Abstract
The most frequent chromosomal rearrangement in childhood B-cell acute lymphoblastic leukemia (B-ALL) is translocation t(12;21)(p13;q22). It results in the fusion of the ETV6::RUNX1 gene, which is active in the regulation of multiple crucial cellular pathways. Recent studies hypothesize that many translocations are influenced by RAG-initiated deletions, as well as defects in the RAS and NRAS pathways. According to a “two-hit” model for the molecular pathogenesis of pediatric ETV6::RUNX1-positive B-ALL, the t(12;21) translocation requires leukemia-causing secondary mutations. Patients with ETV6::RUNX1 express up to 60 different aberrations, which highlights the heterogeneity of this B-ALL subtype and is reflected in differences in patient response to treatment and chances of relapse. Most studies of secondary genetic changes have concentrated on deletions of the normal, non-rearranged ETV6 allele. Other predominant structural changes included deletions of chromosomes 6q and 9p, loss of entire chromosomes X, 8, and 13, duplications of chromosome 4q, or trisomy of chromosomes 21 and 16, but the impact of these changes on overall survival remains unclarified. An equally genetically diverse group is the recently identified new B-ALL subtype ETV6::RUNX1-like ALL. In our review, we provide a comprehensive description of recurrent secondary mutations in pediatric B-ALL with t(12;21) to emphasize the value of investigating detailed molecular mechanisms in ETV6::RUNX1-positive B-ALL, both for our understanding of the etiology of the disease and for future clinical advances in patient treatment and management.
1. Introduction
Acute lymphoblastic leukemia (ALL) is the most common childhood cancer, accounting for more than 20% of all childhood cancers [1]. It develops by malignant transformation of immature lymphocyte precursors in the bone marrow (BM) or thymus [2]. ALL can exist in the B- (B-ALL) or T-lymphoid (T-ALL) lineage and include more than thirty distinct subtypes characterized by inherited and recurrent somatic genetic alterations that coincide with distinct gene expression profiles [3]. Currently, the survival of pediatric patients with ALL exceeds 90% [4]. Chemotherapy is administered based on stratified risk classification, which is determined by clinical factors: age and white blood cell (WBC) counts at diagnosis, genomic and cytogenetic analysis of ALL cells, and response assessment according to minimal residual disease (MRD). Karyotyping, fluorescence in situ hybridization (FISH), multiplex ligation probe-dependent amplification (MLPA), SNP microarray, next-generation sequencing (NGS) technologies such as RNA-Seq, and whole genome/whole exome sequencing (WGS/WES), are increasingly used to define more than 30 genetic subtypes. The majority of precursor B-ALL are classified in the 5th edition of the World Health Organization Classification of Hematolymphoid Tumors (WHO-HAEM5) according to aneuploidy changes, which include high hyperdiploidy (over 50 chromosomes) and hypodiploidy (less than 44 chromosomes), well-known chromosomal rearrangements (KMT2A rearrangements, ETV6::RUNX1 fusion, TCF3::PBX1 fusion, BCR::ABL1 fusion, or IGH::IL3 fusion), or the newly identified other genetic drivers (BCR::ABL1-like ALL, intrachromosomal amplification of chromosome 21 (iAMP21), ETV6::RUNX1-like ALL, TCF3::HLF) [5,6]. These abnormalities are, at present, routinely used in risk stratification for treatment, which has greatly contributed to outstanding improvements in treatment outcomes [3,7,8].
Certain genetic mutations that initiate the development of leukemia are acquired already in prenatal life. These mutations are the first hit, allowing certain blood cells to multiply faster than others, forming their own independent population, but this is not life-threatening. However, as we age, the number of acquired mutations increases, and if another “second hit” mutation occurs, these cells become malignant and multiply rapidly, and the result is leukemic transformation. B-ALL with ETV6::RUNX1 (formerly known as TEL::AML1) is classified as a well-distinguished entity by the World Health Organization. Based on scientific reports, this somatic chromosomal aberration within leukemic blasts is considered a favorable prognostic indicator and is associated with improved treatment results. Nevertheless, this notion is disputed because researchers report predominantly late relapses occurring in up to 20% of patients [9]. The increasing amount of research focuses on the fact that it is not the presence of ETV6::RUNX1 alone, but secondary rearrangements that can affect clinical implications. We focus in this study on the mechanism, significance, and treatment outcome of the most common translocation, t(12;21)(p13;q22), leading to the ETV6::RUNX1 fusion gene that is associated with the recurrent secondary alterations.
1.1. ETV6
The ETV6 gene (formerly known as TEL)consists of eight exons spanning 240 kb and is located on chromosome 12p13.2. It has been identified as a fusion partner in over 30 chromosomal translocation oncogenes. This gene encodes the essential hematopoietic transcriptional repressor erythroblast transformation specific (ETS) variant 6. Six isoforms of the ETV6 are distinguished, ranging in length from 245 to 452 aminoacids [10,11]. The transcript consists of three functional domains: an N-terminal pointed dimerization domain, also termed helix loop helix (HLH), or sterile alpha motif domain, a central linker domain, and an 85-amino acid C-terminal DNA-binding domain enabling the association with the promoter region of target genes [12]. The 57-kDa ETV6 protein belongs to the ETS superfamily of transcription factors associated with numerous biological processes and pathways, such as cell growth and differentiation, hematopoiesis, angiogenesis, and megakaryocyte development. ETV6 is highly expressed in early hematopoietic progenitor cells and is essential for hematopoiesis in the bone marrow [13,14]. At first, ETV6 was identified as the fusion partner of the platelet-derived growth factor receptor beta gene (PDGFRB) in a balanced t(5;12)(q21;p13) translocation from a patient with chronic myelomonocytic leukemia. In translocations involving MN1, BTL, and PAX5 (Figure 1), the DNA-binding domain of ETV6 is part of the leukemogenic fusion protein, which suggests that altered expression of normal ETV6 target genes is involved in the pathogenesis of leukemia [15,16,17]. The somatic or germline mutations in ETV6 have been shown to contribute to malignancies such as hematologic malignancies, including myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), high-risk B-ALL, early T-cell precursor (ETP) ALL, diffuse large B-cell lymphoma (DLBCL), melanoma, and colon, salivary gland, and breast cancer [18,19]. Furthermore, a relationship between heterozygous germline ETV6 mutations to dominantly inherited thrombocytopenia and predisposition to hematologic malignancies was defined [20,21].
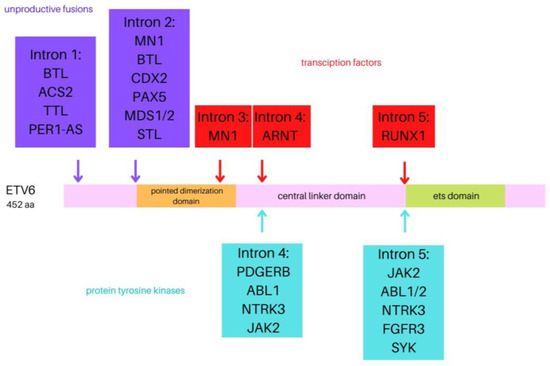
Figure 1.
Visual representation of the ETV6 protein with the position of the breakpoints of the various fusion partner genes.
Melazzini et al.’s study indicated that monoallelic ETV6 mutations result in a frequent form of inherited thrombocytopenia and a mild bleeding tendency in the affected patients, alongside the tendency towards hematologic malignancies, in particular ALL. ETV6-related thrombocytopenia belongs to a group of a few autosomal dominant forms of inherited thrombocytopenia without platelet macrocytosis. Therefore, screening for ETV6 mutations in all patients with these characteristics is recommended [22].
1.2. RUNX1
RUNX1 is a member of the evolutionarily conserved Runt transcription factor family. It is vital for the establishment of definitive hematopoiesis in vertebrates and is also an important regulator of the immune cells. RUNX1 is mapped on chromosome 21q22.12 and contains nine exons spanning 150 kb. The transcript can take the form of 1 of 11 isoforms [23]. The encoded protein, which can play the role of both an activator and a repressor, is a member of the core-binding transcription factors family together with RUNX2, RUNX3, and non-DNA-binding co-factor core-binding factor beta [24,25]. RUNX1 is essential for hematopoiesis. During hematopoietic stem cells (HSCs) differentiation, RUNX1 expression supports cell patterning and maintaining the correct lineage. RUNX1 is one of the most frequently mutated genes in various hematological malignancies. The alterations of RUNX1 can result in the loss of RUNX1 function or a dominant-negative effect. Mono-allelic RUNX1 mutations are found in approximately 15% of T-ALL, mainly in cases with an immature phenotype and a poor prognosis [26]. In de novoAML patients, somatic mutations in RUNX1 are detectable in 3% of pediatric and 15% of adult patients. Patients with MDS who have RUNX1 mutations have a greater chance of developing AML. Additionally, germline mutations in RUNX1 contribute to familial platelet disorder with a predisposition to acute myeloid leukemia (FPD/AML) occurrence. There are no statistical data regarding the frequency of FPD/AML; however, to this day, there have been over 70 families reported with this condition worldwide [27]. In addition, Latger-Cannard et al. obtained molecular characteristics of FPD/AML patients in France and found that germline RUNX1 mutations and deletions were the most widespread alterations found in those patients [28]. Collectively, there are more than 50 different types of chromosomal translocations that affect RUNX1 [29].
1.3. Detection, Mechanism, and Clinical Significance of t(12;21)
The ETV6::RUNX1 fusion transcript is detected in about 17% to 25% of pediatric B-cell precursors in ALL patients, making it one of the most common non-random, recurrent translocations associated with childhood ALL [7]. Exact values vary by country: India (6%), China (18.2%), Czech Republic (22%), Greece (22.3%), Korea (23.1%), Turkey (25.5%), Italy (25.9%), Belgium (25.9%), France (27%), and Iran (34.9%). In the context of the detection of fusion genes, diverse studies have presented a significantly lower prevalence of favorable ETV6::RUNX1 rearrangements in Mexicans (6.9–14.9%) and in Hispanics than in Caucasian or other ethnical backgrounds (25%). These studies suggested that the low occurrence of ETV6::RUNX1 could be the result of valuable geographic and ethnic differences connected with the percentage of European and Native American genetic background in Mexicans. In the study conducted by Mata-Rocha et al., 247 samples of BM were collected from Hispanic children from Mexico City with newly diagnosed ALL. The results showed that the occurrence of the ETV6::RUNX1 fusion gene transcript was at a level of 10.5% in the selected cohort. Although the reason for the variation in rates between nations is unknown, it has been suggested that they may be related to ethnic differences. Nevertheless, no significant differences in the frequency of secondary rearrangements have been observed among ETV6::RUNX1-positive (ETV6::RUNX1+) B-ALL pediatrics of different ethnical backgrounds [30,31,32,33,34,35,36,37,38,39,40]. This chromosomal translocation was first identified in 1994 by Romana et al. using the fluorescence in situ hybridization method (FISH). The t(12;21)(p13;q22) is commonly cryptic and ignored by metaphase cytogenetics, but can be detected using FISH or reverse transcriptase-polymerase chain reaction (RT-PCR) [41]. This translocation is most frequent among pediatric patients between 1 and 12 years of age, with a peak between 2 and 5 years, absent in infancy (age less than 1 year); nevertheless, between adult patients, it is rare (1–4.4%). The blast immunophenotype characteristically shows the expression of a higher intensity CD10 and HLA-DR with lower levels of the CD20, CD45, CD135, and CD34 antigens in comparison with the other genetic subtype B-ALL. In addition, ETV6::RUNX1 cases also co-express the myeloid antigens CD13, CD33, CD65, CD27, and low or lacking expression of CD44 [42,43]. The study of Blunck et al. focused on re-evaluating the CD9 cellular expression using flow cytometry as a screening method to estimate the presence of ETV6::RUNX1. For the analysis, a total of 186 BM aspirates and/or peripheral blood samples have been collected from B-ALL pediatric patients. Subsequently, immunophenotyping of BM aspirates has been performed. The ETV6::RUNX1 translocation has been found in 23.6% of cases. The analysis indicated that the best cut-off point for using the CD9 expression as a prediction tool for the detection of ETV6::RUNX1 was 64% of the cells. Blunk et al. observed a strong association between the level of CD9 expression and the presence of ETV6::RUNX1 [44].
