakumar, M.; Joshi, V.; Psychoyos, D.; Kutlar, A.; Umapathy, N.; Basavarajappa, B. CB1R-Mediated Activation of Caspase-3 Causes Epigenetic and Neurobehavioral Abnormalities in Postnatal Ethanol-Exposed Mice. Front. Mol. Neurosci. 2018, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Nagre, N.; Umapathy, N.; Pace, B.; Basavarajappa, B. Ethanol exposure induces neonatal neurodegeneration by enhancing CB1R Exon1 histone H4K8 acetylation and up-regulating CB1R function causing neurobehavioral abnormalities in adult mice. Int. J. Neuropsychopharmacol. 2015, 18, 1–15. [Google Scholar] [CrossRef]
- Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Xie, S.; Basavarajappa, B. Anandamide-CB1 Receptor Signaling Contributes to Postnatal Ethanol-Induced Neonatal Neurodegeneration, Adult Synaptic and Memory Deficits. J. Neuosci. 2013, 33, 6350–6366. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Shivakumar, M.; Umapathy, N.; Saito, M.; Mohan, P.; Kumar, A.; Nixonc, R.; Verin, A.; Psychoyos, D.; Basavarajappa, B. G9a-Mediated Histone Methylation Regulates Ethanol-Induced Neurodegeneration in the Neonatal Mouse Brain. Neurobiol. Dis. 2013, 54, 475–485. [Google Scholar] [CrossRef]
- Toso, L.; Roberson, R.; Woodard, J.; Abebe, D.; Spong, C. Prenatal alcohol exposure alters GABA(A)alpha5 expression: A mechanism of alcohol-induced learning dysfunction. Am. J. Obstet. Gynecol. 2006, 195, 522–527. [Google Scholar] [CrossRef]
- Zhang, W.; Peng, C.; Zheng, M.; Gao, W.; Zhu, J.; Lv, T.; Liu, L.; Liu, Z.; Li, H.; Xv, Y.; et al. Prenatal alcohol exposure causes the over-expression of DHAND and EHAND by increasing histone H3K14 acetylation in C57 BL/6 mice. Toxicol. Lett. 2014, 228, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Appelbaum, L.G.; Shenasa, M.; Stolz, L.; Daskalakis, Z. Synaptic plasticity and mental health: Methods, challenges and opportunities. Neuropsychopharmacology 2022, 48, 113–120. [Google Scholar] [CrossRef]
- Caporale, N.; Dan, Y. Spike timing-dependent plasticity: A Hebbian learning rule. Annu. Rev. Neurosci. 2008, 31, 25–46. [Google Scholar] [CrossRef]
- Markram, H.; Gerstner, W.; Sjostrom, P. A history of spike-timing-dependent plasticity. Front. Synaptic. Neurosci. 2011, 3, 4. [Google Scholar] [CrossRef]
- Markram, H.; Lubke, J.; Frotscher, M.; Sakmann, B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 1997, 275, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Froemke, R.C.; Dan, Y. Spike-timing-dependent synaptic modification induced by natural spike trains. Nature 2002, 416, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef]
- Chater, T.E.; Goda, Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell Neurosci. 2014, 8, 401. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, M.; Heinemann, S. Cloned glutamate receptors. Annu. Rev. Neurosci. 1994, 17, 31–108. [Google Scholar] [CrossRef] [PubMed]
- Seeburg, P.H. The TiPS/TINS lecture: The molecular biology of mammalian glutamate receptor channels. Trends Pharmacol. Sci. 1993, 14, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Dore, K.; Aow, J.; Malinow, R. The Emergence of NMDA Receptor Metabotropic Function: Insights from Imaging. Front. Synaptic. Neurosci. 2016, 8, 20. [Google Scholar] [CrossRef]
- Valbuena, S.; Lerma, J. Non-canonical Signaling, the Hidden Life of Ligand-Gated Ion Channels. Neuron 2016, 92, 316–329. [Google Scholar] [CrossRef]
- Lau, C.G.; Zukin, R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 413–426. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.; McBain, C.; Menniti, F.; Vance, K.; Ogden, K.; Hansen, K.; Yuan, H.; Myers, S.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Dildy-Mayfield, J.E.; Harris, R.A. Comparison of ethanol sensitivity of rat brain kainate, DL-alpha-amino-3-hydroxy-5-methyl-4-isoxalone proprionic acid and N-methyl-D-aspartate receptors expressed in Xenopus oocytes. J. Pharmacol. Exp. Ther. 1992, 262, 487–494. [Google Scholar] [PubMed]
- Wirkner, K.; Eberts, C.; Poelchen, W.; Allgaier, C.; Illes, P. Mechanism of inhibition by ethanol of NMDA and AMPA receptor channel functions in cultured rat cortical neurons. Naunyn Schmiedebergs Arch. Pharmacol. 2000, 362, 568–576. [Google Scholar] [PubMed]
- Moykkynen, T.; Korpi, E.; Lovinger, D. Ethanol inhibits alpha-amino-3-hydyroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor function in central nervous system neurons by stabilizing desensitization. J. Pharmacol. Exp. Ther. 2003, 306, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Mameli, M.; Zamudio, P.; Carta, M.; Valenzuela, C. Developmentally regulated actions of alcohol on hippocampal glutamatergic transmission. J. Neurosci. 2005, 25, 8027–8036. [Google Scholar] [CrossRef] [PubMed]
- Puglia, M.P.; Valenzuela, C.F. Ethanol acutely inhibits ionotropic glutamate receptor-mediated responses and long-term potentiation in the developing CA1 hippocampus. Alcohol Clin. Exp. Res. 2010, 34, 594–606. [Google Scholar] [CrossRef]
- Durand, G.M.; Kovalchuk, Y.; Konnerth, A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature 1996, 381, 71–75. [Google Scholar] [CrossRef]
- Molnar, E.; Pickard, L.; Duckworth, J. Developmental changes in ionotropic glutamate receptors: Lessons from hippocampal synapses. Neuroscientist 2002, 8, 143–153. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Ninan, I.; Arancio, O. Acute Ethanol Suppresses Glutamatergic Neurotransmission through Endocannabinoids in Hippocampal Neurons. J. Neurochem. 2008, 107, 1001–1013. [Google Scholar] [CrossRef]
- Twitchell, W.; Brown, S.; Mackie, K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 1997, 78, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Maldve, R.E.; Chen, X.; Zhang, T.; Morrisett, R. Ethanol selectively inhibits enhanced vesicular release at excitatory synapses: Real-time visualization in intact hippocampal slices. Alcohol Clin. Exp. Res. 2004, 28, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Joshi, V.; Subbanna, S.; Shivakumar, M.; Basavarajappa, B. CB1R regulates CDK5 signaling and epigenetically controls Rac1 expression contributing to neurobehavioral abnormalities in mice postnatally exposed to ethanol. Neuropsychopharmacology 2019, 44, 514–525. [Google Scholar] [CrossRef]
