
Any Role for Microbiota in Cholangiocarcinoma? A Comprehensive Review

 , and
, and 
Abstract
1. Introduction
2. Classification and Epidemiology of CCA
3. Established, Emerging, and Controversial Risk Factors for CCA
3.1. Chronic Biliary Disease
3.2. Chronic Liver Diseases
3.3. Liver Fluke Infections
3.4. Genetic Factors
3.5. Metabolic Syndrome
3.6. Alcohol Consumption, Smoking, Races
3.7. Professional/Toxic Exposure
4. Microbiota and Biliary Tract
5. Microbiota and CCA
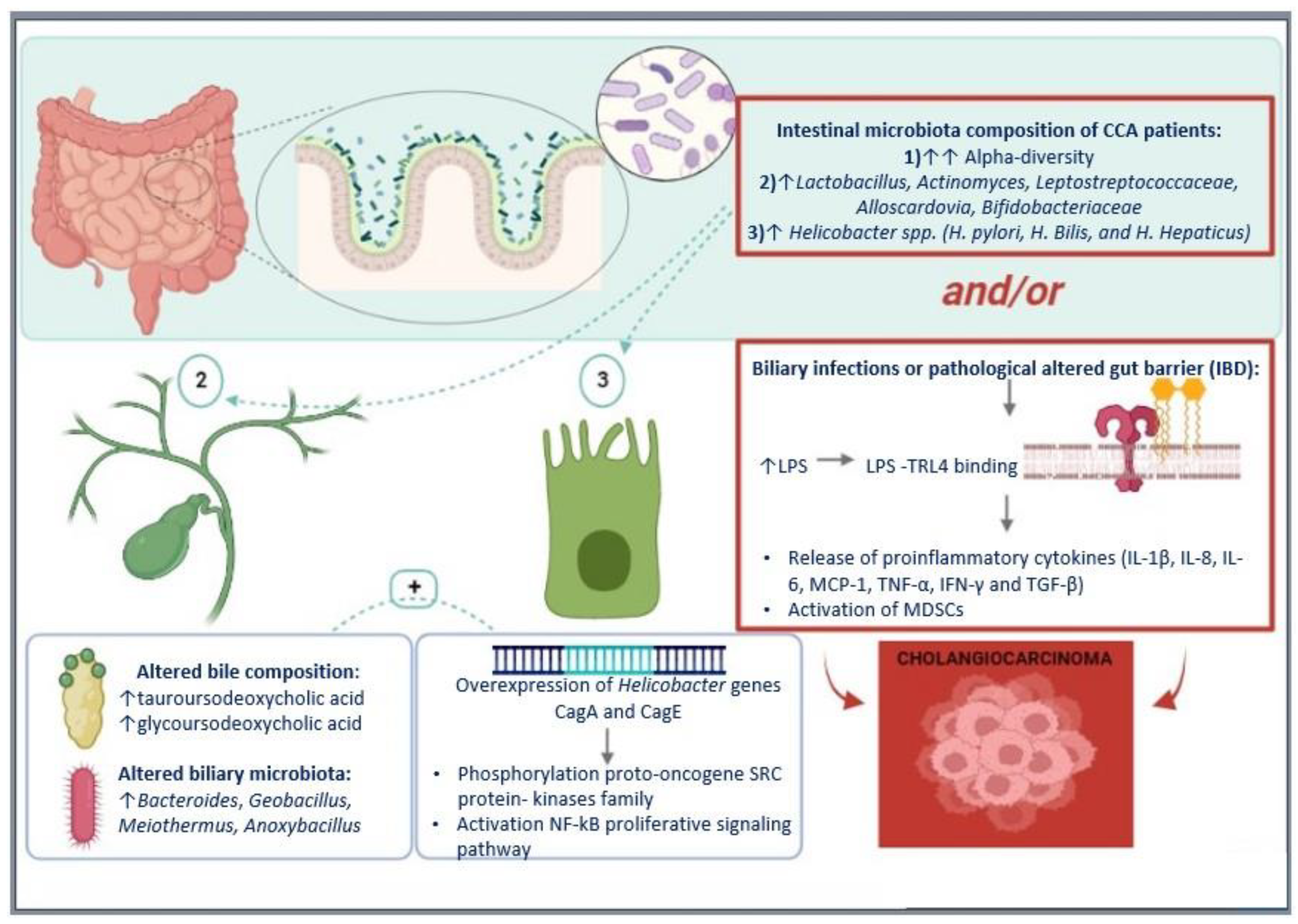
5.1. Microbiota in CCA Pathogenesis
5.2. Microbiota in CCA Diagnosis
5.3. Microbiota in CCA Prophylaxis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef] [PubMed]
- Elvevi, A.; Laffusa, A.; Scaravaglio, M.; Rossi, R.E.; Longarini, R.; Stagno, A.M.; Cristoferi, L.; Ciaccio, A.; Cortinovis, D.L.; Invernizzi, P.; et al. Clinical Treatment of Cholangiocarcinoma: An Updated Comprehensive Review. Ann. Hepatol. 2022, 27, 100737. [Google Scholar] [CrossRef] [PubMed]
- Massarweh, N.N.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. Cancer Control 2017, 24. [Google Scholar] [CrossRef] [PubMed]
- Clements, O.; Eliahoo, J.; Kim, J.U.; Taylor-Robinson, S.D.; Khan, S.A. Risk Factors for Intrahepatic and Extrahepatic Cholangiocarcinoma: A Systematic Review and Meta-Analysis. J. Hepatol. 2020, 72, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next Horizon in Mechanisms and Management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Saab, M.; Mestivier, D.; Sohrabi, M.; Rodriguez, C.; Khonsari, M.R.; Faraji, A.; Sobhani, I. Characterization of Biliary Microbiota Dysbiosis in Extrahepatic Cholangiocarcinoma. PLoS ONE 2021, 16, e0247798. [Google Scholar] [CrossRef]
- Miyabe, K.; Chandrasekhara, V.; Wongjarupong, N.; Chen, J.; Yang, L.; Johnson, S.; Chia, N.; Walther-Antonio, M.; Yao, J.Z.; Harrington, S.C.; et al. Potential Role of Inflammation-Promoting Biliary Microbiome in Primary Sclerosing Cholangitis and Cholangiocarcinoma. Cancers 2022, 14, 2120. [Google Scholar] [CrossRef]
- Valle, J.W.; Borbath, I.; Khan, S.A.; Huguet, F.; Gruenberger, T.; Arnold, D. ESMO Guidelines Committee Biliary Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2016, 27, v28–v37. [Google Scholar] [CrossRef]
- Bertuccio, P.; Malvezzi, M.; Carioli, G.; Hashim, D.; Boffetta, P.; El-Serag, H.B.; La Vecchia, C.; Negri, E. Global Trends in Mortality from Intrahepatic and Extrahepatic Cholangiocarcinoma. J. Hepatol. 2019, 71, 104–114. [Google Scholar] [CrossRef]
- Taylor-Robinson, S.D.; Toledano, M.B.; Arora, S.; Keegan, T.J.; Hargreaves, S.; Beck, A.; Khan, S.A.; Elliott, P.; Thomas, H.C. Increase in Mortality Rates from Intrahepatic Cholangiocarcinoma in England and Wales 1968–1998. Gut 2001, 48, 816–820. [Google Scholar] [CrossRef]
- Barner-Rasmussen, N.; Pukkala, E.; Hadkhale, K.; Färkkilä, M. Risk Factors, Epidemiology and Prognosis of Cholangiocarcinoma in Finland. UEG J. 2021, 9, 1128–1135. [Google Scholar] [CrossRef]