In the course of the past decades, improvements have been made in understanding the precise role of the ETV6::RUNX1 fusion gene product in childhood ALL. ETV6 and RUNX1 encode transcription factors that play important roles in hematopoiesis, and deficiency in either result in the failure of embryonic hematopoiesis. The ETV6::RUNX1 transcript fuses ETV6 exons 1 through 5 with RUNX1 exons 2 through 8. The resulting ETV6::RUNX1 fusion transcript is controlled by the ETV6 promoter and includes the N-terminal HLH domains of ETV6 connected to a large C-terminal part of the RUNX1 coding sequence, containing the Runt and transactivation domains. This contrasts with RUNX1 fusion genes found in AML, which are controlled by the RUNX1 promoter and lack the RUNX1 C-terminus along with the transactivation domain (Figure 2) [45].
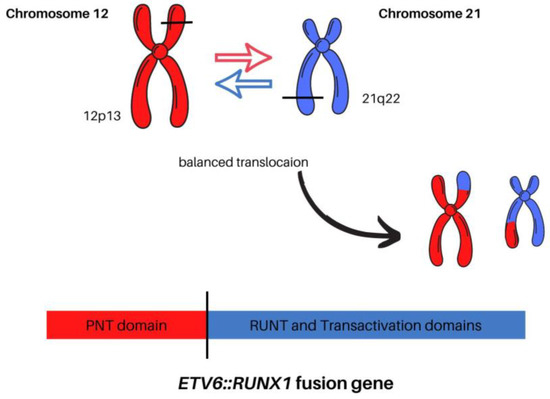
Figure 2.
The comparison between normal chromosomes 12 and 21 versus the t(12;21)(p13;q22) translocation.
Chimeric fusion genes are created by standard, error-prone repair of DNA double-strand breaks. Gene fusions between ETV6 and RUNX1 involve the noncoding introns of both genes, and the breaks are both distributed and diverse within the respective breakpoint cluster regions. The translocation t(12;21)(p13;q22) responsible for the fusion protein cause dimerization of an encoding 5′ region of the epithelium-specific ETS-like transcription factor 1 (ETS-like) and the ETV6 gene with nearly the entire RUNX1 gene, including its DNA-binding region and transactivation domain (Figure 3) [3]. The chromosome 12 breakpoints cluster within a single intron of the ETV6 gene, whereas RUNX1 breaks occur within the large and currently unsized first two introns of the RUNX1 gene on chromosome 21. In the case of other fusion genes in leukemia, each patient’s intronic breakpoints and the following fusion sequence are unique, which provides a stable genomic marker of the derivative clone of cells [46].
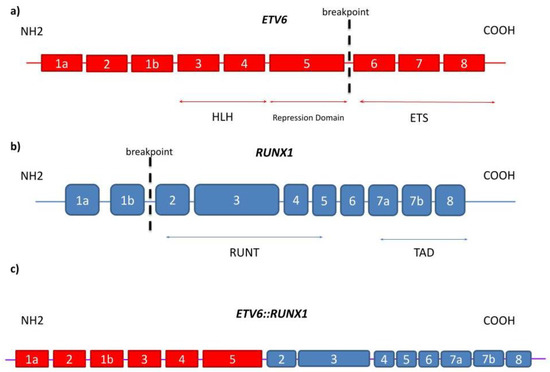
Figure 3.
(a) On ETV6 gene, which consists of nine exons, the translocation occurs between exon 5 and exon 6; (b) On RUNX1 gene, the breakpoint is located between exon 1b and exon 2 out of 10 exons in total; (c) the fusion results in formation of ETV6::RUNX1 translocation which is transcribed into ETV6::RUNX1 fusion protein.
The translocation results in ETV6::RUNX1 fusion gene emergence and is associated with excellent outcomes [47,48]. Reports have shown that among patients with ETV6::RUNX1+ ALL, the 5-year event-free survival (EFS) rate was from 80% to 97%, which was significantly higher than in other subtypes [30,48,49,50]. Nevertheless, the results of different studies indicate a high rate (20–24%) of relapse in ETV6::RUNX1+ patients, casting doubt on the prognostic significance of this genetic mutation [51,52]. In addition, resistance to chemotherapy and relapse concern 10% of patients, which emphasizes the need for further therapy improvements [47]. ETV6::RUNX1+ ALL is thought to arise prenatally and may be preceded by a pre-leukemic phase [53]. The controversially good prognosis of ETV6::RUNX1 and long latency period in identical twins with ALL demonstrated the need for the identification of additional secondary events in disease progression. The analysis of recurrent secondary rearrangements may provide novel prognostic factors to improve current relapse treatment strategies [48].
Despite the high occurrence of t(12;21) in B-ALL, the translocation was shown to be insufficient to induce leukemic transformation on its own [13]. The ETV6::RUNX1 fusion protein induces a silent preleukemic clone. The ETV6::RUNX1+ HSCs are able to self-renew, and they contribute to hematopoiesis; however, they differ from typical HSCs [51]. The translocation is usually acquired in utero in very early progenitor cells, prior to T- or B-cell receptor gene rearrangements [54]. As mentioned above, additional genetic hits are needed for the transition to leukemia. The further alterations are acquired independently with no preferential order, and they may include loss of ETV6 or alterations directed to genes regulating regular B-cell differentiation [52]. These genetic modifications can be obtained after years of latency and lead to ALL development (Figure 4) [29].
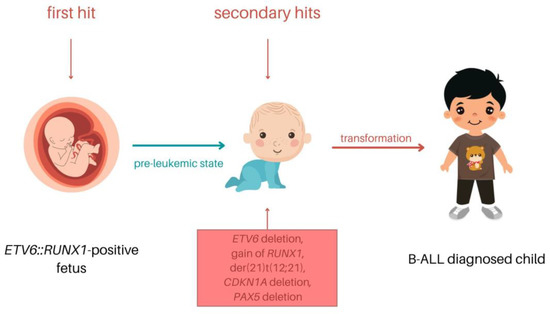
Figure 4.
The model of the pediatric ETV6::RUNX1 B-ALL process.
According to research on monozygotic twins with concordant leukemia, chromosomal translocations, which are typical for pediatric leukemia, often occur prenatally. Based on this hypothesis, preleukemic clones, functional fusion genes, and chromosome translocations should be far more frequent in healthy newborns’ blood than the general risk of developing the associated leukemia. Following the analysis of 253 healthy newborns’ cord blood, Zuna et al. state that 2% of newborns carried ETV6::RUNX1 fusion. In addition, liver and spleen tissue samples from aborted fetuses were tested for the presence of the ETV6::RUNX1 mutation, and the spleen tissue was found to harbor the mutation [55]. To analyze the prevalence of ETV6::RUNX1+ preleukemic cells in newborns, Fueller et al. developed a DNA-based method of genomic inverse PCR for the exploration of ligated breakpoints (GIPFEL) that enables the sensitive recognition of recurrent chromosomal translocations. A total of 50 out of 1000 samples were detected with ETV6::RUNX1 gene fusion, establishing that more than 5% of newborns harbor the ETV6::RUNX1 gene fusion. Translocation has been shown to be present frequently during regular fetal development and is 100 times more common than developing leukemia (1 in 10,000 children) [56]. Mori et al. confirmed previous reports that the frequency of ETV6::RUNX1 fusion genes commonly found in leukemia in cord blood is 100 times higher than the chance of developing the corresponding leukemia in the future. ETV6::RUNX1+ cells were found in more than 1% of cord blood from newborns. This indicates that ETV6::RUNX1 has a very low oncogenic potential and prophylactic screening of newborns carrying ETV6::RUNX1 is limited, considering that 99% of newborns will never develop the disease. These findings emphasize the significance of spontaneous or environmentally induced secondary hits in ETV6::RUNX1+ ALL [57].
2. Secondary Genetic Rearrangements
Numerous studies demonstrated that ETV6::RUNX1 rearranged B-ALL patients harbored a high number of copy number alterations (CNAs).
A study using the array comparative genomic hybridization carried out on eleven pediatric ETV6::RUNX1+ ALL patients by Zakaria et al. showed a significant amount of additional chromosomal aberrations: 119 losses, 36 gains. The number of aberrations per patient ranged from 3 to 58, with a mean of 14 aberrations per patient [58].
Zhang et al. sequenced 120 candidate genes in 187 patients with high-risk B-ALL. A total of eight ETV6 sequence mutations were detected in six patients. When ETV6 sequence mutations were combined with somatic copy number deletions, 12% had loss-of-function mutations in this gene, with 18 of 23 being monoallelic and 5 of 23 biallelic [59].
Rare anomalies in childhood ALL are near-tetraploidy (82–94 chromosomes), which was observed in ETV6::RUNX1+ ALL by Attarbaschi et al. The authors did not explain whether tetraploidization also is such a comparatively early evolutionary step that merely promotes the malignant potential of ETV6::RUNX1+ blast cells, or whether it is actually rather a late event or even the last trigger that sets off the clinical disease manifestation [60].
Borst et al. used SNP arrays to investigate secondary rearrangements found in pediatric Scandinavian patients enrolled in the NOPHO treatment protocol. After analyzing a total of 62 patients, 129 deletions were detected, and 59% of them ranged over 1Mbp. Interestingly, of a total number of 45 gains, 44 of them were above 1 Mbp. In only 13 patients, no CNAs were detected [61].
There is an abundance of research articles regarding secondary rearrangements in B-ALL; however, there is very little research regarding ETV6::RUNX1+ ALL specifically. Certain research contains percentage data on the prevalence of secondary mutations in ETV6::RUNX1+ ALL; however, their clinical relevance has not been established yet. In B-ALL, there are mutations whose presence allows for the stratification of patients upon the beginning of the treatment into low- or high-risk groups. Therefore, based on the existing knowledge, it can be assumed that those secondary mutations that have been associated with another B-ALL subgroup could be present as “second hit” mutations in the ETV6::RUNX group and have similar diagnostic and clinical implications. The described recurrent secondary genetic deletions in ETV6:RUNX1+ B-ALL in order from most to least frequent are deletions of 12p, 6q, 9p, 5q, 3p, 3q, 14q, 7p, 7q, 4q, 19q, 11q, 11p, 1q, 13q, and 8p, and the most common duplications are 4q, Xq [59,62,63,64,65]. The summary of these alterations related to ETV6::RUNX1+ B-ALL is presented in Table 1.

Table 1.
Recurrent secondary genetic changes found in childhood ETV6::RUNX1 B-ALL.