- Shivakumar, M.; Subbanna, S.; Joshi, V.; Basavarajappa, B. Postnatal Ethanol Exposure Activates HDAC-Mediated Histone Deacetylation, Impairs Synaptic Plasticity Gene Expression and Behavior in Mice. Int. J. Neuropsychopharmacol. 2020, 23, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, F.P.; Davidson, M.; Bedi, K.; Wilce, P. Neonatal ethanol exposure reduces AMPA but not NMDA receptor levels in the rat neocortex. Brain Res. Dev. Brain Res. 2002, 136, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, T.S.; Barnes, A.; Iqbal, U.; Bailey, C.; Reynolds, J.; Brien, J.; Valenzuela, C. Chronic prenatal ethanol exposure alters ionotropic glutamate receptor subunit protein levels in the adult guinea pig cerebral cortex. Alcohol Clin. Exp. Res. 2003, 27, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Vaglenova, J.; Pandiella, N.; Wijayawardhane, N.; Vaithianathan, T.; Birru, S.; Breese, C.; Suppiramaniam, V.; Randal, C. Aniracetam reversed learning and memory deficits following prenatal ethanol exposure by modulating functions of synaptic AMPA receptors. Neuropsychopharmacology 2008, 33, 1071–1083. [Google Scholar] [CrossRef]
- Hausknecht, K.; Haj-Dahmane, S.; Shen, Y.; Vezina, P.; Dlugos, C.; Shen, R. Excitatory synaptic function and plasticity is persistently altered in ventral tegmental area dopamine neurons after prenatal ethanol exposure. Neuropsychopharmacology 2015, 40, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Louth, E.L.; Bignell, W.; Taylor, C.; Bailey, C. Developmental Ethanol Exposure Leads to Long-Term Deficits in Attention and Its Underlying Prefrontal Circuitry. eNeuro 2016, 3, 1–21. [Google Scholar] [CrossRef]
- Takahashi, Y.; Yamashita, R.; Okano, H.; Takashima, K.; Ogawa, B.; Ojiro, R.; Tang, Q.; Ozawa, S.; Woo, G.; Yoshida, T.; et al. Aberrant neurogenesis and late onset suppression of synaptic plasticity as well as sustained neuroinflammation in the hippocampal dentate gyrus after developmental exposure to ethanol in rats. Toxicology 2021, 462, 152958. [Google Scholar] [CrossRef]
- Montagud-Romero, S.; Cantacorps, L.; Fernandez-Gomez, F.; Nunez, C.; Minarro, J.; Rodriguez-Arias, M.; Milanes, M.; Valverde, O. Unraveling the molecular mechanisms involved in alcohol intake and withdrawal in adolescent mice exposed to alcohol during early life stages. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 104, 110025. [Google Scholar] [CrossRef]
- Liu, S.J.; Savtchouk, I. Ca(2+) permeable AMPA receptors switch allegiances: Mechanisms and consequences. J. Physiol. 2012, 590, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Cull-Candy, S.G.; Leszkiewicz, D.N. Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004, 2004, re16. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Wollmuth, L.; Bowie, D.; Furukawa, H.; Menniti, F.; Sobolevsky, A.; Swanson, G.; Swanger, S.; Greger, I.; Nakagawa, T.; et al. Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol. Rev. 2021, 73, 298–487. [Google Scholar] [CrossRef] [PubMed]
- Blevins, T.; Mirshahi, T.; Chandler, L.; Woodward, J. Effects of acute and chronic ethanol exposure on heteromeric N-methyl-D-aspartate receptors expressed in HEK 293 cells. J. Neurochem. 1997, 69, 2345–2354. [Google Scholar] [CrossRef]
- Mirshahi, T.; Woodward, J.J. Ethanol sensitivity of heteromeric NMDA receptors: Effects of subunit assembly, glycine and NMDAR1 Mg(2+)-insensitive mutants. Neuropharmacology 1995, 34, 347–355. [Google Scholar] [CrossRef]
- Ariwodola, O.J.; Weiner, J.L. Ethanol potentiation of GABAergic synaptic transmission may be self-limiting: Role of presynaptic GABA(B) receptors. J. Neurosci. 2004, 24, 10679–10686. [Google Scholar] [CrossRef]
- Wright, J.M.; Peoples, R.; Weight, F. Single-channel and whole-cell analysis of ethanol inhibition of NMDA-activated currents in cultured mouse cortical and hippocampal neurons. Brain Res. 1996, 738, 249–256. [Google Scholar] [CrossRef]
- Xu, M.; Smothers, C.; Trudell, J.; Woodward, J. Ethanol inhibition of constitutively open N-methyl-D-aspartate receptors. J. Pharmacol. Exp. Ther. 2012, 340, 218–226. [Google Scholar] [CrossRef]
- Ren, H.; Zhao, Y.; Dwyer, D.; Peoples, R. Interactions among positions in the third and fourth membrane-associated domains at the intersubunit interface of the N-methyl-D-aspartate receptor forming sites of alcohol action. J. Biol. Chem. 2012, 287, 27302–27312. [Google Scholar] [CrossRef] [PubMed]
- Ronald, K.M.; Mirshahi, T.; Woodward, J. Ethanol inhibition of N-methyl-D-aspartate receptors is reduced by site-directed mutagenesis of a transmembrane domain phenylalanine residue. J. Biol. Chem. 2001, 276, 44729–44735. [Google Scholar] [CrossRef] [PubMed]
- Smothers, C.T.; Woodward, J.J. Effects of amino acid substitutions in transmembrane domains of the NR1 subunit on the ethanol inhibition of recombinant N-methyl-D-aspartate receptors. Alcohol Clin. Exp. Res. 2006, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.D.; Montano, C.; Otero, M.; Paxton, L. Prenatal ethanol exposure decreases hippocampal NMDA-sensitive [3H]-glutamate binding site density in 45-day-old rats. Alcohol 1991, 8, 193–201. [Google Scholar] [CrossRef]
- Abdollah, S.; Brien, J.F. Effect of chronic maternal ethanol administration on glutamate and N-methyl-D-aspartate binding sites in the hippocampus of the near-term fetal guinea pig. Alcohol 1995, 12, 377–382. [Google Scholar] [CrossRef]
- Diaz-Granados, J.L.; Spuhler-Phillips, K.; Lilliquist, M.; Amsel, A.; Leslie, S. Effects of prenatal and early postnatal ethanol exposure on [3H]MK-801 binding in rat cortex and hippocampus. Alcohol Clin. Exp. Res. 1997, 21, 874–881. [Google Scholar] [CrossRef]
- Rema, V.; Ebner, F.F. Effect of enriched environment rearing on impairments in cortical excitability and plasticity after prenatal alcohol exposure. J. Neurosci. 1999, 19, 10993–11006. [Google Scholar] [CrossRef]
- Naassila, M.; Daoust, M. Effect of prenatal and postnatal ethanol exposure on the developmental profile of mRNAs encoding NMDA receptor subunits in rat hippocampus. J. Neurochem. 2002, 80, 850–860. [Google Scholar] [CrossRef]
- Nixon, K.; Hughes, P.; Amsel, A.; Leslie, S. NMDA receptor subunit expression following early postnatal exposure to ethanol. Brain Res. Dev. Brain Res. 2002, 139, 295–299. [Google Scholar] [CrossRef]
- Honse, Y.; Nixon, K.; Browning, M.; Leslie, S. Cell surface expression of NR1 splice variants and NR2 subunits is modified by prenatal ethanol exposure. Neuroscience 2003, 122, 689–698. [Google Scholar] [CrossRef]