- Boonstra, K.; Weersma, R.K.; van Erpecum, K.J.; Rauws, E.A.; Spanier, B.W.M.; Poen, A.C.; van Nieuwkerk, K.M.; Drenth, J.P.; Witteman, B.J.; Tuynman, H.A.; et al. Population-Based Epidemiology, Malignancy Risk, and Outcome of Primary Sclerosing Cholangitis: Boonstra et Al. Hepatology 2013, 58, 2045–2055. [Google Scholar] [CrossRef]
- Chapman, M.H.; Thorburn, D.; Hirschfield, G.M.; Webster, G.G.J.; Rushbrook, S.M.; Alexander, G.; Collier, J.; Dyson, J.K.; Jones, D.E.; Patanwala, I.; et al. British Society of Gastroenterology and UK-PSC Guidelines for the Diagnosis and Management of Primary Sclerosing Cholangitis. Gut 2019, 68, 1356–1378. [Google Scholar] [CrossRef]
- Stone, J.H.; Zen, Y.; Deshpande, V. IgG4-Related Disease. N. Engl. J. Med. 2012, 366, 539–551. [Google Scholar] [CrossRef]
- Petrick, J.L.; Yang, B.; Altekruse, S.F.; Van Dyke, A.L.; Koshiol, J.; Graubard, B.I.; McGlynn, K.A. Risk Factors for Intrahepatic and Extrahepatic Cholangiocarcinoma in the United States: A Population-Based Study in SEER-Medicare. PLoS ONE 2017, 12, e0186643. [Google Scholar] [CrossRef]
- Söreide, K.; Körner, H.; Havnen, J.; Söreide, J.A. Bile Duct Cysts in Adults. Br. J. Surg. 2004, 91, 1538–1548. [Google Scholar] [CrossRef]
- Shaib, Y.; El-Serag, H. The Epidemiology of Cholangiocarcinoma. Semin. Liver Dis. 2004, 24, 115–125. [Google Scholar] [CrossRef]
- Kim, H.J. Hepatolithiasis and Intrahepatic Cholangiocarcinoma: A Review. World J. Gastroenterol. 2015, 21, 13418. [Google Scholar] [CrossRef]
- Matsumoto, K.; Onoyama, T.; Kawata, S.; Takeda, Y.; Harada, K.; Ikebuchi, Y.; Ueki, M.; Miura, N.; Yashima, K.; Koda, M.; et al. Hepatitis B and C Virus Infection Is a Risk Factor for the Development of Cholangiocarcinoma. Intern. Med. 2014, 53, 651–654. [Google Scholar] [CrossRef]
- Ralphs, S.; Khan, S.A. The Role of the Hepatitis Viruses in Cholangiocarcinoma. J. Viral Hepat. 2013, 20, 297–305. [Google Scholar] [CrossRef]
- Schwartz, D.A. Helminths in the Induction of Cancer: Opisthorchis Viverrini, Clonorchis Sinensis and Cholangiocarcinoma. Trop. Geogr. Med. 1980, 32, 95–100. [Google Scholar] [PubMed]
- IARC Working Group. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans Biological Agents. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 1–441. [Google Scholar]
- Shin, H.-R.; Oh, J.-K.; Masuyer, E.; Curado, M.-P.; Bouvard, V.; Fang, Y.-Y.; Wiangnon, S.; Sripa, B.; Hong, S.-T. Epidemiology of Cholangiocarcinoma: An Update Focusing on Risk Factors. Cancer Sci. 2010, 101, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Dodson, R.M.; Weiss, M.J.; Cosgrove, D.; Herman, J.M.; Kamel, I.; Anders, R.; Geschwind, J.-F.H.; Pawlik, T.M. Intrahepatic Cholangiocarcinoma: Management Options and Emerging Therapies. J. Am. Coll. Surg. 2013, 217, 736–750.e4. [Google Scholar] [CrossRef] [PubMed]