2.1. Chromosome 12
The most common secondary alteration detected in approximately 70% of ALL patients with ETV6::RUNX1 includes the loss of the normal or non-rearranged ETV6 homolog. ETV6 can function as a tumor suppressor and dimerize with ETV6::RUNX1 to reduce its transforming activity [59].
The majority of studies have reported deletion region 12p in patients with ETV6::RUNX1 [71]. This region is abundant in genes that are vital for B-cell development, such as CDKN1B (Cyclin Dependent Kinase Inhibitor 1B) and BCL2L14 (BCL2-Like 14). CDKN1B is a negative regulator of cell-cycle progression from the G1 to S phases. Deletions of CDKN1B have been observed in pediatric ALL as well as murine models of this disease [72]. BCL2L14 is a pro-apoptotic gene, therefore acting as a tumor suppressor [73,74].
The expression of BTG1 (B-cell translocation gene Anti-Proliferation Factor 1) is located on chromosome 12q21.33. The genotoxic and cellular stress signaling pathways, which both affect the activity of transcriptional co-factors and regulate posttranscriptional mRNA stability, are thought to be mediated by BTG1 and BTG2, respectively [75]. Research conducted on pediatric patients by Waanders et al. showed that deletion of BTG1 was present in 27 out of 142 (19%) ETV6::RUNX1+ cases [76].
Interestingly, ETV6 deletions were detected less frequently among patients with clonal RAS pathway mutation [77]. Nevertheless, there has been a great deal of differing research revealing the scarce, yet definite, presence of RAS mutations in patients with ETV6::RUNX1+ ALL. Nishii et al. analyzed different variants of ETV6 and assessed their transcriptional profile. Germline ETV6 variants were associated with hyperdiploid B-ALL, whereas somatic ETV6 mutations were found in ETV6::RUNX1+ ALL. In cases presenting “damaging ETV6 variants”, the aberrant transcription was driven by hyperdiploidy as well as gene aberrations such as RAS and NRAS pathway mutations. Without NRAS mutation, the cells harboring somatic ETV6 mutation did not undergo oncogenic transformation [78].
2.2. Chromosome 6
CCND3 gene is located on 6p21.1, and mutation of CCND3 results in cell-cycle deregulation. In the study by Ueno et al., with the use of panel sequencing, ETV6::RUNX1+ ALL was shown to be correlated with CCND3 mutation. The alteration was identified in 13 cases of the study, 8 of which (62%) were ETV6::RUNX1+ [64]. Van Delft et al. investigated the clonal origins of relapse by comparing the profiles of genome-wide CNA presented in 21 patients with those in matched relapse. There were identified, in total, 159 copy number alterations at presentation and 231 at relapse. Deletions of CDKN2A/B or CCNC (6q16.2-3), or both, increased from 38% at presentation to 76% in relapse, suggesting that cell-cycle deregulation contributed to the emergence of relapse [79]. RUNX1 gene is not a potent transcriptional regulator by itself. Instead, interactions with other transcriptional regulators such as transcription factors (FOX3, GATA2, SCL1, ERG), co-activators, or co-repressors, as well as post-translational changes that impact influence RUNX1 transcriptional activity [80].
2.3. Chromosome 21
RUNX1 gene has been characterized as a molecular switch that regulates the balance between proliferation and differentiation during development. Pais et al. conducted a large cohort study of 515 patients, in which 77 cases showed ETV6::RUNX1 fusion. In the ETV6::RUNX1+ group, 70% had additional ETV6::RUNX1 homolog changes. The changes that affect the RUNX1 gene are the presence of additional RUNX1 signals and the loss of residual RUNX1 [65].
SPI-B (encoded by SPIB) is an important transcriptional activator of B-cell development and differentiation. SPIB mRNA transcript levels are low in ETV6::RUNX1+ ALL relative to other leukemia subtypes, as the SPIB gene is directly activated by RUNX1 during B-cell development. Xu et al. hypothesized that SPIB is directly transcriptionally repressed by ETV6::RUNX1; therefore, using chromatin immunoprecipitation, they identified a regulatory region in the first intron of SPIB that interacts with ETV6::RUNX1 [81]. Additionally, the research conducted by Pang et al. proves that SPI-B expression is significantly reduced in ETV6::RUNX1+ ALL. Therefore, p21 and SPI-B are important targets of ETV6::RUNX1 in the regulation of B-cell gene expression in leukemogenesis [82].
To evaluate the impact of RUNX1 and ETV6::RUNX1 on RAG1 expression, Jakobczyk et al. used human B-cell precursor Nalm6 cells. In their study, enforced expression of ETV6::RUNX1 or RUNX1 in Nalm6 cells intensified the levels of endogenous RAG1 transcript and protein. Those results validated the direct or indirect upregulation of RAG1 expression by both ETV6::RUNX1 and RUNX1 [83].
2.4. Chromosome 9
Up to 25% of ETV::RUNX1+ children have the 9p deletion, which frequently impacts cyclin-dependent kinase inhibitor 2A or 2B (CDKN2A/CDKN2B) and the B-cell differentiation regulator PAX5 [84]. PAX5 is a gene located in 9p13. The absence of PAX5 activity causes developmental arrest and the lack of typical B-lymphoid lineage commitment. In mature B-cells, the inactivation of PAX5 leads to the loss of their identity. In a physiological complex containing IKZF1 and RUNX1, PAX5 can be discovered. PAX5 works as a metabolic gatekeeper with IKZF1 to regulate the distribution of glucose and energy. This limitation is lifted by heterozygous PAX5 deletion, which also boosts ATP levels and glucose absorption [85]. Agarwal et al. observed CNAs in the population of Indian pediatric patients. In a cohort of 91 B-ALL patients, 30.7% showed significant cytogenetic alterations. Four out of nine ETV6::RUNX1+ patients had ETV6 deletion, one patient had BTG1 deletion, and one patient had PAX5 deletion. It was stated that 12 months after induction chemotherapy, patients who were found with deletions in PAX5 and ETV6 had better survival outcomes as compared to those without gene alterations [86].
2.5. Chromosome 5
NR3C1 (Nuclear Receptor Subfamily 3 Group C Member 1) is a protein-coding gene that encodes a glucocorticoid receptor. Kuster et al. examined single nucleotide polymorphism arrays for DNA CNAs in 18 matched diagnoses and relapse leukemias to comprehend the molecular background of relapse mechanisms. Only 21.4% of patients had totally new mutations at relapse, whereas the additional 78.6% had a common prototype and later acquired distinctive CNA. Researchers detected recurring, generally nonoverlapping deletions related to apoptosis caused by glucocorticoids. It is targeted by the Bcl2 modifying factor (BMF) and the glucocorticoid receptor NR3C1. In the study by Kuster et al., 24 patients with relapse and 72 without relapse of ETV6::RUNX1+ were tested by FISH, and the results showed that BMF deletions were always present at diagnosis, while NR3C1 and mismatch repair aberrations predominated in relapse. The above-mentioned genes’ alteration was connected to leukemias’ treatment failures [87]. In Grausenburger et al. study, deletions involving the glucocorticoid receptor gene NR3C1 were particularly associated with poor response to induction therapy [88].
2.6. Chromosome 7
IKZF1 (Ikaros zinc finger transcription factor1), located on 7p12.2, regulates hematopoiesis and is essential for lymphoid differentiation [89]. Mullighan et al. proved that deletion or mutation of IKZF1 is related to poor treatment outcomes and a high risk of relapse among pediatric ALL patients [90]. Enshaei et al. investigated 368 ETV6::RUNX1+ and stated that six IKZF1-deleted patients have remained in complete remission for over 9.5 years [73].
IGF2BP (Insulin-like growth factor 2 mRNA-binding protein), located on 7p15.3, is a family of oncofetal RNA-binding protein genes consisting of IGF2B1, IGF2B2,and IGF2B3 [91]. In recent years, it has been proposed as a novel marker for ETV6::RUNX1 by Sharma et al., who observed that the expression of both ETV6::RUNX1 and IGF2BP1 correlated significantly with the blast percentage. Interestingly, the relative expression of ETV6::RUNX1 was much lower than IGF2BP1 levels contributing to the higher sensitivity of IGF2BP1 expression [92]. Research conducted by Stoskus et al. showed that among patients with ETV6::RUNX1 fusion, IGF2BP1 and IGF2BP3 were expressed at a higher level than IGF2BP2 [93]. Similar results were obtained by Mäkinen et al., who detected high levels of IGF2BP3 in patients with ETV6::RUNX1 [94].
2.7. Chromosome 11
KMT2A (lysine methyltransferase 2A) located in the 11q23 region gene encodes a transcriptional co-activator, which is crucial for controlling gene expression throughout hematopoiesis and early development [95]. Pais et al. reported a unique finding, which is the co-existence of ETV6::RUNX1 and KMT2A (previously known as MLL) aberrations. Translocation, monoallelic deletion, or rearrangement of KMT2A were identified in 9% of patients. Results suggested that KMT2A aberrations in the ETV6::RUNX1+ group had no prognostic significance [65].
Attarbaschi et al. classified KMT2A as a non-random and highly specific secondary aberration in ETV6::RUNX1+ ALL, rather than a disease-relevant primary abnormality. Their results provided evidence that ALL with KMT2A abnormalities represents a highly distinct and separate subgroup of ALL. Based thereon, they suggested that in future clinical ALL studies, cases with a cytogenetically evident KMT2A should not be evaluated together with KMT2A translocations anymore, but should be screened for the ETV6::RUNX1 fusion gene right away [60].
Papaemmanuil et al. performed a genomic analysis on 57 patients with ETV6::RUNX1+ ALL, which showed that RAG-initiated deletions, characterized by recombination signal sequence motifs near breakpoint junctions, appear as the predominant mutational process. A common target of deletions was the RAG locus on 11p12. What is more, Papaemmanuil et al. state that the major secondary events leading to leukemic transformation in ETV6::RUNX1+ ALL are often caused by genomic rearrangements mediated by aberrant RAG recombinase activity, and rarely by point mutations [96].
3. ETV6::RUNX1+ Prognosis and Treatment
The prognostic significance of the ETV6::RUNX1 fusion is still analyzed by scientists, as the results of the prognosis studies of this subgroup are ambiguous. Already in the 1990s, there were discrepancies regarding the prognosis of patients with ETV6::RUNX1 in B-ALL. The reports noted that as many as 25% of children on BFM protocols who suffer from relapses were ETV6::RUNX1+ [97].
Loh et al. conducted a prospective study to determine the incidence and outcomes of children with ETV6::RUNX1+ ALL. Patients were risk stratified primarily by current National Cancer Institute (NCI)–Rome risk criteria. According to their results, the 5-year OS rate was 97% among ETV6::RUNX1+ patients compared with 89% among ETV6::RUNX1− patients. The analysis concludes that patients with ETV6::RUNX1+ usually have excellent outcomes. However, factors such as age at diagnosis and presented leukocyte count should be considered during treatment [51].
Ampatzidou et al. studied, in a pediatric cohort of 119 B-ALLs, the relation between the ETV6::RUNX1 aberration and the co-existing subclones with clinical and biological features (sex, WBC, organ infiltration, karyotype, additional molecular aberrations), early response to treatment (MRD) and long-term outcome. In the study group, 22.7% of patients were ETV6::RUNX1+ and 70.4% harbored additional genetic abnormalities, while 33.3% presented clonal heterogeneity. A common feature of all recurrences was subclonal heterogeneity, FCM-MRD d15-positivity, and additional del(9p21). There were no altered recurrences among ETV6::RUNX1+ patients. In this study, the presence of clonal heterogeneity and impaired FCM-MRD clearance among ETV6::RUNX1+ patients influenced prognosis, but the author suggested that a longer follow-up was needed [62].