- Nixon, K.; Hughes, P.; Amsel, A.; Leslie, S. NMDA receptor subunit expression after combined prenatal and postnatal exposure to ethanol. Alcohol Clin. Exp. Res. 2004, 28, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Toso, L.; Poggi, S.; Abebe, D.; Roberson, R.; Dunlap, V.; Park, J.; Spong, C. N-methyl-D-aspartate subunit expression during mouse development altered by in utero alcohol exposure. Am. J. Obstet. Gynecol. 2005, 193, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, U.; Brien, J.; Kapoor, A.; Matthews, S.; Reynolds, J. Chronic prenatal ethanol exposure increases glucocorticoid-induced glutamate release in the hippocampus of the near-term foetal guinea pig. J. Neuroendocrinol. 2006, 18, 826–834. [Google Scholar] [CrossRef]
- Barbier, E.; Pierrefiche, O.; Vaudry, D.; Vaudry, H.; Daoust, M.; Naassila, M. Long-term alterations in vulnerability to addiction to drugs of abuse and in brain gene expression after early life ethanol exposure. Neuropharmacology 2008, 55, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Staples, M.C.; Porch, M.; Savage, D. Impact of combined prenatal ethanol and prenatal stress exposures on markers of activity-dependent synaptic plasticity in rat dentate gyrus. Alcohol 2014, 48, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.D.; Kim, Y.; Randall, P.; Leslie, S. Effect of prenatal ethanol exposure on the developmental profile of the NMDA receptor subunits in rat forebrain and hippocampus. Alcohol Clin. Exp. Res. 1998, 22, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Sickmann, H.M.; Patten, A.; Morch, K.; Sawchuk, S.; Zhang, C.; Parton, R.; Szlavik, L.; Christie, B. Prenatal ethanol exposure has sex-specific effects on hippocampal long-term potentiation. Hippocampus 2014, 24, 54–64. [Google Scholar] [CrossRef]
- Samudio-Ruiz, S.L.; Allan, A.; Sheema, S.; Caldwell, K. Hippocampal N-methyl-D-aspartate receptor subunit expression profiles in a mouse model of prenatal alcohol exposure. Alcohol Clin. Exp. Res. 2010, 34, 342–353. [Google Scholar] [CrossRef]
- Bird, C.W.; Candelaria-Cook, F.; Magcalas, C.; Davies, S.; Valenzuela, C.; Savage, D.; Hamilton, D. Moderate prenatal alcohol exposure enhances GluN2B containing NMDA receptor binding and ifenprodil sensitivity in rat agranular insular cortex. PLoS One 2015, 10, e0118721. [Google Scholar] [CrossRef]
- Kervern, M.; de Ferron, B.S.; Alaux-Cantin, S.; Fedorenko, O.; Antol, J.; Naassila, M.; Pierrefiche, O. Aberrant NMDA-dependent LTD after perinatal ethanol exposure in young adult rat hippocampus. Hippocampus 2015, 25, 912–923. [Google Scholar] [CrossRef]
- Zink, M.; Ferbert, T.; Frank, S.; Seufert, P.; Gebicke-Haerter, P.; Spanagel, R. Perinatal exposure to alcohol disturbs spatial learning and glutamate transmission-related gene expression in the adult hippocampus. Eur. J. Neurosci. 2011, 34, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Castellani, G.C.; Quinlan, E.; Bersani, F.; Cooper, L.; Shouval, H. A model of bidirectional synaptic plasticity: From signaling network to channel conductance. Learn Mem. 2005, 12, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.D.; Wilson, W.; Leslie, S. Effect of gestational ethanol exposure on the NMDA receptor complex in rat forebrain: From gene transcription to cell surface. Brain Res. Dev. Brain Res. 2001, 129, 135–145. [Google Scholar] [CrossRef]
- Krapivinsky, G.; Krapivinsky, L.; Manasian, Y.; Ivanov, A.; Tyzio, R.; Pellegrino, C.; Ben-Ari, Y.; Clapham, D.; Medina, I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 2003, 40, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Samudio-Ruiz, S.L.; Allan, A.; Valenzuela, C.; Perrone-Bizzozero, N.; Caldwell, K. Prenatal ethanol exposure persistently impairs NMDA receptor-dependent activation of extracellular signal-regulated kinase in the mouse dentate gyrus. J. Neurochem. 2009, 109, 1311–1323. [Google Scholar] [CrossRef]
- Sweatt, J.D. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr. Opin. Neurobiol. 2004, 14, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.M.; Selcher, J.; Petraitis, J.; Trzaskos, J.; Sweatt, J. The MAPK cascade is required for mammalian associative learning. Nat. Neurosci. 1998, 1, 602–609. [Google Scholar] [CrossRef] [PubMed]
- English, J.D.; Sweatt, J.D. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J. Biol. Chem. 1996, 271, 24329–24332. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.D.; Becher, M.; de la Torre, A.; Sutherland, R. Dose-dependent effects of prenatal ethanol exposure on synaptic plasticity and learning in mature offspring. Alcohol Clin. Exp. Res. 2002, 26, 1752–1758. [Google Scholar] [CrossRef]
- Sutherland, R.J.; McDonald, R.; Savage, D. Prenatal exposure to moderate levels of ethanol can have long-lasting effects on hippocampal synaptic plasticity in adult offspring. Hippocampus 1997, 7, 232–238. [Google Scholar] [CrossRef]
- Allan, A.M.; Chynoweth, J.; Tyler, L.; Caldwell, K. A mouse model of prenatal ethanol exposure using a voluntary drinking paradigm. Alcohol Clin. Exp. Res. 2003, 27, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Gardoni, F.; Mauceri, D.; Malinverno, M.; Polli, F.; Costa, C.; Tozzi, A.; Siliquini, S.; Picconi, B.; Cattabeni, F.; Calabresi, P.; et al. Decreased NR2B subunit synaptic levels cause impaired long-term potentiation but not long-term depression. J. Neurosci. 2009, 29, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Titterness, A.K.; Christie, B.R. Prenatal ethanol exposure enhances NMDAR-dependent long-term potentiation in the adolescent female dentate gyrus. Hippocampus 2012, 22, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Licheri, V.; Chandrasekaran, J.; Bird, C.; Valenzuela, C.; Brigman, J. Sex-specific effect of prenatal alcohol exposure on N-methyl-D-aspartate receptor function in orbitofrontal cortex pyramidal neurons of mice. Alcohol Clin. Exp. Res. 2021, 45, 1994–2005. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Briceno, W.; Estay, S.; de la Fuente-Ortega, E.; Gutierrez, C.; Sanchez, G.; Hidalgo, C.; Chavez, A.; Haeger, P. N-Methyl-d-Aspartate Receptor Modulation by Nicotinamide Adenine Dinucleotide Phosphate Oxidase Type 2 Drives Synaptic Plasticity and Spatial Memory Impairments in Rats Exposed Pre- and Postnatally to Ethanol. Antioxid. Redox. Signal. 2020, 32, 602–617. [Google Scholar] [CrossRef] [PubMed]