- Braconi, C.; Roessler, S.; Kruk, B.; Lammert, F.; Krawczyk, M.; Andersen, J.B. Molecular Perturbations in Cholangiocarcinoma: Is It Time for Precision Medicine? Liver Int. 2019, 39, 32–42. [Google Scholar] [CrossRef]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and Risk Factors. Liver Int. 2019, 39, 19–31. [Google Scholar] [CrossRef]
- Sia, D.; Losic, B.; Moeini, A.; Cabellos, L.; Hao, K.; Revill, K.; Bonal, D.; Miltiadous, O.; Zhang, Z.; Hoshida, Y.; et al. Massive Parallel Sequencing Uncovers Actionable FGFR2–PPHLN1 Fusion and ARAF Mutations in Intrahepatic Cholangiocarcinoma. Nat. Commun. 2015, 6, 6087. [Google Scholar] [CrossRef]
- Chan-on, W.; Nairismägi, M.-L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome Sequencing Identifies Distinct Mutational Patterns in Liver Fluke–Related and Non-Infection-Related Bile Duct Cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef]
- Nepal, C.; O’Rourke, C.J.; Oliveira, D.V.N.P.; Taranta, A.; Shema, S.; Gautam, P.; Calderaro, J.; Barbour, A.; Raggi, C.; Wennerberg, K.; et al. Genomic Perturbations Reveal Distinct Regulatory Networks in Intrahepatic Cholangiocarcinoma. Hepatology 2018, 68, 949–963. [Google Scholar] [CrossRef]
- Gingras, M.-C.; Covington, K.R.; Chang, D.K.; Donehower, L.A.; Gill, A.J.; Ittmann, M.M.; Creighton, C.J.; Johns, A.L.; Shinbrot, E.; Dewal, N.; et al. Ampullary Cancers Harbor ELF3 Tumor Suppressor Gene Mutations and Exhibit Frequent WNT Dysregulation. Cell Rep. 2016, 14, 907–919. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic Spectra of Biliary Tract Cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef]
- Mazzaferro, V.; El-Rayes, B.F.; Droz Dit Busset, M.; Cotsoglou, C.; Harris, W.P.; Damjanov, N.; Masi, G.; Rimassa, L.; Personeni, N.; Braiteh, F.; et al. Derazantinib (ARQ 087) in Advanced or Inoperable FGFR2 Gene Fusion-Positive Intrahepatic Cholangiocarcinoma. Br. J. Cancer 2019, 120, 165–171. [Google Scholar] [CrossRef]
- O’Rourke, C.J.; Munoz-Garrido, P.; Aguayo, E.L.; Andersen, J.B. Epigenome Dysregulation in Cholangiocarcinoma. Biochim. Biophys. Acta–Mol. Basis Dis. 2018, 1864, 1423–1434. [Google Scholar] [CrossRef]
- O’Rourke, C.J.; Lafuente-Barquero, J.; Andersen, J.B. Epigenome Remodeling in Cholangiocarcinoma. Trends Cancer 2019, 5, 335–350. [Google Scholar] [CrossRef]
- Laffusa, A.; Ciaccio, A. Impact of Metformin on the Incidence of Human Cholangiocarcinoma in Diabetic Patients: A Systematic Review and Meta-Analysis. Eur. J. Gastroenterol. Hepatol. 2022; in press. [Google Scholar]
- Labib, P.L.; Goodchild, G.; Pereira, S.P. Molecular Pathogenesis of Cholangiocarcinoma. BMC Cancer 2019, 19, 185. [Google Scholar] [CrossRef]
- Alvaro, D.; Barbaro, B.; Franchitto, A.; Onori, P.; Glaser, S.S.; Alpini, G.; Francis, H.; Marucci, L.; Sterpetti, P.; Ginanni-Corradini, S.; et al. Estrogens and Insulin-like Growth Factor 1 Modulate Neoplastic Cell Growth in Human Cholangiocarcinoma. Am. J. Pathol. 2006, 169, 877–888. [Google Scholar] [CrossRef]