Qiu et al. conducted a multicenter, retrospective study on 2530 children to analyze the outcomes of ETV6::RUNX1+ pediatric B-ALL in South China, with the aim of identifying significant prognostic variables. ETV6::RUNX1+ translocation was present in 18.2% of cases. Qiu et al.’s study results demonstrate that chemotherapy protocol, WBC, age, and mixed-lineage leukemia gene rearrangement (MLL-r) status are all independent, significant predictors of the outcome among childhood B-ALL. Notably, this study mentioned that a large percentage of ETV6::RUNX1+ patients had moderate anemia and thrombocytopenia at the diagnosis onset [30].
The group of Piette et al. evaluated the long-term prognostic and predictive value of ETV6::RUNX1 in BCR::ABL1 negative de novo B-ALL children. Particular attention was given to the effects of the randomized treatments in the ETV6::RUNX1+ subgroup as compared with those observed in the high hyperdiploid and other analyzed B-ALL subgroups, in order to reveal specific drug response profiles related to distinct oncogenic processes. The 10-year EFS rate was 73.1–82.7%, but it was significantly higher in the ETV6::RUNX1+ than in the ETV6::RUNX1− group. ETV6::RUNX1+ patients also had a significantly higher 10-year OS rate exceeding 95.3%. It was remarkable that in the ETV6::RUNX1+ group, EFS events mostly occurred after the end of the maintenance therapy, with very few events before, and virtually no event after 6 years from diagnosis [37]. Pui et al. conducted a study where they incorporated more stringent risk classification, early intensification of intrathecal chemotherapy, reinduction treatment, and the use of prednisone instead of the commonly used dexamethasone in post-remission therapy. They found that sex, age, race, immunophenotype, CNS status, and the t(1;19)/E2A-PBX1 or ETV6::RUNX1 lacked prognostic significance [98].
One of the first examinations of the chemosensitivity of primary leukemic cells from children with newly diagnosed t(12;21)-positive B-ALL was conducted by Narla et al., whose results provided unprecedented evidence that ETV6::RUNX1+ ALL cells are more sensitive to dexamethasone and vincristine than ALL cells without ETV6::RUNX1 [99].
Ramakers-van Woerden et al. investigated the relationship between the presence of ETV6::RUNX1 and in vitro drug resistance in pediatric B-ALL using cells obtained from the BM or peripheral blood of 192 children with newly diagnosed ALL. The ETV6::RUNX1+ patients were slightly, but significantly, more resistant to vincristine and cytarabine, and comparatively more sensitive to l-asparaginase (L-ASP). These results imply that ETV6::RUNX1+ patients could benefit from therapy regimens with significant use of L-ASP [100].
To estimate whether outcomes for ETV6::RUNX1+ ALL are improved by contemporary risk-directed therapy, Bhojwani et al. studied clinical features, response, and adverse events of 168 children with newly diagnosed ETV6::RUNX1+ ALL in St. Jude Total Therapy studies XIIIA, XIIIB, and XV. Results were compared with 494 ETV6::RUNX1-negative (ETV6::RUNX1-) B-ALL patients. The incidence of ETV6::RUNX1 was associated with a patient age between 1 and 9 years, pre-treatment classification as low risk, and lower levels of MRD at 19 days of therapy. EFS or OS did not differ between patients with or without ETV6::RUNX1 in Total XIIIA or XIIIB. By contrast, in Total XV, patients with ETV6::RUNX1 had significantly better results than those without ETV6::RUNX1 fusion EFS (97% vs. 88%) and OS (99% vs. 94%). The MRD-guided treatment schema that included intensive asparaginase and high-dose methotrexate in the Total XV study produced significantly better outcomes than previous regimens, and demonstrated that nearly all children with ETV6::RUNX1+ ALL can be cured with risk-directed therapy including intensive asparaginase, vincristine, dexamethasone, and high-dose methotrexate [50].
A study conducted by Wang et al. proved a good prognosis for the ETV6::RUNX1+ ALL subgroup based on the clinical characteristics and treatment outcomes of 77 pediatric patients with ETV6::RUNX1+ ALL. The 5-year EFS and the disease-free survival (DFS) were reported to be 90% and 96%, respectively [49].
In general, maintenance therapy lasts over a year and consists of daily mercaptopurine and weekly methotrexate intake, and its role is to eradicate residual leukemic cells [79]. Shortening of maintenance therapy (terminated at 1 year after initiation of treatment) in ALL patients is thought to result in a high relapse rate [101]. However, studies indicate that some genetic subgroups, including ETV6::RUNX1, were associated with excellent DFS despite completing maintenance therapy at one year after diagnosis [102].
Kato et al. conducted a study to identify groups of patients who can be cured even with a shortened duration of maintenance therapy. After the comparison of genetic analysis results with clinical data, Kato suggested that short-duration therapy can cure more than 50% of pediatric ALL, especially in females and patients with TCF3::PBX1 or ETV6::RUNX1 mutations [103].
The translocation (12;21) had a strong impact on OS after relapse. Gandemer et al. conducted a long-term, follow-up retrospective study based on patients included in the French group for the childhood ALL 93 trial (FRALLE 93) protocol to evaluate the outcomes of patients with relapsed ETV6::RUNX1+ ALL. Among the 713 children with ALL tested for ETV6::RUNX1, 191 patients were ETV6::RUNX1+, of which 19.4% had a relapse. The three-year survival rate for ETV6::RUNX1+ patients totaled 64.7%, compared to 46.5% for ETV6::RUNX1-. ETV6::RUNX1+ ALL male patients had a longer remission-free period compared to patients with other B-ALL. However, in ETV6::RUNX1+ male patients, a significant increase was noticed in testicular cancer relapses. The authors point out that among patients with ETV6::RUNX1 mutation, more attention should be focused on long-term follow up to relapses in gonads or usage of chemotherapeutics that do not cross the testicular barrier [38].
4. ETV6::RUNX1+ Future Treatment Perspective
Despite significant improvements in the treatment of children with ALL, it is emphasized that the intensity of conventional chemotherapy has reached the limit of tolerance. Therefore, cytostatic dosing can no longer be increased with the hope of achieving better results. This reinforces the need to search for a new solution to toxic chemotherapy, such as molecular and immune therapies. Pui suggested that among different ALL subgroups, children with ETV6::RUNX1 notably appear to be suitable candidates for reduced standard chemotherapy treatment [98]. In the Associazione Italiana di Ematologia e Oncologia Pediatrica (AIEOP) and BFM ALL 2000 protocol, patients from 1to 17 years of age with standard-risk B-ALL were randomized to receive standard treatment or reduced delayed intensification. With the exception of individuals with ETV6::RUNX1+ ALL or patients of one to six years of age, who responded equally well toall treatment regimens, this modification influenced total eight-year DFS, which varied from 89.2% to 92.3% between the experimental reduced-intensity group and standard treatment group [104].
It has been established that cytotoxic chemotherapeutics, frequently applied in the treatment of leukemia, act by promoting apoptosis in sensitive target cells. Faulty apoptosis may contribute to the development of leukemia resistance to chemotherapy. Several positive and negative regulators of apoptosis have been identified, including the pro-apoptotic receptorCD95/Fas and the anti-apoptotic protein BCL2. Narla et al. conducted an in vitro study on the chemosensitivity of primary leukemic cells from newly diagnosed ETV6::RUNX1+ ALL patients vs. ETV6::RUNX1- ALL cells. Their results indicated that ETV6::RUNX1+ ALL cells expressed higher levels of the pro-apoptotic protein Fas and lower levels of the anti-apoptotic protein Bcl2 than ETV6::RUNX1− ALL cells. Moreover, the ETV6::RUNX1+ ALL cells were more sensitive to the apoptosis-inducing effects of serum deprivation, dexamethasone, and vincristine than ETV6::RUNX1− ALL cells [99].
Target therapy is not currently available for children with ETV6::RUNX1+ ALL. To identify the molecular mechanisms underlying ETV6::RUNX1+ ALL, Polak et al. performed gene expression profiling in healthy hematopoietic progenitors ectopically expressing ETV6::RUNX1. In that process, they revealed a transcriptional network driven by ETV6::RUNX1 that induces proliferation, survival, and cellular homeostasis. Moreover, Vps34, an important regulator of autophagy, was found to be induced by ETV6::RUNX1 and upregulated in ETV6::RUNX1+ cells. Inhibition of Vps34 in ETV6::RUNX1+ cell lines severely reduced proliferation and survival. Hydroxychloroquine was applied to inhibit autophagy, which reduced cell viability in both ETV6::RUNX1+ cell lines and primary ALL samples. In addition, it selectively sensitized primary ETV6::RUNX1+ ALL samples to L-asparaginase [53].
Serafin et al. aimed to assess the role of spleen tyrosine kinase (SYK) in cells with ETV6::RUNX1 translocation from B-ALL patients. In order to sensitize the resistant primary cells to conventional medications, Serafin treated cells from patients both at the time of diagnosis and after relapse with the combination of entospletinib and chemotherapeutics. Their results suggested thatentospletinib is able to induce cell death and enhance the efficacy of conventional chemotherapeutics. However, more extensive studies considering other SYKs are required [105].
Akbari et al. proposed an application of homologous recombination (HR) using an in vivo model of ETV6::RUNX1 as a way of targeted therapy in ETV6::RUNX1+ ALL. The results showed that the expression of ETV6::RUNX1 was significantly decreased in the HR-edited cells. Moreover, the edited cells had decreased viability in comparison with non-edited cells. Hopefully, in the future, modification by the HR technique may have the effect of reducing the leukemic effect of the fusion associated with the ETV6 and RUNX1 genes; however, it is still too early to introduce this method into clinical practice [106].
5. ETV6::RUNX1-like
In recent years, multiple groups from the USA, Europe, Japan, and China have generated or used B-ALL RNA-seq data to identify new targets of recurring rearrangement (e.g., DUX4, MEF2D, and ZNF384) associated with distinct gene expression profiles and the presence of cases with alterations that phenocopy additional canonical B-ALL drivers, e.g., ETV6::RUNX1-like ALL [107,108]. ETV6::RUNX1-like occurs in about 2–3% of children and less than 1% of adult patients with B-ALL. ETV6::RUNX1-like is a subtype of B-ALL defined by the ETV6::RUNX1-specific gene expression profile harboring concurrent ETV6 and IKZF1 lesions, but no exact ETV6::RUNX1 gene fusion. Both IKZF1 and RUNX1 encode transcription factors crucial for B-cell maturation, and there is speculation that the loss of IKZF1 may replace the altered function of RUNX1 in the ETV6::RUNX1 fusion protein. Consistent with this, Lilljebjörn et al. note that IKZF1 deletions are rare in ETV6::RUNX1+ patients and occur in approximately 3% of them. Among the changes in ETV6::RUNX1-like ALL patients, the following fusions are included: ETV6 and PMEL; IKZF1 and CDK2; ETV6 and BORCS5; SETD5 and IKZF1; ETV6 and NID1; ETV6 and CREBBP; ETV6 and BCL2L14; and ETV6 and MSH6. In addition, deletions of the second ETV6 allele and 7p were detected [107].