- Ieraci, A.; Herrera, D.G. Early Postnatal Ethanol Exposure in Mice Induces Sex-Dependent Memory Impairment and Reduction of Hippocampal NMDA-R2B Expression in Adulthood. Neuroscience 2020, 427, 105–115. [Google Scholar] [CrossRef]
- Goodfellow, M.J.; Abdulla, K.; Lindquist, D. Neonatal Ethanol Exposure Impairs Trace Fear Conditioning and Alters NMDA Receptor Subunit Expression in Adult Male and Female Rats. Alcohol Clin. Exp. Res. 2016, 40, 309–318. [Google Scholar] [CrossRef]
- Pressey, J.C.; de Saint-Rome, M.; Raveendran, V.; Woodin, M. Chloride transporters controlling neuronal excitability. Physiol. Rev. 2022, 103, 1095–1135. [Google Scholar] [CrossRef]
- Sigel, E.; Steinmann, M.E. Structure, function, and modulation of GABA(A) receptors. J. Biol. Chem. 2012, 287, 40224–40231. [Google Scholar] [CrossRef]
- Allan, A.M.; Wu, H.; Paxton, L.; Savage, D. Prenatal ethanol exposure alters the modulation of the gamma-aminobutyric acidA1 receptor-gated chloride ion channel in adult rat offspring. J. Pharmacol. Exp. Ther. 1998, 284, 250–257. [Google Scholar]
- Bailey, C.D.; Brien, J.; Reynolds, J. Chronic prenatal ethanol exposure increases GABA(A) receptor subunit protein expression in the adult guinea pig cerebral cortex. J. Neurosci. 2001, 21, 4381–4389. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, S.H.; Parrish, A.; Nahm, S.; Abbott, L.; McCool, B.; Frye, G. Effects of early postnatal ethanol intubation on GABAergic synaptic proteins. Brain Res. Dev. Brain Res. 2002, 138, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Sebe, J.Y.; Eggers, E.; Berger, A. Differential effects of ethanol on GABA(A) and glycine receptor-mediated synaptic currents in brain stem motoneurons. J. Neurophysiol. 2003, 90, 870–875. [Google Scholar] [CrossRef]
- Spear, L.P.; Penson, J.; Linville, D. GABA and behavioral inhibition in the neonatal rat pup. Psychopharmacology 1986, 90, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Rawat, A.K. Developmental changes in the brain levels of neurotransmitters as influenced by maternal ethanol consumption in the rat. J. Neurochem. 1977, 28, 1175–1182. [Google Scholar] [CrossRef]
- Ledig, M.; Ciesielski, L.; Simler, S.; Lorentz, J.; Mandel, P. Effect of pre- and postnatal alcohol consumption on GABA levels of various brain regions in the rat offspring. Alcohol Alcohol 1988, 23, 63–67. [Google Scholar]
- Lee, K.; Kentroti, S.; Vernadakis, A. Differential sensitivity of cholinergic and GABAergic neurons in chick embryos treated intracerebrally with ethanol at 8 days of embryonic age. Neurochem. Res. 1992, 17, 565–569. [Google Scholar] [CrossRef]
- Kentroti, S.; Vernadakis, A. Effects of early in ovo administration of ethanol on expression of the GABAergic neuronal phenotype in the chick embryo. Brain Res. Dev. Brain Res. 1991, 61, 290–292. [Google Scholar] [CrossRef]
- Janiri, L.; Gobbi, G.; Persico, A.; Santarelli, M.; Minciacchi, D.; Tempesta, E. Alterations of neocortical neuronal responses to acetylcholine and GABA in rats born to alcohol-dependent mothers. Alcohol Alcohol 1994, 29, 611–619. [Google Scholar]
- Moore, D.B.; Ruygrok, A.; Walker, D.; Heaton, M. Effects of prenatal ethanol exposure on parvalbumin-expressing GABAergic neurons in the adult rat medial septum. Alcohol Clin. Exp. Res. 1997, 21, 849–856. [Google Scholar] [CrossRef]
- Moore, D.B.; Quintero, M.; Ruygrok, A.; Walker, D.; Heaton, M. Prenatal ethanol exposure reduces parvalbumin-immunoreactive GABAergic neuronal number in the adult rat cingulate cortex. Neurosci. Lett. 1998, 249, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.D.; Brien, J.; Reynolds, J. Chronic prenatal ethanol exposure alters the proportion of GABAergic neurons in layers II/III of the adult guinea pig somatosensory cortex. Neurotoxicol. Teratol. 2004, 26, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W. Effect of prenatal exposure to ethanol on glutamate and GABA immunoreactivity in macaque somatosensory and motor cortices: Critical timing of exposure. Neuroscience 2006, 138, 97–107. [Google Scholar] [CrossRef]
- Cuzon, V.C.; Yeh, P.; Yanagawa, Y.; Obata, K.; Yeh, H. Ethanol consumption during early pregnancy alters the disposition of tangentially migrating GABAergic interneurons in the fetal cortex. J. Neurosci. 2008, 28, 1854–1864. [Google Scholar] [CrossRef] [PubMed]
- Skorput, A.G.; Gupta, V.; Yeh, P.; Yeh, H. Persistent Interneuronopathy in the Prefrontal Cortex of Young Adult Offspring Exposed to Ethanol In Utero. J. Neurosci. 2015, 35, 10977–10988. [Google Scholar] [CrossRef]
- Skorput, A.G.; Lee, S.; Yeh, P.; Yeh, H. The NKCC1 antagonist bumetanide mitigates interneuronopathy associated with ethanol exposure in utero. Elife 2019, 8, e48648. [Google Scholar] [CrossRef]
- Skorput, A.G.; Yeh, H.H. Chronic Gestational Exposure to Ethanol Leads to Enduring Aberrances in Cortical Form and Function in the Medial Prefrontal Cortex. Alcohol Clin. Exp. Res. 2016, 40, 1479–1488. [Google Scholar] [CrossRef]
- Larsen, Z.H.; Chander, P.; Joyner, J.; Floruta, C.; Demeter, T.; Weick, J. Effects of Ethanol on Cellular Composition and Network Excitability of Human Pluripotent Stem Cell-Derived Neurons. Alcohol Clin. Exp. Res. 2016, 40, 2339–2350. [Google Scholar] [CrossRef]
- Kenton, J.A.; Ontiveros, T.; Bird, C.; Valenzuela, C.; Brigman, J. Moderate prenatal alcohol exposure alters the number and function of GABAergic interneurons in the murine orbitofrontal cortex. Alcohol 2020, 88, 33–41. [Google Scholar] [CrossRef]
- Marquardt, K.; Sigdel, R.; Caldwell, K.; Brigman, J. Prenatal ethanol exposure impairs executive function in mice into adulthood. Alcohol Clin. Exp. Res. 2014, 38, 2962–2968. [Google Scholar] [CrossRef]
- Granato, A. Altered organization of cortical interneurons in rats exposed to ethanol during neonatal life. Brain Res. 2006, 1069, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Nirgudkar, P.; Taylor, D.; Yanagawa, Y.; Valenzuela, C. Ethanol exposure during development reduces GABAergic/glycinergic neuron numbers and lobule volumes in the mouse cerebellar vermis. Neurosci. Lett. 2016, 632, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Bird, C.W.; Taylor, D.; Pinkowski, N.; Chavez, G.; Valenzuela, C. Long-term Reductions in the Population of GABAergic Interneurons in the Mouse Hippocampus following Developmental Ethanol Exposure. Neuroscience 2018, 383, 60–73. [Google Scholar] [CrossRef]