- Chen, H.-F.; Chen, P.; Li, C.-Y. Risk of Malignant Neoplasms of Liver and Biliary Tract in Diabetic Patients with Different Age and Sex Stratifications. Hepatology 2010, 52, 155–163. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kubo, S.; Hai, S.; Uenishi, T.; Yamamoto, T.; Shuto, T.; Takemura, S.; Tanaka, H.; Yamazaki, O.; Hirohashi, K.; et al. Hepatitis C Virus Infection as a Likely Etiology of Intrahepatic Cholangiocarcinoma. Cancer Sci. 2004, 95, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Jin, G.; Zhou, X.; Zhou, Y.; Zhang, Y.; Shao, C.; Liu, R.; Hu, X. Diabetes Mellitus and Increased Risk of Cholangiocarcinoma: A Meta-Analysis. Eur. J. Cancer Prev. 2012, 21, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Chaiteerakij, R.; Yang, J.D.; Harmsen, W.S.; Slettedahl, S.W.; Mettler, T.A.; Fredericksen, Z.S.; Kim, W.R.; Gores, G.J.; Roberts, R.O.; Olson, J.E.; et al. Risk Factors for Intrahepatic Cholangiocarcinoma: Association between Metformin Use and Reduced Cancer Risk. Hepatology 2013, 57, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Baidoun, F.; Sarmini, M.T.; Merjaneh, Z.; Moustafa, M.A. Controversial Risk Factors for Cholangiocarcinoma. Eur. J. Gastroenterol. Hepatol. 2022, 34, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.H.; Tseleni-Balafouta, S.; Moon, H.-S.; Chamberland, J.P.; Liu, X.; Kavantzas, N.; Mantzoros, C.S. Adiponectin Receptor Expression in Human Malignant Tissues. Horm. Cancer 2010, 1, 136–145. [Google Scholar] [CrossRef]
- Menon, S.; Mathew, R. Association between Metabolic Syndrome and Hepatobiliary Cancers: A Case-Control Study. Indian J. Gastroenterol. 2019, 38, 61–68. [Google Scholar] [CrossRef]
- Palmer, W.C.; Patel, T. Are Common Factors Involved in the Pathogenesis of Primary Liver Cancers? A Meta-Analysis of Risk Factors for Intrahepatic Cholangiocarcinoma. J. Hepatol. 2012, 57, 69–76. [Google Scholar] [CrossRef]
- Welzel, T.M.; Mellemkjaer, L.; Gloria, G.; Sakoda, L.C.; Hsing, A.W.; Ghormli, L.E.; Olsen, J.H.; McGlynn, K.A. Risk Factors for Intrahepatic Cholangiocarcinoma in a Low-Risk Population: A Nationwide Case-Control Study. Int. J. Cancer 2007, 120, 638–641. [Google Scholar] [CrossRef]
- Choi, J.; Ghoz, H.M.; Peeraphatdit, T.; Baichoo, E.; Addissie, B.D.; Harmsen, W.S.; Therneau, T.M.; Olson, J.E.; Chaiteerakij, R.; Roberts, L.R. Aspirin Use and the Risk of Cholangiocarcinoma. Hepatology 2016, 64, 785–796. [Google Scholar] [CrossRef]
- Osataphan, S.; Mahankasuwan, T.; Saengboonmee, C. Obesity and Cholangiocarcinoma: A Review of Epidemiological and Molecular Associations. J. Hepato-Biliary-Pancreat. Sci. 2021, 28, 1047–1059. [Google Scholar] [CrossRef]
- Welzel, T.M.; Graubard, B.I.; Zeuzem, S.; El-Serag, H.B.; Davila, J.A.; McGlynn, K.A. Metabolic Syndrome Increases the Risk of Primary Liver Cancer in the United States: A Study in the SEER-Medicare Database. Hepatology 2011, 54, 463–471. [Google Scholar] [CrossRef]