At the early stages of B-cell development, the precursors undergo immunoglobulin gene rearrangement, which is necessary for BCR formation on mature B-cells. Studies on mice have shown that recombination-activating genes (RAGs) are essential for this process. Chen et al. analyzed 1582 ALL patients, using both microarray and RNAseq data sets, to determine which subtypes express RAG1 or RAG2 and if any genes are co-expressed whose presence could identify new subtypes. Chen et al. analyzed the expression of RAG1 and RAG2 genes. Both genes were detected in the ETV6::RUNX1 subtype. In particular, RAG1 expression was consistently higher in the ETV6::RUNX1 compared to all other genetic subtypes, except the B-other ALL group. In a genomic screen for genes co-expressed with RAG1 and RAG2, none appeared consistently with RAG2, but an expression set of 31 genes was identified with RAG1. Using RAG1 along with the co-expression genes as an identifier distinguishes ETV6::RUNX1 from all other B-ALL subtypes, except for five cases. ETV6 target genes CLIC5, WBP1L, ANGPTL2, and BIRC7 were expressed at very high levels in both ETV6::RUNX1 and ETV6::RUNX1-like samples, with a consistent pattern and levels. The results of the Chen et al. study suggest that the RAG1 signature identifies a new ETV6::RUNX1-like subtype, since there are no definitive genetic markers that identify this new subtype [109].
A recent study by Zaliova et al. aimed to answer the question of whether ETV6::RUNX1-like can be distinguished by a specific immunophenotype. During diagnostic immunophenotyping of 573 pediatric B-ALL, researchers, among the B-other ALL patient group, identified eight cases with characteristic immunophenotypes for ETV6::RUNX1+ patients. The results suggest that the expression pattern of two surface proteins, CD27-positive and CD44-low-negative, distinguish ETV6::RUNX1-like ALL from other B-ALL. In addition, in genetic studies, the two most common alterations associated with ETV6 loss were various IKZF1 mutations and ARPP21 deletions [110].
6. Conclusions
The most prevalent genetic aberration in pediatric B-ALL is the t(12;21)(p13;q22) translocation. Accurate characterization of the disease genotype remains a priority, as it forms the basis for diagnosis, prognosis, risk assessment, and choice of therapy. Studies indicate that depending on the nature of the second hit, ETV6::RUNX1 ALL can be divided into subtypes based on the gene expression profile. Integrating the knowledge about ETV6::RUNX1 fusion and secondary molecular rearrangements into a treatment approach could reduce patients’ chance of relapse and improve outcomes.
Author Contributions
M.L. was responsible for the conception; M.L. and M.N. were responsible for the design of the study; A.K, J.D. and M.P. were responsible for the acquisition of the literature for the manuscript; A.K., J.D. and M.L. wrote the original draft of the manuscript; M.L. and M.N. reviewed and edited; M.L. supervised the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AIEOP | AssociazioneItaliana di Ematologia e OncologiaPediatrica |
| AIM1 | Absent in melanoma 1 protein |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| ANGPTL2 | Angiopoietin-like 2 |
| ARPP21 | CAMP-regulated phosphoprotein 21 |
| B-ALL | B-cell acute lymphoblastic leukemia |
| BCR::ABL1 | BCR and ABL fusion gene |
| BCR::ABL1-like ALL | BCR and ABL fusion gene-like acute lymphoblastic leukemia |
| BFM | Berlin–Frankfurt–Münster |
| BIRC7 | Baculoviral IAP repeat containing 7 |
| BM | Bone marrow |
| BMF | Bcl2 modifying factor |
| BTG1 | BTG-anti-proliferation factor 1 |
| CD95 | Cluster of differentiation 95 |
| CDKN1A | Cyclin-dependent kinase inhibitor 1A |
| CDKN1B | Cyclin-dependent kinase inhibitor 1B |
| CLIC5 | Chloride intracellular channel 5 |
| CNAs | Copy number alteration |
| CNS1 | CNS1 = patients with white blood cell count in cerebrospinal fluid < 5 and having no blasts in the cerebrospinal fluid |
| DLBCL | Diffuse large B-cell lymphoma |
| EBF1 | EBF transcription factor 1 |
| EFS | Event-free survival |
| ETS | Erythroblast transformation specific |
| ETV6 | ETS variant transcription factor 6 |
| ETV6::RUNX1 | ETS variant transcription factor 6 and RUNX family transcription factor 1 fusion gene |
| ETV6::RUNX1-like ALL | ETS variant transcription factor 6 and RUNX family transcription factor 1 fusion gene-like acute lymphoblastic leukemia |
| ETV6::RUNX1+ | ETV6::RUNX1-positive |
| ETV6::RUNX1− | ETV6::RUNX1-negative |
| FISH | Fluorescence in situ hybridization (FISH) |
| FLT3 | Fms-related receptor tyrosine kinase 3 |
| FOXO3A | Forkheadbox O3 |
| FPD/AML | familial platelet disorder with predisposition to acute myeloid leukemia (FPD/AML) |
| FRALLE 93 | French group for childhood ALL 93 trial (FRALLE 93 |
| GD-ALL-2008 | Guangdong Children’s Leukemia Group-ALL-2008 |
| GDP | Guanosine diphosphate |
| GIPFEL | Genomic inverse PCR for exploration of ligated breakpoints |
| GTP | Bound and inactive guanosine diphosphate |
| HLH | Helix loop helix |
| HRAS | HRasproto-oncogene, GTPase |
| HSCs | Hematopoietic stem cells |
| iAMP21 | Intrachromosomal amplification of chromosome 21 |
| IGF2BP | Insulin-like growth factor 2 mRNA-binding protein |
| IGF2BP1 | Insulin-like growth factor 2 MRNA-Binding Protein 1 |
| IGF2BP2 | Insulin-like growth factor 2 MRNA-Binding Protein 2 |
| IGF2BP3 | Insulin-like growth factor 2 MRNA-Binding Protein 3 |
| IGH::IL3 | Immunoglobulin heavy locus and interleukin 3 fusion gene |
| IL7R | Interleukin 7 receptor |
| KMT2A | lysine methyltransferase 2A |
| KRAS | KRAS proto-oncogene, GTPase |
| L-ASP | L-asparaginase |
| MDS | Myelodysplastic syndrome |
| MLL-r | mixed-lineage leukemia gene rearrangement |
| MLPA | Multiplex ligation probe-dependent amplification |
| MRD | Minimal residual disease |
| NF1 | Neurofibromin 1 |
| NGS | Next-generation sequencing |
| NOPHO | Nordic Society of Pediatric Hematology and Oncology |
| NR3C1 | Nuclear receptor subfamily 3 group C member 1 |
| NRAS | NRAS proto-oncogene, GTPase |
| OS | Overall survival |
| PAX5 | Paired box 5 |
| PTPN11 | Protein tyrosine phosphatase non-receptor type 11 |
| PTPN11 | Protein tyrosine phosphatase non-receptor type 11 |
| RAGs | Recombination-activating genes |
| RAS | Protein family include |
| RHD | DNA-binding Runt-domain |
| RT-PCR | Reverse transcriptase-polymerase chain reaction |
| RUNX1 | RUNX family transcription factor 1 |
| SPI-B | Spi-B transcription factor |
| SYK | Spleen tyrosine kinase |
| T-ALL | T-cell acute lymphoblastic leukemia |
| TCF3::HLF | Transcription factor 3 and HLF transcription factor fusion gene |
| TCF3::PBX1 | Transcription factor 3 and PBX homeobox 1 |
| WBC | White blood cell |
| WBP1L | WW domain-binding protein 1-like |
| WGS/WES | Whole genome/whole exome sequencing |
| WHO-HAEM5 | 5th edition of the World Health Organization Classification of HaematolymphoidTumours |
| ZFP423 | Zinc finger protein 423 |
References
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA: A Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef]
- Lejman, M.; Chałupnik, A.; Chilimoniuk, Z.; Dobosz, M. Genetic Biomarkers and Their Clinical Implications in B-Cell Acute Lymphoblastic Leukemia in Children. Int. J. Mol. Sci. 2022, 23, 2755. [Google Scholar] [CrossRef]
- Inaba, H.; Pui, C.-H. Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia. J. Clin. Med. 2021, 10, 1926. [Google Scholar] [CrossRef]
- Jeha, S.; Pei, D.; Choi, J.; Cheng, C.; Sandlund, J.T.; Coustan-Smith, E.; Campana, D.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J. Clin. Oncol. 2019, 37, 3377–3391. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.D.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of HaematolymphoidTumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Duffield, A.S.; Mullighan, C.G.; Borowitz, M.J. International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Arch. 2022, 1. Available online: https://link.springer.com/article/10.1007/s00428-022-03448-8#citeas (accessed on 24 November 2022). [CrossRef]
- Szczepański, T.; Harrison, C.J.; van Dongen, J.J. Genetic aberrations in paediatric acute leukaemias and implications for management of patients. Lancet Oncol. 2010, 11, 880–889. [Google Scholar] [CrossRef]
- Tran, T.H.; Tasian, S.K. Has Ph-like ALL Superseded Ph+ ALL as the Least Favorable Subtype? BestPract. Res. Clin. Haematol. 2021, 34, 101331. [Google Scholar] [CrossRef]
- Forestier, E.; Heyman, M.; Andersen, M.K.; Autio, K.; Blennow, E.; Borgström, G.; Golovleva, I.; Heim, S.; Heinonen, K.; Hovland, R.; et al. Outcome of ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia in the NOPHO-ALL-1992 protocol: Frequent late relapses but good overall survival. Br. J. Haematol. 2008, 140, 665–672. [Google Scholar] [CrossRef]
- ETV6 ETS Variant Transcription Factor 6 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=2120 (accessed on 14 January 2023).