- Coleman, L.G., Jr.; Oguz, I.; Lee, J.; Styner, M.; Crews, F. Postnatal day 7 ethanol treatment causes persistent reductions in adult mouse brain volume and cortical neurons with sex specific effects on neurogenesis. Alcohol 2012, 46, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Sadrian, B.; Lopez-Guzman, M.; Wilson, D.; Saito, M. Distinct neurobehavioral dysfunction based on the timing of developmental binge-like alcohol exposure. Neuroscience 2014, 280, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Smiley, J.; Hui, M.; Masiello, K.; Betz, J.; Ilina, M.; Saito, M.; Wilson, D. Neonatal Ethanol Disturbs the Normal Maturation of Parvalbumin Interneurons Surrounded by Subsets of Perineuronal Nets in the Cerebral Cortex: Partial Reversal by Lithium. Cereb. Cortex. 2019, 29, 1383–1397. [Google Scholar] [CrossRef]
- Smiley, J.F.; Saito, M.; Bleiwas, C.; Masiello, K.; Ardekani, B.; Guilfoyle, D.; Gerum, S.; Wilson, D.; Vadasz, C. Selective reduction of cerebral cortex GABA neurons in a late gestation model of fetal alcohol spectrum disorder. Alcohol 2015, 49, 571–580. [Google Scholar] [CrossRef]
- Bird, C.W.; Chavez, G.; Barber, M.; Valenzuela, C. Enhancement of parvalbumin interneuron-mediated neurotransmission in the retrosplenial cortex of adolescent mice following third trimester-equivalent ethanol exposure. Sci. Rep. 2021, 11, 1716. [Google Scholar] [CrossRef]
- De Giorgio, A.; Comparini, S.; Intra, F.; Granato, A. Long-term alterations of striatal parvalbumin interneurons in a rat model of early exposure to alcohol. J. Neurodev. Disord. 2012, 4, 18. [Google Scholar] [CrossRef]
- Swartzwelder, H.S.; Farr, K.; Wilson, W.; Savage, D. Prenatal exposure to ethanol decreases physiological plasticity in the hippocampus of the adult rat. Alcohol 1988, 5, 121–124. [Google Scholar] [CrossRef]
- Varaschin, R.K.; Akers, K.; Rosenberg, M.; Hamilton, D.; Savage, D. Effects of the cognition-enhancing agent ABT-239 on fetal ethanol-induced deficits in dentate gyrus synaptic plasticity. J. Pharmacol. Exp. Ther. 2010, 334, 191–198. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Zhang, T. Spatial cognition and sexually dimorphic synaptic plasticity balance impairment in rats with chronic prenatal ethanol exposure. Behav. Brain Res. 2013, 256, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Patten, A.R.; Brocardo, P.; Sakiyama, C.; Wortman, R.; Noonan, A.; Gil-Mohapel, J.; Christie, B. Impairments in hippocampal synaptic plasticity following prenatal ethanol exposure are dependent on glutathione levels. Hippocampus 2013, 23, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Skinkle, K.; Hicks, T. Age-dependent, steroid-specific effects of oestrogen on long-term potentiation in rat hippocampal slices. J. Physiol. 1999, 515 Pt 1, 209–220. [Google Scholar] [CrossRef]
- Uban, K.A.; Herting, M.; Wozniak, J.; Sowell, E.; Cifasd. Sex differences in associations between white matter microstructure and gonadal hormones in children and adolescents with prenatal alcohol exposure. Psychoneuroendocrinology 2017, 83, 111–121. [Google Scholar] [CrossRef]
- Sliwowska, J.H.; Comeau, W.; Bodnar, T.; Ellis, L.; Weinberg, J. Prenatal Alcohol Exposure and Pair Feeding Differentially Impact Puberty and Reproductive Development in Female Rats: Role of the Kisspeptin System. Alcohol Clin. Exp. Res. 2016, 40, 2368–2376. [Google Scholar] [CrossRef]
- Richardson, D.P.; Byrnes, M.; Brien, J.; Reynolds, J.; Dringenberg, H. Impaired acquisition in the water maze and hippocampal long-term potentiation after chronic prenatal ethanol exposure in the guinea-pig. Eur. J. Neurosci. 2002, 16, 1593–1598. [Google Scholar] [CrossRef]
- Izumi, Y.; Kitabayashi, R.; Funatsu, M.; Izumi, M.; Yuede, C.; Hartman, R.; Wozniak, D.; Zorumski, C. A single day of ethanol exposure during development has persistent effects on bi-directional plasticity, N-methyl-D-aspartate receptor function and ethanol sensitivity. Neuroscience 2005, 136, 269–279. [Google Scholar] [CrossRef]
- Puglia, M.P.; Valenzuela, C.F. Repeated third trimester-equivalent ethanol exposure inhibits long-term potentiation in the hippocampal CA1 region of neonatal rats. Alcohol 2010, 44, 283–290. [Google Scholar] [CrossRef]
- Subbanna, S.; Basavarajappa, B.S. Pre-administration of G9a/GLP inhibitor during Synaptogenesis Prevents Postnatal Ethanol-induced LTP Deficits and Neurobehavioral Abnormalities in Adult Mice. Exp. Neurol. 2014, 261, 34–43. [Google Scholar] [CrossRef]
- Subbanna, S.; Basavarajappa, B.S. Binge-like Prenatal Ethanol Exposure Causes Impaired Cellular Differentiation in the Embryonic Forebrain and Synaptic and Behavioral Defects in Adult Mice. Brain Sci. 2022, 12, 793. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente-Ortega, E.; Plaza-Briceno, W.; Vargas-Robert, S.; Haeger, P. Prenatal Ethanol Exposure Misregulates Genes Involved in Iron Homeostasis Promoting a Maladaptation of Iron Dependent Hippocampal Synaptic Transmission and Plasticity. Front. Pharmacol. 2019, 10, 1312. [Google Scholar] [CrossRef] [PubMed]
- Helfer, J.L.; White, E.; Christie, B. Enhanced deficits in long-term potentiation in the adult dentate gyrus with 2nd trimester ethanol consumption. PLoS ONE 2012, 7, e51344. [Google Scholar] [CrossRef] [PubMed]
- Patten, A.R.; Brocardo, P.; Christie, B. Omega-3 supplementation can restore glutathione levels and prevent oxidative damage caused by prenatal ethanol exposure. J. Nutr. Biochem. 2013, 24, 760–769. [Google Scholar] [CrossRef]
- Varaschin, R.K.; Rosenberg, M.; Hamilton, D.; Savage, D. Differential effects of the histamine H(3) receptor agonist methimepip on dentate granule cell excitability, paired-pulse plasticity and long-term potentiation in prenatal alcohol-exposed rats. Alcohol Clin. Exp. Res. 2014, 38, 1902–1911. [Google Scholar] [CrossRef]
- Silvestre de Ferron, B.; Vilpoux, C.; Kervern, M.; Robert, A.; Antol, J.; Naassila, M.; Pierrefiche, O. Increase of KCC2 in hippocampal synaptic plasticity disturbances after perinatal ethanol exposure. Addict. Biol. 2017, 22, 1870–1882. [Google Scholar] [CrossRef]
- Fontaine, C.J.; Pinar, C.; Yang, W.; Pang, A.; Suesser, K.; Choi, J.; Christie, B. Impaired Bidirectional Synaptic Plasticity in Juvenile Offspring Following Prenatal Ethanol Exposure. Alcohol Clin. Exp. Res. 2019, 43, 2153–2166. [Google Scholar] [CrossRef]