- Kan, H.-P.; Huang, Y.-Q.; Tan, Y.-F.; Zhou, J. Meta-Analysis of Alcohol Consumption and Risk of Extrahepatic Bile System Cancer: Alcohol and Risk of Extrahepatic Bile Cancer. Hepatol. Res. 2011, 41, 746–753. [Google Scholar] [CrossRef]
- Kono, S.; Shinchi, K.; Todoroki, I.; Honjo, S.; Sakurai, Y.; Wakabayashi, K.; Imanishi, K.; Nishikawa, H.; Ogawa, S.; Katsurada, M. Gallstone Disease among Japanese Men in Relation to Obesity, Glucose Intolerance, Exercise, Alcohol Use, and Smoking. Scand. J. Gastroenterol. 1995, 30, 372–376. [Google Scholar] [CrossRef]
- McGee, E.E.; Jackson, S.S.; Petrick, J.L.; Van Dyke, A.L.; Adami, H.-O.; Albanes, D.; Andreotti, G.; Beane-Freeman, L.E.; Berrington de Gonzalez, A.; Buring, J.E.; et al. Smoking, Alcohol, and Biliary Tract Cancer Risk: A Pooling Project of 26 Prospective Studies. J. Natl. Cancer Inst. 2019, 111, 1263–1278. [Google Scholar] [CrossRef]
- Ye, X.-H. Smoking, Alcohol Consumption, and the Risk of Extrahepatic Cholangiocarcinoma: A Meta-Analysis. World J. Gastroenterol. 2013, 19, 8780. [Google Scholar] [CrossRef]
- Saha, S.K.; Zhu, A.X.; Fuchs, C.S.; Brooks, G.A. Forty-Year Trends in Cholangiocarcinoma Incidence in the U.S.: Intrahepatic Disease on the Rise. Oncologist 2016, 21, 594–599. [Google Scholar] [CrossRef]
- Patel, N.; Benipal, B. Incidence of Cholangiocarcinoma in the USA from 2001 to 2015: A US Cancer Statistics Analysis of 50 States. Cureus 2019, 11, e3962. [Google Scholar] [CrossRef]
- Brandi, G.; Di Girolamo, S.; Farioli, A.; de Rosa, F.; Curti, S.; Pinna, A.D.; Ercolani, G.; Violante, F.S.; Biasco, G.; Mattioli, S. Asbestos: A Hidden Player behind the Cholangiocarcinoma Increase? Findings from a Case–Control Analysis. Cancer Causes Control 2013, 24, 911–918. [Google Scholar] [CrossRef]
- Luberto, F.; Ferrante, D.; Silvestri, S.; Angelini, A.; Cuccaro, F.; Nannavecchia, A.M.; Oddone, E.; Vicentini, M.; Barone-Adesi, F.; Cena, T.; et al. Cumulative Asbestos Exposure and Mortality from Asbestos Related Diseases in a Pooled Analysis of 21 Asbestos Cement Cohorts in Italy. Environ. Health 2019, 18, 71. [Google Scholar] [CrossRef]
- Farioli, A.; Straif, K.; Brandi, G.; Curti, S.; Kjaerheim, K.; Martinsen, J.I.; Sparen, P.; Tryggvadottir, L.; Weiderpass, E.; Biasco, G.; et al. Occupational Exposure to Asbestos and Risk of Cholangiocarcinoma: A Population-Based Case–Control Study in Four Nordic Countries. Occup. Environ. Med. 2018, 75, 191–198. [Google Scholar] [CrossRef]
- Ceci, L.; Zhou, T.; Lenci, I.; Meadows, V.; Kennedy, L.; Li, P.; Ekser, B.; Milana, M.; Zhang, W.; Wu, C.; et al. Molecular Mechanisms Linking Risk Factors to Cholangiocarcinoma Development. Cancers 2022, 14, 1442. [Google Scholar] [CrossRef] [PubMed]
- Tyson, G.L.; El-Serag, H.B. Risk Factors for Cholangiocarcinoma. Hepatology 2011, 54, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Kato, I.; Kido, C. Increased Risk of Death in Thorotrast-Exposed Patients during the Late Follow-up Period. Jpn. J. Cancer Res. 1987, 78, 1187–1192. [Google Scholar] [PubMed]