- Bohlander, S.K. ETV6: A versatile player in leukemogenesis. Semin. Cancer Biol. 2005, 15, 162–174. [Google Scholar] [CrossRef]
- Fisher, M.H.; Kirkpatrick, G.D.; Stevens, B.; Jones, C.; Callaghan, M.; Rajpurkar, M.; Fulbright, J.; Cooper, M.A.; Rowley, J.; Porter, C.C.; et al. ETV6 Germline Mutations Cause HDAC3/NCOR2 Mislocalization and Upregulation of Interferon Response Genes. JCI Insight 2020, 5, e140332. [Google Scholar] [CrossRef] [PubMed]
- Neveu, B.; Richer, C.; Cassart, P.; Caron, M.; Jimenez-Cortes, C.; St-Onge, P.; Fuchs, C.; Garnier, N.; Gobeil, S.; Sinnett, D. Identification of new ETV6 modulators through a high-throughput functional screening. Iscience 2022, 25, 103858. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Rispoli, R.; Patient, R.; Ciau-Uitz, A.; Porcher, C. Etv6 activates vegfa expression through positive and negative transcriptional regulatory networks in Xenopus embryos. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- van Wely, K.H.M.; Meester-Smoor, M.A.; Janssen, M.J.F.W.; Aarnoudse, A.-J.; Grosveld, G.C.; Zwarthoff, E.C. The MN1-TEL myeloid leukemia-associated fusion protein has a dominant-negative effect on RAR-RXR-mediated transcription. Oncogene 2007, 26, 5733–5740. [Google Scholar] [CrossRef] [PubMed]
- Cools, J.; Bilhou-Nabera, C.; Wlodarska, I.; Cabrol, C.; Talmant, P.; Bernard, P.; Hagemeijer, A.; Marynen, P. Fusion of a Novel Gene, BTL, to ETV6 in Acute Myeloid Leukemias with a t(4;12)(Q11-Q12;P13). Blood 1999, 94, 1820–1824. [Google Scholar] [CrossRef]
- Fazio, G.; Palmi, C.; Rolink, A.; Biondi, A.; Cazzaniga, G. PAX5/TEL Acts as a Transcriptional Repressor Causing down-Modulation of CD19, Enhances Migration to CXCL12, and Confers Survival Advantage in Pre-BI Cells. Cancer Res. 2008, 68, 181–189. [Google Scholar] [CrossRef]
- Zhang, M.; E Churpek, J.; Keel, S.B.; Walsh, T.; Lee, M.K.; Loeb, K.R.; Gulsuner, S.; Pritchard, C.C.; Sanchez-Bonilla, M.; Delrow, J.J.; et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat. Genet. 2015, 47, 180–185. [Google Scholar] [CrossRef]
- Wang, M.; Gu, D.; Du, M.; Xu, Z.; Zhang, S.; Zhu, L.; Lu, J.; Zhang, R.; Xing, J.; Miao, X.; et al. Common genetic variation in ETV6 is associated with colorectal cancer susceptibility. Nat. Commun. 2016, 7, 11478. [Google Scholar] [CrossRef]
- Noetzli, L.; Lo, R.W.; Lee-Sherick, A.B.; Callaghan, M.; Noris, P.; Savoia, A.; Rajpurkar, M.; Jones, K.; Gowan, K.; Balduini, C.; et al. Germline Mutations in ETV6 Are Associated with Thrombocytopenia, Red Cell Macrocytosis and Predisposition to Lymphoblastic Leukemia. Nat. Genet. 2015, 47, 535–538. [Google Scholar] [CrossRef]
- Di Paola, J.; Porter, C.C. ETV6-related thrombocytopenia and leukemia predisposition. Blood 2019, 134, 663–667. [Google Scholar] [CrossRef]
- Melazzini, F.; Palombo, F.; Balduini, A.; De Rocco, D.; Marconi, C.; Noris, P.; Gnan, C.; Pippucci, T.; Bozzi, V.; Faleschini, M.; et al. Clinical and pathogenic features of ETV6 -related thrombocytopenia with predisposition to acute lymphoblastic leukemia. Haematologica 2016, 101, 1333–1342. [Google Scholar] [CrossRef]
- RUNX1 RUNX Family Transcription Factor 1 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=861 (accessed on 14 January 2023).
- Lie-A-Ling, M.; Mevel, R.; Patel, R.; Blyth, K.; Baena, E.; Kouskoff, V.; Lacaud, G. RUNX1 Dosage in Development and Cancer. Mol. Cells 2020, 43, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Hass, M.R.; Brissette, D.; Parameswaran, S.; Pujato, M.; Donmez, O.; Kottyan, L.C.; Weirauch, M.T.; Kopan, R. Runx1 shapes the chromatin landscape via a cascade of direct and indirect targets. PLoS Genet. 2021, 17, e1009574. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Kern, W.; Harbich, S.; Alpermann, T.; Jeromin, S.; Schnittger, S.; Haferlach, C.; Haferlach, T.; Kohlmann, A. Prognostic relevance of RUNX1 mutations in T-cell acute lymphoblastic leukemia. Haematologica 2011, 96, 1874–1877. [Google Scholar] [CrossRef]
- Silva, M.C.D.A.; Krepischi, A.C.V.; Kulikowski, L.D.; Zanardo, É.A.; Nardinelli, L.; Leal, A.M.; Costa, S.S.; Muto, N.H.; Rocha, V.; Velloso, E.D.R.P. Deletion of RUNX1 exons 1 and 2 associated with familial platelet disorder with propensity to acute myeloid leukemia. Cancer Genet. 2018, 222–223, 32–37. [Google Scholar] [CrossRef]
- Latger-Cannard, V.; Philippe, C.; Bouquet, A.; Baccini, V.; Alessi, M.-C.; Ankri, A.; Bauters, A.; Bayart, S.; Cornillet-Lefebvre, P.; Daliphard, S.; et al. Haematological spectrum and genotype-phenotype correlations in nine unrelated families with RUNX1 mutations from the French network on inherited platelet disorders. Orphanet J. Rare Dis. 2016, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Seitz, V.; Kleo, K.; Dröge, A.; Schaper, S.; Elezkurtaj, S.; Bedjaoui, N.; Dimitrova, L.; Sommerfeld, A.; Berg, E.; von der Wall, E.; et al. Evidence for a role of RUNX1 as recombinase cofactor for TCRβ rearrangements and pathological deletions in ETV6-RUNX1 ALL. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Qiu, K.Y.; Xu, H.G.; Luo, X.Q.; Mai, H.R.; Liao, N.; Yang, L.H.; Zheng, M.C.; Wan, W.Q.; Wu, X.D.; Liu, R.Y.; et al. Prognostic Value and Outcome for ETV6/RUNX1-Positive Pediatric Acute Lymphoblastic Leukemia: A Report From the South China Children’s Leukemia Group. Front. Oncol. 2021, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kim, S.; Jang, P.-S.; Chung, N.-G.; Cho, B. Differing Outcomes of Patients with High Hyperdiploidy and ETV6-RUNX1 Rearrangement in Korean Pediatric Precursor B Cell Acute Lymphoblastic Leukemia. Cancer Res. Treat. 2021, 53, 567–575. [Google Scholar] [CrossRef]
- Jarosova, M.; Volejnikova, J.; Porizkova, I.; Holzerova, M.; Pospisilova, D.; Novak, Z.; Vrbkova, J.; Mihal, V. Chromosomal aberrations in childhood acute lymphoblastic leukemia: 15-year single center experience. Cancer Genet. 2016, 209, 340–347. [Google Scholar] [CrossRef]
- Ampatzidou, Μ.; Florentin, L.; Papadakis, V.; Paterakis, G.; Tzanoudaki, M.; Bouzarelou, D.; Papadhimitriou, S.I.; Polychronopoulou, S. Copy Number Alteration Profile Provides Additional Prognostic Value for Acute Lymphoblastic Leukemia Patients Treated on BFM Protocols. Cancers 2021, 13, 3289. [Google Scholar] [CrossRef] [PubMed]
- Aydin, C.; Cetin, Z.; Manguoglu, A.E.; Tayfun, F.; Clark, O.A.; Kupesiz, A.; Akkaya, B.; Karauzum, S.B. Evaluation of ETV6/RUNX1 Fusion and Additional Abnormalities Involving ETV6 and/or RUNX1 Genes Using FISH Technique in Patients with Childhood Acute Lymphoblastic Leukemia. Indian J. Hematol. Blood Transfus 2016, 32, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-J.; Zhu, X.-H.; Yang, Y.; Wu, Y.; Lu, F.-J.; Zhai, X.-W.; Wang, H.-S. Prevalence of ETV6–RUNX1 fusion gene in children with acute lymphoblastic leukemia in China. Cancer Genet. Cytogenet. 2007, 178, 57–60. [Google Scholar] [CrossRef]
- Inamdar, N.; A Kumar, S.; Banavali, S.D.; Advani, S.; Magrath, I.; Bhatia, K. Comparative incidence of the rearrangements of TEL/AML1 and ALL1 genes in pediatric precursor B acute lymphoblastic leukemias in India. Int. J. Oncol. 1998, 13, 1319–1341. [Google Scholar] [CrossRef]
- Piette, C.; Suciu, S.; Clappier, E.; Bertrand, Y.; Drunat, S.; Girard, S.; Yakouben, K.; Plat, G.; Dastugue, N.; Mazingue, F.; et al. Differential impact of drugs on the outcome of ETV6-RUNX1 positive childhood B-cell precursor acute lymphoblastic leukaemia: Results of the EORTC CLG 58881 and 58951 trials. Leukemia 2017, 32, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Gandemer, V.; Chevret, S.; Petit, A.; Vermylen, C.; Leblanc, T.; Michel, G.; Schmitt, C.; Lejars, O.; Schneider, P.; Demeocq, F.; et al. Excellent Prognosis of Late Relapses of ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukemia: Lessons from the FRALLE 93 Protocol. Haematologica 2012, 97, 1743–1750. [Google Scholar] [CrossRef]
- Conter, V.; Bartram, C.R.; Valsecchi, M.G.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Aricò, M.; Zimmermann, M.; Mann, G.; De Rossi, G.; et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: Results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010, 115, 3206–3214. [Google Scholar] [CrossRef]
- Mata-Rocha, M.; Rangel-López, A.; Jimenez-Hernandez, E.; Nuñez-Enríquez, J.C.; Morales-Castillo, B.A.; Sánchez-Escobar, N.; Sepúlveda-Robles, O.A.; Bravata-Alcántara, J.C.; Nájera-Cortés, A.S.; Pérez-Saldivar, M.L.; et al. Low Prevalence of ETV6::RUNX1 Fusion Gene in a Hispanic Population. Front. Pediatr. 2022, 10, 837656. [Google Scholar] [CrossRef]
- Romana, S.P.; Mauchauffé, M.; Le Coniat, M.; Chumakov, I.; Le Paslier, D.; Berger, R.; Bernard, A.O. The t(12;21) of Acute Lymphoblastic Leukemia Results in a Tel-AML1 Gene Fusion. Blood 1995, 85, 3662–3670. [Google Scholar]
- Vaskova, M.; Fronkova, E.; Starkova, J.; Kalina, T.; Mejstrikova, E.; Hrusak, O. CD44 and CD27 delineate B-precursor stages with different recombination status and with an uneven distribution in nonmalignant and malignant hematopoiesis. Tissue Antigens 2007, 71, 57–66. [Google Scholar] [CrossRef]