- Grafe, E.L.; Wade, M.; Hodson, C.; Thomas, J.; Christie, B. Postnatal Choline Supplementation Rescues Deficits in Synaptic Plasticity Following Prenatal Ethanol Exposure. Nutrients 2022, 14, 2004. [Google Scholar] [CrossRef]
- Titterness, A.K.; Christie, B.R. Long-term depression in vivo: Effects of sex, stress, diet, and prenatal ethanol exposure. Hippocampus 2008, 18, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.J.; Wang, Y.; Liu, F. Direct receptor cross-talk can mediate the modulation of excitatory and inhibitory neurotransmission by dopamine. J. Mol. Neurosci. 2005, 26, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Lucchi, L.; Covelli, V.; Petkov, V.; Spano, P.; Trabucchi, M. Effects of ethanol, given during pregnancy, on the offspring dopaminergic system. Pharmacol. Biochem. Behav. 1983, 19, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Lucchi, L.; Covelli, V.; Spano, P.; Trabucchi, M. Acute ethanol administration during pregnancy: Effects on central dopaminergic transmission in rat offspring. Neurobehav. Toxicol. Teratol. 1984, 6, 19–21. [Google Scholar] [PubMed]
- Rathbun, W.; Druse, M.J. Dopamine, serotonin, and acid metabolites in brain regions from the developing offspring of ethanol-treated rats. J. Neurochem. 1985, 44, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Druse, M.J.; Tajuddin, N.; Kuo, A.; Connerty, M. Effects of in utero ethanol exposure on the developing dopaminergic system in rats. J. Neurosci. Res. 1990, 27, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Boggan, W.O.; Xu, W.; Shepherd, C.; Middaugh, L. Effects of prenatal ethanol exposure on dopamine systems in C57BL/6J mice. Neurotoxicol. Teratol. 1996, 18, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Nowak, P.; Dabrowska, J.; Bortel, A.; Izabela, B.; Kostrzewa, R.; Brus, R. Prenatal cadmium and ethanol increase amphetamine-evoked dopamine release in rat striatum. Neurotoxicol. Teratol. 2006, 28, 563–572. [Google Scholar] [CrossRef]
- Tattoli, M.; Cagiano, R.; Gaetani, S.; Ghiglieri, V.; Giustino, A.; Mereu, G.; Trabace, L.; Cuomo, V. Neurofunctional effects of developmental alcohol exposure in alcohol-preferring and alcohol-nonpreferring rats. Neuropsychopharmacology 2001, 24, 691–705. [Google Scholar] [CrossRef]
- Shen, R.Y.; Hannigan, J.; Chiodo, L. The effects of chronic amphetamine treatment on prenatal ethanol-induced changes in dopamine receptor function: Electrophysiological findings. J. Pharmacol. Exp. Ther. 1995, 274, 1054–1060. [Google Scholar]
- Shen, R.Y.; Chiodo, L.A. The effects of in utero ethanol administration on the electrophysiological activity of rat nigrostriatal dopaminergic neurons. Brain Res. 1993, 624, 216–222. [Google Scholar] [CrossRef]
- Xu, C.; Shen, R.Y. Amphetamine normalizes the electrical activity of dopamine neurons in the ventral tegmental area following prenatal ethanol exposure. J. Pharmacol. Exp. Ther. 2001, 297, 746–752. [Google Scholar]
- Choong, K.; Shen, R. Prenatal ethanol exposure alters the postnatal development of the spontaneous electrical activity of dopamine neurons in the ventral tegmental area. Neuroscience 2004, 126, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Haj-Dahmane, S.; Shen, R. Effects of prenatal ethanol exposure on the excitability of ventral tegmental area dopamine neurons in vitro. J. Pharmacol. Exp. Ther. 2006, 319, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Wang, S.; Zhu, X. Prenatal ethanol exposure alters synaptic plasticity in the dorsolateral striatum of rat offspring via changing the reactivity of dopamine receptor. PLoS One 2012, 7, e42443. [Google Scholar] [CrossRef] [PubMed]
- Kroner, S.; Rosenkranz, J.; Grace, A.; Barrionuevo, G. Dopamine modulates excitability of basolateral amygdala neurons in vitro. J. Neurophysiol. 2005, 93, 1598–1610. [Google Scholar] [CrossRef]
- Diaz, M.R.; Chappell, A.; Christian, D.; Anderson, N.; McCool, B. Dopamine D3-like receptors modulate anxiety-like behavior and regulate GABAergic transmission in the rat lateral/basolateral amygdala. Neuropsychopharmacology 2011, 36, 1090–1103. [Google Scholar] [CrossRef]
- Diaz, M.R.; Jotty, K.; Locke, J.; Jones, S.; Valenzuela, C. Moderate Alcohol Exposure during the Rat Equivalent to the Third Trimester of Human Pregnancy Alters Regulation of GABAA Receptor-Mediated Synaptic Transmission by Dopamine in the Basolateral Amygdala. Front. Pediatr. 2014, 2, 46. [Google Scholar] [CrossRef]
- Hausknecht, K.; Shen, Y.; Wang, R.; Haj-Dahmane, S.; Shen, R. Prenatal Ethanol Exposure Persistently Alters Endocannabinoid Signaling and Endocannabinoid-Mediated Excitatory Synaptic Plasticity in Ventral Tegmental Area Dopamine Neurons. J. Neurosci. 2017, 37, 5798–5808. [Google Scholar] [CrossRef]
- Cuzon Carlson, V.C.; Gremel, C.; Lovinger, D. Gestational alcohol exposure disrupts cognitive function and striatal circuits in adult offspring. Nat. Commun. 2020, 11, 2555. [Google Scholar] [CrossRef]
- Oubraim, S.; Wang, R.; Hausknecht, K.; Kaczocha, M.; Shen, R.; Haj-Dahmane, S. Prenatal ethanol exposure causes anxiety-like phenotype and alters synaptic nitric oxide and endocannabinoid signaling in dorsal raphe nucleus of adult male rats. Transl. Psychiatry 2022, 12, 440. [Google Scholar] [CrossRef]
- Druse, M.J.; Kuo, A.; Tajuddin, N. Effects of in utero ethanol exposure on the developing serotonergic system. Alcohol Clin. Exp. Res. 1991, 15, 678–684. [Google Scholar] [CrossRef]
- Sari, Y.; Powrozek, T.; Zhou, F. Alcohol deters the outgrowth of serotonergic neurons at midgestation. J. Biomed. Sci. 2001, 8, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.C.; Sari, Y.; Powrozek, T. Fetal alcohol exposure reduces serotonin innervation and compromises development of the forebrain along the serotonergic pathway. Alcohol Clin. Exp. Res. 2005, 29, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.C.; Sari, Y.; Zhang, J.; Goodlett, C.; Li, T. Prenatal alcohol exposure retards the migration and development of serotonin neurons in fetal C57BL mice. Brain Res. Dev. Brain Res. 2001, 126, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Lee, M.; Kim, H.; Sim, Y.; Shin, M.; Lee, S.; Yang, H.; Chang, H.; Lee, T.; Jang, M.; et al. Maternal ethanol administration inhibits 5-hydroxytryptamine synthesis and tryptophan hydroxylase expression in the dorsal raphe of rat offspring. Brain Dev. 2005, 27, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Madarnas, C.; Villalba, N.; Soriano, D.; Brusco, A. Anxious Behavior of Adult CD1 Mice Perinatally Exposed to Low Concentrations of Ethanol Correlates With Morphological Changes in Cingulate Cortex and Amygdala. Front. Behav. Neurosci. 2020, 14, 92. [Google Scholar] [CrossRef]