- Ketpueak, T.; Thiennimitr, P.; Apaijai, N.; Chattipakorn, S.C.; Chattipakorn, N. Association of Chronic Opisthorchis Infestation and Microbiota Alteration on Tumorigenesis in Cholangiocarcinoma. Clin. Transl. Gastroenterol. 2020, 12, e00292. [Google Scholar] [CrossRef] [PubMed]
- Tomkovich, S.; Jobin, C. Microbiota and Host Immune Responses: A Love-Hate Relationship. Immunology 2016, 147, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut Microbiota, Metabolites and Host Immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Chagani, S.; Kwong, L.N. Cholangiocarcinoma Risk Factors Open the Floodgates for Gut Microbes and Immunosuppressive Myeloid Cells. Cancer Discov. 2021, 11, 1014–1015. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, C.; Duan, Y.; Heinrich, B.; Rosato, U.; Diggs, L.P.; Ma, L.; Roy, S.; Fu, Q.; Brown, Z.J.; et al. Gut Microbiome Directs Hepatocytes to Recruit MDSCs and Promote Cholangiocarcinoma. Cancer Discov. 2021, 11, 1248–1267. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Weeding, E.; Jorgensen, R.A.; Petz, J.L.; Keach, J.C.; Talwalkar, J.A.; Lindor, K.D. Randomised Clinical Trial: Vancomycin or Metronidazole in Patients with Primary Sclerosing Cholangitis—A Pilot Study. Aliment. Pharmacol. Ther. 2013, 37, 604–612. [Google Scholar] [CrossRef]
- Ji, J.; Wu, L.; Wei, J.; Wu, J.; Guo, C. The Gut Microbiome and Ferroptosis in MAFLD. J. Clin. Transl. Hepatol. 2022, 11, 174–187. [Google Scholar] [CrossRef]
- Harada, K.; Nakanuma, Y. Innate Immunity in the Pathogenesis of Cholangiopathy: A Recent Update. Inflamm. Allergy-Drug Targets 2012, 11, 478–483. [Google Scholar] [CrossRef]
- Syal, G.; Fausther, M.; Dranoff, J.A. Advances in Cholangiocyte Immunobiology. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1077–G1086. [Google Scholar] [CrossRef]
- Moriyama, K.; Nishida, O. Targeting Cytokines, Pathogen-Associated Molecular Patterns, and Damage-Associated Molecular Patterns in Sepsis via Blood Purification. Int. J. Mol. Sci. 2021, 22, 8882. [Google Scholar] [CrossRef]
- Greuter, T.; Vavricka, S.; König, A.O.; Beaugerie, L.; Scharl, M.; on behalf of Swiss IBDnet, an official working group of the Swiss Society of Gastroenterology. Malignancies in Inflammatory Bowel Disease. Digestion 2020, 101, 136–145. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary Sclerosing Cholangitis—A Comprehensive Review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef]
- Di Giorgio, A.; Tulone, A.; Nicastro, E.; Norsa, L.; Sonzogni, A.; D’Antiga, L. Use of Oral Vancomycin in Children with Autoimmune Liver Disease: A Single Centre Experience. World J. Hepatol. 2021, 13, 2113–2127. [Google Scholar] [CrossRef]