- de Zen, L.; Orfao, A.; Cazzaniga, G.; Masiero, L.; Cocito, M.G.; Spinelli, M.; Rivolta, A.; Biondi, A.; Zanesco, L.; Basso, G. Quantitative Multiparametric Immunophenotyping in Acute Lymphoblastic Leukemia: Correlation with Specific Genotype. I. ETV6/AML1 ALLs Identification. Leukemia 2000, 14, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Blunck, C.B.; Terra-Granado, E.; Noronha, E.P.; Wajnberg, G.; Passetti, F.; Pombo-De-Oliveira, M.S.; Emerenciano, M. CD9 predicts ETV6-RUNX1 in childhood B-cell precursor acute lymphoblastic leukemia. Hematol. Transfus. Cell Ther. 2019, 41, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Ohira, M.; Shimizu, K.; Mitani, K.; Hirai, H.; Imai, T.; Yokoyama, K.; Soceda, E.; Ohkl, M. Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucleic Acids Res. 1995, 23, 2762–2769. [Google Scholar] [CrossRef] [PubMed]
- Wiemels, J.L.; Cazzaniga, G.; Daniotti, M.; Eden, O.B.; Addison, G.M.; Masera, G.; Saha, V.; Biondi, A.; Greaves, M.F. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet 1999, 354, 1499–1503. [Google Scholar] [CrossRef]
- Golub, T.R.; Barker, G.F.; Bohlander, S.K.; Hiebert, S.W.; Ward, D.C.; Bray-Ward, P.; Morgan, E.; Raimondi, S.C.; Rowley, J.D.; Gilliland, D.G. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 1995, 92, 4917–4921. [Google Scholar] [CrossRef]
- Ford, A.M.; Bennett, C.A.; Price, C.M.; Bruin, M.C.A.; Van Wering, E.R.; Greaves, M. Fetal origins of the TEL-AML1 fusion gene in identical twins with leukemia. Proc. Natl. Acad. Sci.USA 1998, 95, 4584–4588. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, H.M.; Zhang, L.P. ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukemia in China: Excellent Prognosis with Improved BFM Protocol. Ital. J. Pediatr. 2018, 44, 94. [Google Scholar] [CrossRef]
- Bhojwani, D.; Pei, D.; Sandlund, J.T.; Jeha, S.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; A Shurtleff, S.; Onciu, M.; Cheng, C.; et al. ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: Improved outcome with contemporary therapy. Leukemia 2011, 26, 265–270. [Google Scholar] [CrossRef]
- Loh, M.L.; Goldwasser, M.A.; Silverman, L.B.; Poon, W.-M.; Vattikuti, S.; Cardoso, A.; Neuberg, D.S.; Shannon, K.M.; Sallan, S.E.; Gilliland, D.G. Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood 2006, 107, 4508–4513. [Google Scholar] [CrossRef]
- Li, X.-Y.; Li, J.-Q.; Luo, X.-Q.; Wu, X.-D.; Sun, X.; Xu, H.-G.; Li, C.-G.; Liu, R.-Y.; Sun, X.-F.; Chen, H.-Q.; et al. Reduced intensity of early intensification does not increase the risk of relapse in children with standard risk acute lymphoblastic leukemia—A multi-centric clinical study of GD-2008-ALL protocol. BMC Cancer 2021, 21, 1–11. [Google Scholar] [CrossRef]
- Polak, R.; Bierings, M.B.; Van Der Leije, C.S.; Sanders, M.A.; Roovers, O.; Marchante, J.R.M.; Boer, J.M.; Cornelissen, J.J.; Pieters, R.; den Boer, M.L.; et al. Autophagy inhibition as a potential future targeted therapy for ETV6-RUNX1-driven B-cell precursor acute lymphoblastic leukemia. Haematologica 2018, 104, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Burjanivova, T.; Madzo, J.; Muzikova, K.; Meyer, C.; Schneider, B.; Votava, F.; Marschalek, R.; Stary, J.; Trka, J.; Zuna, J. Prenatal origin of childhood AML occurs less frequently than in childhood ALL. BMC Cancer 2006, 6, 100. [Google Scholar] [CrossRef] [PubMed]
- Zuna, J.; Madzo, J.; Krejci, O.; Zemanova, Z.; Kalinova, M.; Muzikova, K.; Zapotocky, M.; Starkova, J.; Hrusak, O.; Horak, J.; et al. ETV6/RUNX1 (TEL/AML1) Is a Frequent Prenatal First Hit in Childhood Leukemia. Blood 2011, 117, 368–369. [Google Scholar] [CrossRef] [PubMed]
- Fueller, E.; Schaefer, D.; Fischer, U.; Krell, P.F.I.; Stanulla, M.; Borkhardt, A.; Slany, R.K. Genomic Inverse PCR for Exploration of Ligated Breakpoints (GIPFEL), a New Method to Detect Translocations in Leukemia. PLoS ONE 2014, 9, e104419. [Google Scholar] [CrossRef]
- Mori, H.; Colman, S.M.; Xiao, Z.; Ford, A.M.; Healy, L.E.; Donaldson, C.; Hows, J.M.; Navarrete, C.; Greaves, M. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc. Natl. Acad. Sci. USA 2002, 99, 8242–8247. [Google Scholar] [CrossRef]
- Zakaria, Z.; Ahid, M.F.M.D.; Ismail, A.; Keoh, T.S.; Nor, N.M.; Kamaluddin, N.R.; Esa, E.; Yuen, L.K.; Rahman, E.J.A.; Osman, R. Chromosomal Aberrations in ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukemia Using 244K Oligonucleotide Array Comparative Genomic Hybridization. Mol. Cytogenet. 2012, 5, 41. [Google Scholar] [CrossRef]
- Zhang, J.; Mullighan, C.; Harvey, R.; Wu, G.; Chen, X.; Edmonson, M.; Buetow, K.H.; Carroll, W.L.; Chen, I.-M.; Devidas, M.; et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Blood 2011, 118, 3080–3087. [Google Scholar] [CrossRef]
- Attarbaschi, A.; Mann, G.; Strehl, S.; König, M.; Steiner, M.; Jeitler, V.; Lion, T.; Dworzak, M.N.; Gadner, H.; Haas, O.A. Deletion of 11q23 Is a Highly Specific Nonrandom Secondary Genetic Abnormality of ETV6/RUNX1-Rearranged Childhood Acute Lymphoblastic Leukemia. Leukemia 2007, 21, 584–586. [Google Scholar] [CrossRef]
- Borst, L.; Wesolowska, A.; Joshi, T.; Borup, R.; Nielsen, F.C.; Andersen, M.K.; Jonsson, O.G.; Wehner, P.S.; Wesenberg, F.; Frost, B.M.; et al. Genome-Wide Analysis of Cytogenetic Aberrations in ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2012, 157, 476–482. [Google Scholar] [CrossRef]
- Ampatzidou, M.; Papadhimitriou, S.I.; Paterakis, G.; Pavlidis, D.; Tsitsikas; Kostopoulos, I.V.; Papadakis, V.; Vassilopoulos, G.; Polychronopoulou, S. ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukemia (ALL): The Spectrum of Clonal Heterogeneity and Its Impact on Prognosis. Cancer Genet. 2018, 224–225, 1–11. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, G.; Casado-García, A.; Isidro-Hernández, M.; Picard, D.; Raboso-Gallego, J.; Alemán-Arteaga, S.; Orfao, A.; Blanco, O.; Riesco, S.; Prieto-Matos, P.; et al. The Second Oncogenic Hit Determines the Cell Fate of ETV6-RUNX1 Positive Leukemia. Front. Cell Dev. Biol. 2021, 9, 1834. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Yoshida, K.; Shiozawa, Y.; Nannya, Y.; Iijima-Yamashita, Y.; Kiyokawa, N.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Isobe, T.; et al. Landscape of driver mutations and their clinical impacts in pediatric B-cell precursor acute lymphoblastic leukemia. Blood Adv. 2020, 4, 5165–5173. [Google Scholar] [CrossRef]
- Pais, A.P.; Amare Kadam, P.S.; Raje, G.C.; Banavali, S.; Parikh, P.; Kurkure, P.; Arora, B.; Gujral, S.; Kumar, S.A.; Badrinath, Y. RUNX1 Aberrations in ETV6/RUNX1-Positive and ETV6/RUNX1-Negative Patients: Its Hemato-Pathological and Prognostic Significance in a Large Cohort (619 Cases) of ALL. Pediatr. Hematol. Oncol. 2008, 25, 582–597. [Google Scholar] [CrossRef]
- Nordlund, J.; Marincevic-Zuniga, Y.; Cavelier, L.; Raine, A.; Martin, T.; Lundmark, A.; Abrahamsson, J.; Norén-Nyström, U.; Lönnerholm, G.; Syvänen, A.-C. Refined detection and phasing of structural aberrations in pediatric acute lymphoblastic leukemia by linked-read whole-genome sequencing. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Chang, L.; Zhu, X. Pathogenesis of ETV6/RUNX1 -Positive Childhood Acute Lymphoblastic Leukemia and Mechanisms Underlying Its Relapse. Oncotarget 2017, 8, 35445–35459. [Google Scholar] [CrossRef] [PubMed]
- Bokemeyer, A.; Eckert, C.; Meyr, F.; Koerner, G.; von Stackelberg, A.; Ullmann, R.; Türkmen, S.; Henze, G.; Seeger, K. Copy Number Genome Alterations Are Associated with Treatment Response and Outcome in Relapsed Childhood ETV6/RUNX1-Positive Acute Lymphoblastic Leukemia. Haematologica 2014, 99, 706–714. [Google Scholar] [CrossRef]
- Forero, R.M.; Hernández, M.; Hernández- Rivas, J.M. Genetics of Acute Lymphoblastic Leukemia. In Leukemia; Guenova, M., Balatzenko, G., Eds.; IntechOpen: London, UK, 2013. [Google Scholar]
- Dun, K.A.; Vanhaeften, R.; Batt, T.J.; Riley, L.A.; Diano, G.; Williamson, J. BCR-ABL1 gene rearrangement as a subclonal change in ETV6-RUNX1–positive B-cell acute lymphoblastic leukemia. Blood Adv. 2016, 1, 132–138. [Google Scholar] [CrossRef]
- Raynaud, S.; Cavé, H.; Baens, M.; Bastard, C.; Cacheux, V.; Grosgeorge, J.; Guidal-Giroux, C.; Guo, C.; Vilmer, E.; Marynen, P.; et al. The 12;21 translocation involving TEL and deletion of the other TEL allele: Two frequently associated alterations found in childhood acute lymphoblastic leukemia. Blood 1996, 87, 2891–2899. [Google Scholar] [CrossRef]
- Komuro, H.; Valentine, M.B.; Rubnitz, J.E.; Saito, M.; Raimondi, S.C.; Carroll, A.J.; Yi, T.; Sherr, C.J.; Look, A.T. p27KIP1 Deletions in Childhood Acute Lymphoblastic Leukemia. Neoplasia 1999, 1, 253–261. [Google Scholar] [CrossRef]
- Enshaei, A.; Schwab, C.J.; Konn, Z.J.; Mitchell, C.D.; Kinsey, S.E.; Wade, R.; Vora, A.; Harrison, C.J.; Moorman, A.V. Long-term follow-up of ETV6-RUNX1 ALL reveals that NCI risk, rather than secondary genetic abnormalities, is the key risk factor. Leukemia 2013, 27, 2256–2259. [Google Scholar] [CrossRef]
- Guo, B.; Godzik, A.; Reed, J.C. Bcl-G, a Novel Pro-apoptotic Member of the Bcl-2 Family. J. Biol. Chem. 2001, 276, 2780–2785. [Google Scholar] [CrossRef] [PubMed]
- Yuniati, L.; Scheijen, B.; van der Meer, L.T.; van Leeuwen, F.N. Tumor suppressors BTG1 and BTG2: Beyond growth control. J Cell Physiol. 2019, 234, 5379–5389. [Google Scholar] [CrossRef]
- Waanders, E.; Scheijen, B.; van der Meer, L.T.; van Reijmersdal, S.V.; van Emst, L.; Kroeze, Y.; Sonneveld, E.; Hoogerbrugge, P.M.; van Kessel, A.G.; van Leeuwen, F.N.; et al. The origin and nature of tightly clustered BTG1 deletions in precursor B-cell acute lymphoblastic leukemia support a model of multiclonal evolution. PLoS Genet. 2012, 8, e1002533. [Google Scholar] [CrossRef]