- Marsicano, G.; Lutz, B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur. J. Neurosci. 1999, 11, 4213–4225. [Google Scholar] [CrossRef]
- Soria-Gomez, E.; Bellocchio, L.; Reguero, L.; Lepousez, G.; Martin, C.; Bendahmane, M.; Ruehle, S.; Remmers, F.; Desprez, T.; Matias, I.; et al. The endocannabinoid system controls food intake via olfactory processes. Nat. Neurosci. 2014, 17, 407–415. [Google Scholar] [CrossRef]
- Ohno-Shosaku, T.; Maejima, T.; Kano, M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminal. Neuron 2001, 29, 729–738. [Google Scholar] [CrossRef]
- Wilson, R.I.; Nicoll, R.A. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 2001, 410, 588–592. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Joshi, V.; Shivakumar, M.; Subbanna, S. Distinct Functions of Endogenous Cannabinoid System in Alcohol Abuse Disorders. Br. J. Pharmacol. 2019, 176, 3085–3109. [Google Scholar] [CrossRef]
- Hoffman, A.F.; Hwang, E.; Lupica, C. Impairment of Synaptic Plasticity by Cannabis, Delta(9)-THC, and Synthetic Cannabinoids. Cold Spring Harb. Perspect. Med. 2021, 11, a039743. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, J.; Berrendero, F.; Hernandez, M.; Ramos, J. The endogenous cannabinoid system and brain development. Trends Neurosci. 2000, 23, 14–20. [Google Scholar] [CrossRef] [PubMed]
- de Salas-Quiroga, A.; Diaz-Alonso, J.; Garcia-Rincon, D.; Remmers, F.; Vega, D.; Gomez-Canas, M.; Lutz, B.; Guzman, M.; Galve-Roperh, I. Prenatal exposure to cannabinoids evokes long-lasting functional alterations by targeting CB1 receptors on developing cortical neurons. Proc. Natl. Acad. Sci. USA 2015, 112, 13693–13698. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, N.C.; Delespaul, P.; Henquet, C.; Bakoula, C.; Stefanis, C.; Van Os, J. Early adolescent cannabis exposure and positive and negative dimensions of psychosis. Addiction 2004, 99, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Jew, C.; Lu, H. Lasting impacts of prenatal cannabis exposure and the role of endogenous cannabinoids in the developing brain. Future Neurol. 2011, 6, 459–480. [Google Scholar] [CrossRef]
- Hansen, H.H.; Krutz, B.; Sifringer, M.; Stefovska, V.; Bittigau, P.; Pragst, F.; Marsicano, G.; Lutz, B.; Ikonomidou, C. Cannabinoids enhance susceptibility of immature brain to ethanol neurotoxicity. Ann. Neurol. 2008, 64, 42–52. [Google Scholar] [CrossRef]
- Noel, M.; Norris, E.; Strickland, S. Tissue plasminogen activator is required for the development of fetal alcohol syndrome in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 5069–5074. [Google Scholar] [CrossRef]
- Sadrian, B.; Subbanna, S.; Wilson, D.; Basavarajappa, B.; Saito, M. Lithium prevents long-term neural and behavioral pathology induced by early alcohol exposure. Neuroscience 2012, 206, 122–135. [Google Scholar] [CrossRef]
- Wilson, D.A.; Peterson, J.; Basavaraj, B.; Saito, M. Local and regional network function in behaviorally relevant cortical circuits of adult mice following postnatal alcohol exposure. Alcohol. Clin. Exp. Res. 2011, 35, 1974–1984. [Google Scholar] [CrossRef]
- Boa-Amponsem, O.; Zhang, C.; Mukhopadhyay, S.; Ardrey, I.; Cole, G. Ethanol and cannabinoids interact to alter behavior in a zebrafish fetal alcohol spectrum disorder model. Birth Defects Res. 2019, 111, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Fish, E.W.; Murdaugh, L.; Zhang, C.; Boschen, K.; Boa-Amponsem, O.; Mendoza-Romero, H.; Tarpley, M.; Chdid, L.; Mukhopadhyay, S.; Cole, G.; et al. Cannabinoids Exacerbate Alcohol Teratogenesis by a CB1-Hedgehog Interaction. Sci. Rep. 2019, 9, 16057. [Google Scholar] [CrossRef] [PubMed]
- McCorvy, J.D.; Roth, B.L. Structure and function of serotonin G protein-coupled receptors. Pharmacol. Ther. 2015, 150, 129–142. [Google Scholar] [CrossRef]
- Ugedo, L.; De Deurwaerdere, P. Serotonergic control of the glutamatergic neurons of the subthalamic nucleus. Prog. Brain Res. 2021, 261, 423–462. [Google Scholar] [PubMed]
- Rahaman, S.M.; Chowdhury, S.; Mukai, Y.; Ono, D.; Yamaguchi, H.; Yamanaka, A. Functional Interaction Between GABAergic Neurons in the Ventral Tegmental Area and Serotonergic Neurons in the Dorsal Raphe Nucleus. Front. Neurosci. 2022, 16, 877054. [Google Scholar] [CrossRef] [PubMed]
- Frederick, A.L.; Stanwood, G.D. Drugs, biogenic amine targets and the developing brain. Dev. Neurosci. 2009, 31, 7–22. [Google Scholar] [CrossRef]



| Alcohol Model | BAC | Tissue/Region | Effects |
|---|---|---|---|
| GD1–14 (Alc liquid diet) (A Rats) | ND | WB | Increased GABA (GD18&21) [76,136]. |
| GD1–PD14 (10% Alc sol, Oral (WS Rats) | 360 mg/dL (PD21) | THA, Pons, CBL, FC, OLB, AC | Increased GABA (FC, OLB, AC&Amy); Reduced GABA (THA, Pons, CBL&HP); (PD21) [137]. |
| E1–E3 (10 mg/50 µL) (Chick embryo) | 12 mM | WB | Increased GAD activity (E8) [138,139]. |
| GD15–18 (2.4 g/kg/4 times/day) (SD rats) | ND | Cortical neurons | Increased GABAergic responses (PD90) [140]. |
| GD1–20 (5% Alc liquid diet) (SD Rats) | 83 mg/dL, maternal | mFC&HP | Increased GABAA receptor-stimulated Cl-flux [131]. |
| GD0–21 (Alc liquid diet) (LE Rats) | 161 mg/dL, maternal | MS&ACC | Reduced GABAergic (PV+) IN (PD60) [141,142]. |
| GD2–67 (4 g/kg, oral) (Pigs) | 327 mg/dL, maternal | SC | Reduced GAD+ cells (PD220) [143] |
| GD3–42 &3-168 (1.8 g/kg/day, intragastric) (Pigs) | 234 mg/dL, maternal | SC and MC | Reduced GABA+ ve neurons [144]. |
| GD1–14.5 (Alc liquid diet) (C57 mice) | 25 mg/dL, maternal | Corticle anlage | Premature GABAergic interneuron tangential migration; Increased GABA and GABA sensitivity [145]. |
| GD13.5–16.5 (5% Alc liquid diet) (C57mice) | 80 mg/dL, maternal | Embryo and adult mPFC | Increased median ganglionic eminence-derived IN (E16.5) [146]; Increased PV+ IN (mPFC) & potentiated GABAAR transmission (PN) [146]. |
| GD13.5–16.5 (5% Alc liquid diet) (Nkx2.1-Cre mice) | 80 mg/dL, maternal | PFC | Increased depolarizing action of GABAAR in migrating neurons (PD58) [146,147]. |