- Damman, J.L.; Rodriguez, E.A.; Ali, A.H.; Buness, C.W.; Cox, K.L.; Carey, E.J.; Lindor, K.D. Review Article: The Evidence That Vancomycin Is a Therapeutic Option for Primary Sclerosing Cholangitis. Aliment. Pharmacol. Ther. 2018, 47, 886–895. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, S.; Jin, C.; Lin, Z.; Deng, T.; Xie, X.; Deng, L.; Li, X.; Ma, J.; Ding, X.; et al. A Predictive Model Based on the Gut Microbiota Improves the Diagnostic Effect in Patients With Cholangiocarcinoma. Front. Cell. Infect. Microbiol. 2021, 11, 751795. [Google Scholar] [CrossRef]
- Deenonpoe, R.; Mairiang, E.; Mairiang, P.; Pairojkul, C.; Chamgramol, Y.; Rinaldi, G.; Loukas, A.; Brindley, P.J.; Sripa, B. Elevated Prevalence of Helicobacter Species and Virulence Factors in Opisthorchiasis and Associated Hepatobiliary Disease. Sci. Rep. 2017, 7, 42744. [Google Scholar] [CrossRef]
- Boonyanugomol, W.; Chomvarin, C.; Baik, S.-C.; Song, J.-Y.; Hahnvajanawong, C.; Kim, K.-M.; Cho, M.-J.; Lee, W.-K.; Kang, H.-L.; Rhee, K.-H.; et al. Role of CagA-Positive Helicobacter Pylori on Cell Proliferation, Apoptosis, and Inflammation in Biliary Cells. Dig. Dis. Sci. 2011, 56, 1682–1692. [Google Scholar] [CrossRef]
- Wheatley, R.C.; Kilgour, E.; Jacobs, T.; Lamarca, A.; Hubner, R.A.; Valle, J.W.; McNamara, M.G. Potential Influence of the Microbiome Environment in Patients with Biliary Tract Cancer and Implications for Therapy. Br. J. Cancer 2022, 126, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-Derived Suppressor Cells Coming of Age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Chaisaingmongkol, J.; Budhu, A.; Dang, H.; Rabibhadana, S.; Pupacdi, B.; Kwon, S.M.; Forgues, M.; Pomyen, Y.; Bhudhisawasdi, V.; Lertprasertsuke, N.; et al. Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017, 32, 57–70.e3. [Google Scholar] [CrossRef]
- Deng, T.; Li, J.; He, B.; Chen, B.; Liu, F.; Chen, Z.; Zheng, J.; Shi, Z.; Zhang, T.; Deng, L.; et al. Gut Microbiome Alteration as a Diagnostic Tool and Associated with Inflammatory Response Marker in Primary Liver Cancer. Hepatol. Int. 2022, 16, 99–111. [Google Scholar] [CrossRef]
- Jia, X.; Lu, S.; Zeng, Z.; Liu, Q.; Dong, Z.; Chen, Y.; Zhu, Z.; Hong, Z.; Zhang, T.; Du, G.; et al. Characterization of Gut Microbiota, Bile Acid Metabolism, and Cytokines in Intrahepatic Cholangiocarcinoma. Hepatology 2020, 71, 893–906. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, K.; Liu, Y.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Jia, H.; Han, W. Phase I Study of Chimeric Antigen Receptor-Modified T Cells in Patients with EGFR-Positive Advanced Biliary Tract Cancers. Clin. Cancer Res. 2018, 24, 1277–1286. [Google Scholar] [CrossRef]
- Feng, K.; Guo, Y.; Liu, Y.; Dai, H.; Wang, Y.; Lv, H.; Huang, J.; Yang, Q.; Han, W. Cocktail Treatment with EGFR-Specific and CD133-Specific Chimeric Antigen Receptor-Modified T Cells in a Patient with Advanced Cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef]
- Loeuillard, E.; Yang, J.; Buckarma, E.; Wang, J.; Liu, Y.; Conboy, C.; Pavelko, K.D.; Li, Y.; O’Brien, D.; Wang, C.; et al. Targeting Tumor-Associated Macrophages and Granulocytic Myeloid-Derived Suppressor Cells Augments PD-1 Blockade in Cholangiocarcinoma. J. Clin. Investig. 2020, 130, 5380–5396. [Google Scholar] [CrossRef]