- Jerchel, I.S.; Hoogkamer, A.Q.; Ariës, I.M.; Steeghs, E.M.P.; Boer, J.M.; Besselink, N.J.M.; Boeree, A.; Van De Ven, C.; De Groot-Kruseman, H.A.; De Haas, V.; et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia 2018, 32, 931–940. [Google Scholar] [CrossRef]
- Nishii, R.; Baskin-Doerfler, R.; Yang, W.; Oak, N.; Zhao, X.; Yang, W.; Hoshitsuki, K.; Bloom, M.; Verbist, K.C.; Burns, M.A.; et al. Molecular basis of ETV6-mediated predisposition to childhood acute lymphoblastic leukemia. Blood 2021, 137, 364–373. [Google Scholar] [CrossRef] [PubMed]
- van Delft, F.W.; Horsley, S.; Colman, S.; Anderson, K.; Bateman, C.; Kempski, H.; Zuna, J.; Eckert, C.; Saha, V.; Kearney, L.; et al. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 2011, 117, 6247–6254. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.K.; Foster, S.D.; Wang, X.; Knezevic, K.; Schütte, J.; Kaimakis, P.; Chilarska, P.M.; Kinston, S.; Ouwehand, W.H.; Dzierzak, E.; et al. Combinatorial Transcriptional Control In Blood Stem/Progenitor Cells: Genome-wide Analysis of Ten Major Transcriptional Regulators. Cell Stem Cell 2010, 7, 532–544. [Google Scholar] [CrossRef]
- Xu, L.S.; Francis, A.; Turkistany, S.; Shukla, D.; Wong, A.; Batista, C.R.; DeKoter, R.P. ETV6-RUNX1 interacts with a region in SPIB intron 1 to regulate gene expression in pre-B-cell acute lymphoblastic leukemia. Exp. Hematol. 2019, 73, 50–63.e2. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Minnich, M.; Gangatirkar, P.; Zheng, Z.; Ebert, A.; Song, G.; Dickins, R.; Corcoran, L.M.; Mullighan, C.G.; Busslinger, M.; et al. PU.1 cooperates with IRF4 and IRF8 to suppress pre-B-cell leukemia. Leukemia 2016, 30, 1375–1387. [Google Scholar] [CrossRef]
- Jakobczyk, H.; Jiang, Y.; Debaize, L.; Soubise, B.; Avner, S.; Sérandour, A.A.; Rouger-Gaudichon, J.; Rio, A.-G.; Carroll, J.S.; Raslova, H.; et al. ETV6-RUNX1 and RUNX1 directly regulate RAG1 expression: One more step in the understanding of childhood B-cell acute lymphoblastic leukemia leukemogenesis. Leukemia 2021, 36, 549–554. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Soneson, C.; Andersson, A.; Heldrup, J.; Behrendtz, M.; Kawamata, N.; Ogawa, S.; Koeffler, H.P.; Mitelman, F.; Johansson, B.; et al. The Correlation Pattern of Acquired Copy Number Changes in 164 ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukemias. Hum. Mol. Genet. 2010, 19, 3150. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Gu, Z. PAX5 alterations in B-cell acute lymphoblastic leukemia. Front. Oncol. 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M.; Shukla, R.; Dwivedi, S.; Saxena, R.; Luthra, K.; Kabra, M.; Seth, R. Gene copy number alterations in Indian children with B-acute Lymphoblastic Leukemia: Correlation with survival outcome. Pediatr. Hematol. Oncol. J. 2021, 6, 151–157. [Google Scholar] [CrossRef]
- Kuster, L.; Grausenburger, R.; Fuka, G.; Kaindl, U.; Krapf, G.; Inthal, A.; Mann, G.; Kauer, M.; Rainer, J.; Kofler, R.; et al. ETV6/RUNX1-Positive Relapses Evolve from an Ancestral Clone and Frequently Acquire Deletions of Genes Implicated in Glucocorticoid Signaling. Blood 2011, 117, 2658–2667. [Google Scholar] [CrossRef]
- Grausenburger, R.; Bastelberger, S.; Eckert, C.; Kauer, M.; Stanulla, M.; Frech, C.; Bauer, E.; Stoiber, D.; von Stackelberg, A.; Attarbaschi, A.; et al. Genetic Alterations in Glucocorticoid Signaling Pathway Components Are Associated with Adverse Prognosis in Children with Relapsed ETV6/RUNX1-Positive Acute Lymphoblastic Leukemia. Leuk Lymphoma 2016, 57, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Kelley, C.M.; Ikeda, T.; Koipally, J.; Avitahl, N.; Wu, L.; Georgopoulos, K.; Morgan, B.A. Helios, a novel dimerization partner of Ikaros expressed in the earliest hematopoietic progenitors. Curr. Biol. 1998, 8, 508-S1. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Su, X.; Zhang, J.; Zhang, J.; Radtke, I.; Phillips, L.A.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C. Children’s Oncology Group: Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef]
- Ramesh-Kumar, D.; Guil, S. The IGF2BP family of RNA binding proteins links epitranscriptomics to cancer. Semin. Cancer Biol. 2022, 86, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Boby, E.; Nidhi, T.; Jain, A.; Singh, J.; Singh, A.; Chattopadhyay, P.; Bakhshi, S.; Chopra, A.; Palanichamy, J.K. Diagnostic Utility of IGF2BP1 and Its Targets as Potential Biomarkers in ETV6-RUNX1 Positive B-Cell Acute Lymphoblastic Leukemia. Front. Oncol. 2021, 11, 588101. [Google Scholar] [CrossRef]
- Stoskus, M.; Gineikiene, E.; Valceckiene, V.; Valatkaite, B.; Pileckyte, R.; Griskevicius, L. Identification of characteristic IGF2BP expression patterns in distinct B-ALL entities. Blood Cells Mol. Dis. 2011, 46, 321–326. [Google Scholar] [CrossRef]
- Mäkinen, A.; Nikkilä, A.; Haapaniemi, T.; Oksa, L.; Mehtonen, J.; Vänskä, M.; Heinäniemi, M.; Paavonen, T.; Lohi, O. IGF2BP3 Associates with Proliferative Phenotype and Prognostic Features in B-Cell Acute Lymphoblastic Leukemia. Cancers 2021, 13, 1505. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.-F.; Smadbeck, J.B.; Sharma, N.; Blackburn, P.R.; Benevides, J.D.; Akkari, Y.M.N.; Jaroscak, J.J.; Znoyko, I.; Wolff, D.J.; Schandl, C.A.; et al. Apparent coexistence of ETV6::RUNX1 and KMT2A::MLLT3 fusions due to a nonproductive KMT2A rearrangement in B-ALL. Leuk. Lymphoma 2022, 63, 2243–2246. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Rapado, I.; Li, Y.; E Potter, N.; Wedge, D.; Tubio, J.; Alexandrov, L.B.; Van Loo, P.; Cooke, S.L.; Marshall, J.; et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Seeger, K.; Adams, H.P.; Buchwald, D.; Beyermann, B.; Kremens, B.; Niemeyer, C.; Ritter, J.; Schwabe, D.; Harms, D.; Schrappe, M.; et al. TEL-AML1 Fusion Transcript in Relapsed Childhood Acute Lymphoblastic Leukemia The Berlin-Frankfurt-Münster Study 1035 Group. Blood 1998, 91, 1716–1722. [Google Scholar] [CrossRef]
- Pui, C.H.; Sandlund, J.T.; Pei, D.; Campana, D.; Rivera, G.K.; Ribeiro, R.C.; Rubnitz, J.E.; Razzouk, B.I.; Howard, S.C.; Hudson, M.M.; et al. Improved outcome for children with acute lymphoblastic leukemia: Results of Total Therapy Study XIIIB at St Jude Children’s Research Hospital. Blood 2004, 104, 2690–2696. [Google Scholar] [CrossRef]
- Narla, R.K.; Navara, C.; Sarquis, M.; Uckun, F.M. Chemosensitivity of TEL-AML1 Fusion Transcript Positive Acute Lymphoblastic Leukemia Cells. Leuk. Lymphoma 2001, 41, 615–623. [Google Scholar] [CrossRef]
- Ramakers-van Woerden, N.L.; Pieters, R.; Loonen, A.H.; Hubeek, I.; van Drunen, E.; Beverloo, H.B.; Slater, R.M.; Harbott, J.; Seyfarth, J.; van Wering, E.R.; et al. TEL/AML1 gene fusion is related to in vitro drug sensitivity for L-asparaginase in childhood acute lymphoblastic leukemia. Blood 2000, 96, 1094–1099. [Google Scholar]
- Teachey, D.T.; Hunger, S.P.; Loh, M.M.L. Optimizing therapy in the modern age: Differences in length of maintenance therapy in acute lymphoblastic leukemia. Blood 2021, 137, 168–177. [Google Scholar] [CrossRef]
- Inaba, H.; Mullighan, C.G. Pediatric Acute Lymphoblastic Leukemia. Haematologica 2020, 105, 2524. [Google Scholar] [CrossRef]
- Kato, M.; Ishimaru, S.; Seki, M.; Yoshida, K.; Shiraishi, Y.; Chiba, K.; Kakiuchi, N.; Sato, Y.; Ueno, H.; Tanaka, H.; et al. Long-term outcome of 6-month maintenance chemotherapy for acute lymphoblastic leukemia in children. Leukemia 2017, 31, 580–584. [Google Scholar] [CrossRef]
- Schrappe, M.; Bleckmann, K.; Zimmermann, M.; Biondi, A.; Möricke, A.; Locatelli, F.; Cario, G.; Rizzari, C.; Attarbaschi, A.; Valsecchi, M.G.; et al. Reduced-Intensity Delayed Intensification in Standard-Risk Pediatric Acute Lymphoblastic Leukemia Defined by Undetectable Minimal Residual Disease: Results of an International Randomized Trial (AIEOP-BFM ALL 2000). J. Clin. Oncol. 2018, 36, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Serafin, V.; Porcù, E.; Cortese, G.; Mariotto, E.; Veltri, G.; Bresolin, S.; Basso, G.; Accordi, B. SYK Targeting Represents a Potential Therapeutic Option for Relapsed Resistant Pediatric ETV6-RUNX1 B-Acute Lymphoblastic Leukemia Patients. Int. J. Mol. Sci. 2019, 20, 6175. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Ebrahimabadi, S.; Golalipour, M.; Shahbazi, M.; Farazmandfar, T. The correction of ETV6/RUNX1 translocation in acute lymphocytic leukemia cells: A new gene targeting system by homologous recombination mechanism. J. Appl. Genet. 2020, 61, 67–73. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Fioretos, T. New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood 2017, 130, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dai, Y.; Wu, L.; Zhang, M.; Ouyang, W.; Huang, J.; Chen, S. Emerging molecular subtypes and therapeutic targets in B-cell precursor acute lymphoblastic leukemia. Front. Med. 2021, 15, 347–371. [Google Scholar] [CrossRef]
- Chen, D.; Camponeschi, A.; Nordlund, J.; Marincevic-Zuniga, Y.; Abrahamsson, J.; Lönnerholm, G.; Fogelstrand, L.; Mårtensson, I.L. RAG1 Co-Expression Signature Identifies ETV6-RUNX1-like B-Cell Precursor Acute Lymphoblastic Leukemia in Children. Cancer Med. 2021, 10, 3997–4003. [Google Scholar] [CrossRef]
- Zaliova, M.; Kotrova, M.; Bresolin, S.; Stuchly, J.; Stary, J.; Hrusak, O.; teKronnie, G.; Trka, J.; Zuna, J.; Vaskova, M. ETV6/RUNX1-like Acute Lymphoblastic Leukemia: A Novel B-Cell Precursor Leukemia Subtype Associated with the CD27/CD44 Immunophenotype. Genes Chromosomes Cancer 2017, 56, 608–616. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).