| GD1–20 (4 g/kg/day) (C57 mice) | 80 mg/dL, maternal | OFC | Increased spontaneous IPSCs amplitude and area in PYNs (PD60) [150]. |
| PD2–PD6 (Alc vapor) (WS Rats) | 206 mg/dL, maternal | MC&SC | Increased CR+ ve INs; Reduced CB+ ve INs (PD60) [152]; Reduced PV+ ve Ins (ST) [152]. |
| GD12–19 & PD2–9 (Alc vapor) (Venus-VGAT-mouse line) | 330 mg/dL, PD7–8 | CBL | Reduced INs (PD16) [153]. |
| PD2–9 (Alc vapor) (Venus-VGAT-mouse) | 221 mg/dL, PD9 | HP | Reduced INs (PD90) [154]. |
| PD7 (Alc vapor) (Venus-VGAT-mouse line) | 297 mg/dL, PD7 | HP | Reduced INs (PD90) [154]. |
| PD7 (2.5 g Alc × 2times) (C57 Mice)) | 500 mg/dL, PD7 | mPFC, HP&PC | Reduced PV+ ve INs (PD90) [155,156,157]. |
| PD7 (2.5 g Alc × 2times) (C57 Mice)) | 500 mg/dL, PD7 | NC | Reduced PV+ ve & CA+ ve INs (PD90) [158]. |
| PD7 (Alc vapor) (Venus-VGAT-mouse line) | 297 mg/dL, PD7 | RSC | Enhancement of PV+ ve INs-mediated Neurotransmission (PD40–60) [159]. |
| Alcohol Model | BAC | HP Slices | Effects |
|---|---|---|---|
| GD1–22 (Alc liquid diet) (Rats) | 31 mg/dL, maternal | CA1 | Reduced LTP (PD50–70) [161]. |
| GD2–67 (Oral Alc, 4 g/kg/day) (Pigs) | 416 mg/dL, maternal | CA1 | Reduced LTP (PD40–50) [168]. |
| PD7 (2.5 g/kg × 2/day) (C57 mice) | 500 mg/dL, PD7 | CA1 | Reduced LTP (PD30–32) [169]. |
| PD2–9 (Alc vapor) (SD Rats) | 395 mg/dL, PD9 | CA1 | Reduced LTP (PD7–9) [170] |
| GD1–22 (Alc liquid diet) (LE Rats) | 87 mg/dL, maternal | CA1 | Reduced LTP in males; Increased LTP in females (PD30–35) [162]. |
| GD0–22 (Alc 4 g/kg/day) (W Rats) | ND | CA1 | Reduced LTP in males and increased in females (PD36) [163]. |
| GD1–22 (Alc liquid diet) (SD Rats) | 135 mg/dL, maternal | CA1 | Reduced LTP in males and no change in females (PD55–65) [164]. |
| PD7 (2.5 g/kg/twice) (C57 mice) | 490 mg/dL, PD7 | CA1 | Reduced LTP (PD90) [43,45,74,75,171]. |
| GD8&12 (1.75 g/kg/twice) (C57 mice) | 300 mg/dL, maternal | CA1 | Reduced LTP (PD90) [172]. |
| GD5–PD7 (10% Alc sol) (SD rats) | ND | CA1 | Reduced LTP (PD17–30) [173]. |
| PD7 (2.5 g/kg × 2/day) (C57 mice) | 500 mg/dL, PD7 | CA1 | Abolished LTD (PD30–32) [169]. |
| GD1–22&lactation (10% alcohol sol) (SD rats) | ND | CA1 | Increased LTD (PD50–52) [111]. |
| GD0–22 (Alc 4 g/kg/day) (WS Rats) | ND | CA1 | Reduced LTD in males and increased in females (PD36) [163]. |
| GD8&12 (1.75 g/kg/twice) (C57 mice) | 300 mg/dL, maternal | CA1 | Reduced LTD in males (PD90) [172]. |
| GD1–22 (Alc liquid diet) (LE Rats) | 84 mg/dL, maternal | DG | Reduced LTP (PD105–140) [162]. |
| GD11–21 (Alc liquid diet) | 142 mg/dL | DG | Reduced LTP in males(PD50–70) [174]. |
| GD1–22 (Alc liquid diet) (SD Rats) | 135 mg/dL, maternal | DG | Reduced LTP in males and no change in females (PD55–70) [175]. |
| GD1–22 (Alc 5%liquid diet) (SD Rats) | 146 mg/dL, maternal | DG | Reduced LTP in males and no change in females (PD55–70) [108]. |
| GD1–22 (5% Alc in water) (LE Rats) | 84 mg/dL, maternal | DG | Reduced LTP (PD105–140) [176]. |
| GD1–PD14 (10% Alc sol.) (SD Rats) | 100 mg/dL, PD7 | DG | Reduced LTP (PD45–55) [177]. |
| GD1–22 (Alc liquid diet) (SD Rats) | 80–180 mg/dL, maternal | DG | Reduced LTP in males and females (PD21–28) [178]. |
| GD1–PD7 (10% Alc sol) (SD Rats) | 62 mg/dL, maternal | CA1 & DG | Reduced LTP (PD21–60) [126]. |
| GD1–22 (Alc liquid diet) (SD Rats) | 80–180 mg/dL, maternal | DG | Reduced LTP in males only (PD31–35) [179]. |
| GD1–PD14 (10% Alc sol.)(SD Rats) | 100 mg/dL, PD7 | DG | Facilitated LTD (PD45–55) [177]. |
| GD1–22 (Alc liquid diet) (SD Rats) | 80–180 mg/dL, maternal | DG | Reduced LTD in females only (PD21–28) [178]. |
| Alcohol Model | BAC | Tissue/Region | Effects |
|---|---|---|---|
| GD6–20 (alc liquid diet) (LE Rats) | ND | VTA | Supersensitive DA autoreceptors (PD90–120) [189]. |
| GD6–20 (alc liquid diet) (LE Rats) | ND | NSDA | Reduced DA receptor functions (P90–120) [190]. |
| GD8–20 (3 g/kg/twice/day) (SD rats) | 281–341 mg/dL, maternal | VTA | Reduced the number of spontaneously active DA neurons (P90) [191]. |
| GD8–20 (3 g/kg/twice a day) (SD rats) | 281–341 mg/dL, maternal | VTA-DA | Enhanced GluA3 (PD14–84) [79]; Enhanced EPSCs strength (PD14–84) [79]. |
| GD7–20 (6g/kg/day) (SD Rats) | 302–331 mg/dL, maternal | DL-ST | Enhanced D1R function (PD30) [194]. |
| PD2–12 (Alc vapor, 4 h/day) (SD Rats) | 23 mg/dL, pups | BLA | Decreased DA; Reduced D1R-mediated potentiation of sIPSCs; Impaired D3R-mediated suppression of mIPSCs (PD40–50) [197]. |
| PD7 (2.5g/kg/twice) (C57 mice) | 490 mg/dL, PD7 | HP&NC | Enhanced AEA levels, NAPE-PLD, GDE & CB1 expression (PD7) [45]. |
| GD8–20 (3 g/kg/twice a day) (SD rats) | 281–341 mg/dL, maternal | VTA-DA | Impaired eCB-LTD (PD28–70) [198]. |
| GD0–20 (Alc vapor) (C57 mice) | 84 mg/dL, maternal | DLS-MSNs | Increased excitability of MS neurons; Increased eCB tone (PD90) [199]. |
| GD8–20 & PD0–10 (3 g/kg/twice a day) (SD rats) | 281–341 mg/dL, maternal | VTA-DA | Reduced CB1 mRNA expression (PD60–70) [200]. |
| GD1–20 (Alc liquid diet) (SD Rats) | ND | MC & SSC | Reduced 5-HT, 5-HIAA &5-HT1 receptors (PD19) [184,201]. |
| GD8–15 (Alc liquid diet) (C57 mice) | ND | MFB, MR, DR, HP, etc. | Reduced 5-HT (E15/18) [202,203,204]. |
| GD15–20 (0.5–2g/kg/day) (SD Rats) | 3.32–106 mg/dL, maternal | DR | Reduced 5-HT and TPH (PD37) [205]. |
| GD1–20-PD20 (6% Alc) (CD1 mice) | 73–102 mg/dL (PD21) | Amy&CC | Reduced 5-HT (Amy&CC); Reduced 5-HT1R (CC); Increased 5-HTR (Amy) (PD77–84) [206]. |
| GD8–20 & PD0–10 (3 g/kg/twice a day) (SD rats) | 281–341 mg/dL, maternal | VTA-DA | Enhanced the electrical activity of DRn 5-HT neurons (PD60–70) [200]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basavarajappa, B.S.; Subbanna, S. Synaptic Plasticity Abnormalities in Fetal Alcohol Spectrum Disorders. Cells 2023, 12, 442. https://doi.org/10.3390/cells12030442
Basavarajappa BS, Subbanna S. Synaptic Plasticity Abnormalities in Fetal Alcohol Spectrum Disorders. Cells. 2023; 12(3):442. https://doi.org/10.3390/cells12030442
Chicago/Turabian StyleBasavarajappa, Balapal S., and Shivakumar Subbanna. 2023. "Synaptic Plasticity Abnormalities in Fetal Alcohol Spectrum Disorders" Cells 12, no. 3: 442. https://doi.org/10.3390/cells12030442
APA StyleBasavarajappa, B. S., & Subbanna, S. (2023). Synaptic Plasticity Abnormalities in Fetal Alcohol Spectrum Disorders. Cells, 12(3), 442. https://doi.org/10.3390/cells12030442