{kind=link}
| Factor | Type of Study | Calculated Risk |
|---|---|---|
| PSC | Population-based study | OR 22 for iCCA, OR 41 for eCCA |
| Bile duct cysts | Meta-analysis | OR 26.71 for iCCA, OR 34.94 for eCCA |
| Caroli’s disease | Population-based study | OR 38 for iCCA, OR 97 for eCCA |
| Hepatolithiasis | Population-based study | 69% of the patients who underwent surgery for CCA had hepatolithiasis |
| Choledocholithiasis | Meta-analysis | OR 10.08 for iCCA, OR 18.58 for eCCA |
| Chronic hepatitis C | Meta-analysis | OR 4.28 for iCCA, OR 1.98 for eCCA |
| Chronic hepatitis B | Meta-analysis | OR 4.57 for iCCA, OR 2.11 for eCCA |
| Cirrhosis | Meta-analysis | OR 15.32 for iCCA, OR 3.82 for eCCA |
| MAFLD and NASH | Meta-analysis | OR 2.2 for iCCA, OR 1.5 for eCCA |
| Hemochromatosis | Population-based study | OR 2.1 for iCCA |
| Liver fluke (Opisthorchis Viverrini, Clonorchis Sinensis) | Meta-analysis | OR 5 iCCA > eCCA |
| Genetic factors | GWAS | IDH, EPHA2, BAP1 mutations, and FGFR2 fusions are more common in iCCA, and PRKACA and PRKACB fusions are more common in eCCA. Epigenetic hypermethylation of CpG early appears in CCA carcinogenesis. |
| Diabetes | Meta-analysis | OR 1.73 for iCCA, OR 1.5 for eCCA |
| Obesity | Meta-analysis | OR 1.14 for iCCA, OR 1.2 for eCCA |
| Hypertension | Meta-analysis | OR 1.10 for iCCA, OR 1.21 for eCCA |
| Alcohol consumption | Meta-analysis | OR 3.15 for iCCA, OR 1.75 for eCCA |
| Smoking | Meta-analysis | OR 1.25 for iCCA, OR 1.69 for eCCA |
| Races | Population-based study | Hispanics and Asian/Pacific Islanders are more likely to have CCA compared with Caucasians, and African American race is inversely associated with CCA |
| Asbestos | Case-control study | OR 4.8 for iCCA, OR 2.1 for eCCA |
| Case-control study | OR 1.1–1.7 for iCCA, no association with eCCA | |
| Thorotrast | Retrospective study | RR > 300 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elvevi, A.; Laffusa, A.; Gallo, C.; Invernizzi, P.; Massironi, S. Any Role for Microbiota in Cholangiocarcinoma? A Comprehensive Review. Cells 2023, 12, 370. https://doi.org/10.3390/cells12030370
Elvevi A, Laffusa A, Gallo C, Invernizzi P, Massironi S. Any Role for Microbiota in Cholangiocarcinoma? A Comprehensive Review. Cells. 2023; 12(3):370. https://doi.org/10.3390/cells12030370
Chicago/Turabian StyleElvevi, Alessandra, Alice Laffusa, Camilla Gallo, Pietro Invernizzi, and Sara Massironi. 2023. "Any Role for Microbiota in Cholangiocarcinoma? A Comprehensive Review" Cells 12, no. 3: 370. https://doi.org/10.3390/cells12030370
APA StyleElvevi, A., Laffusa, A., Gallo, C., Invernizzi, P., & Massironi, S. (2023). Any Role for Microbiota in Cholangiocarcinoma? A Comprehensive Review. Cells, 12(3), 370. https://doi.org/10.3390/cells12030370