
Human RTEL1 Interacts with KPNB1 (Importin β) and NUP153 and Connects Nuclear Import to Nuclear Envelope Stability in S-Phase

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell lines and Culture Conditions
2.2. Constructs
2.3. Transfections
2.4. Drug Treatments
2.5. Anti-hRTEL1 Antibody Production
2.6. Commercial Antibodies
2.7. Immunofluorescence
2.8. Circularity
2.9. Western Blot Analyses
2.10. Immunoprecipitations
2.11. Generation of CRISPR/Cas9 RTEL1 Knockout
2.12. Statistical Analysis
3. Results
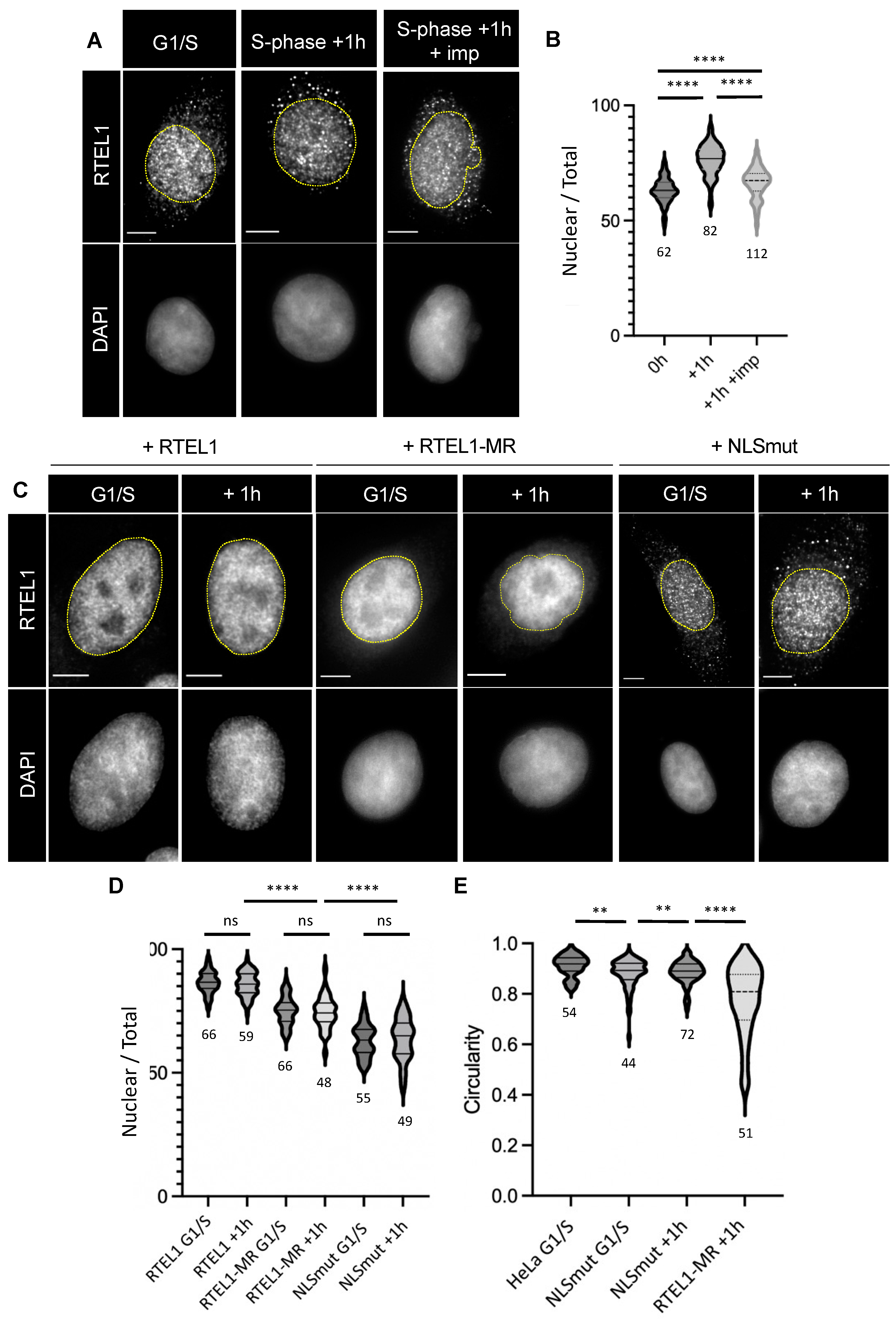
3.1. RTEL1 Deficiency Is Associated with Nuclear Envelope Deformations That Specifically Occur in S-Phase
3.2. RTEL1 Is Imported to the Nucleus When Cells Enter S-Phase
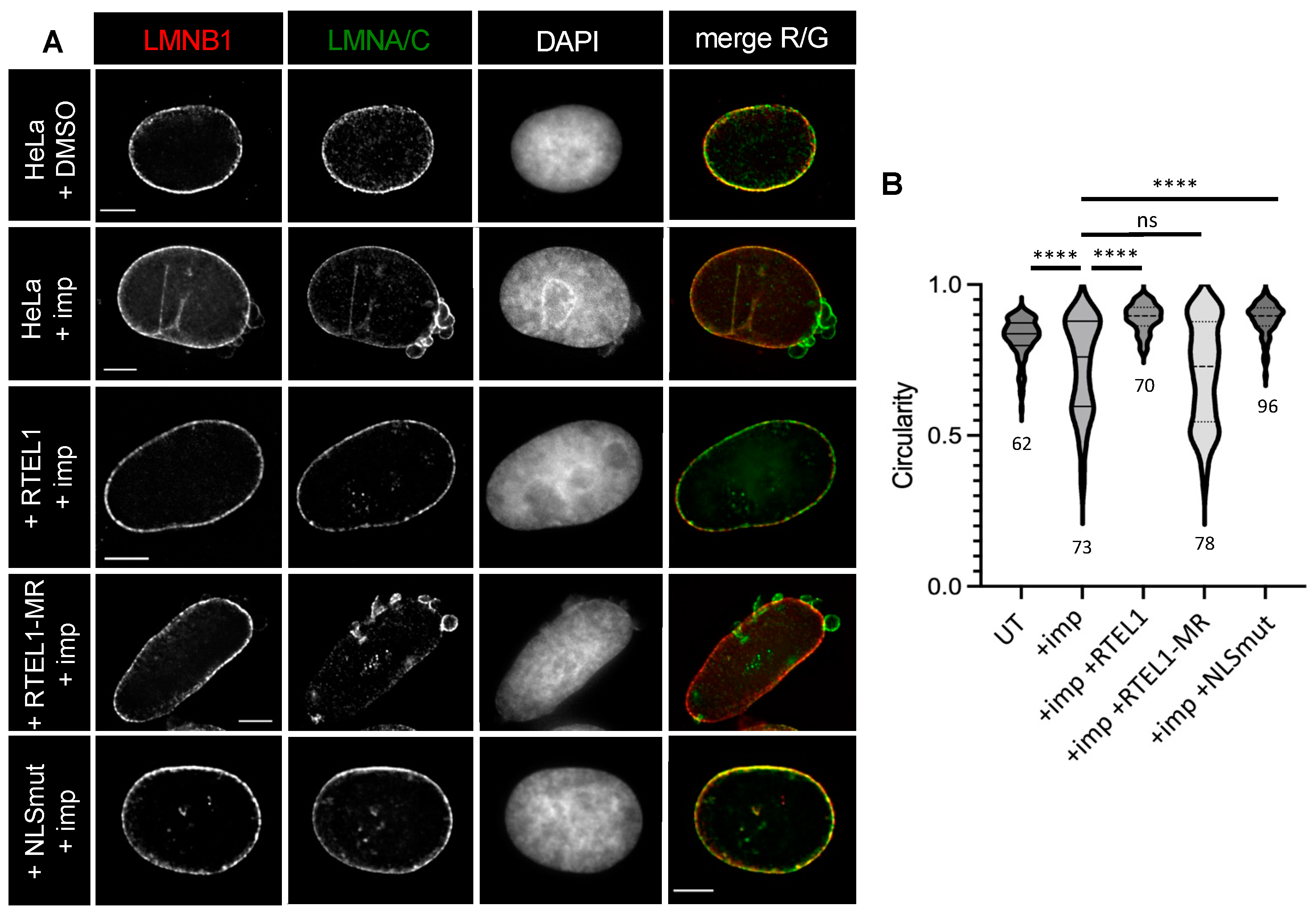
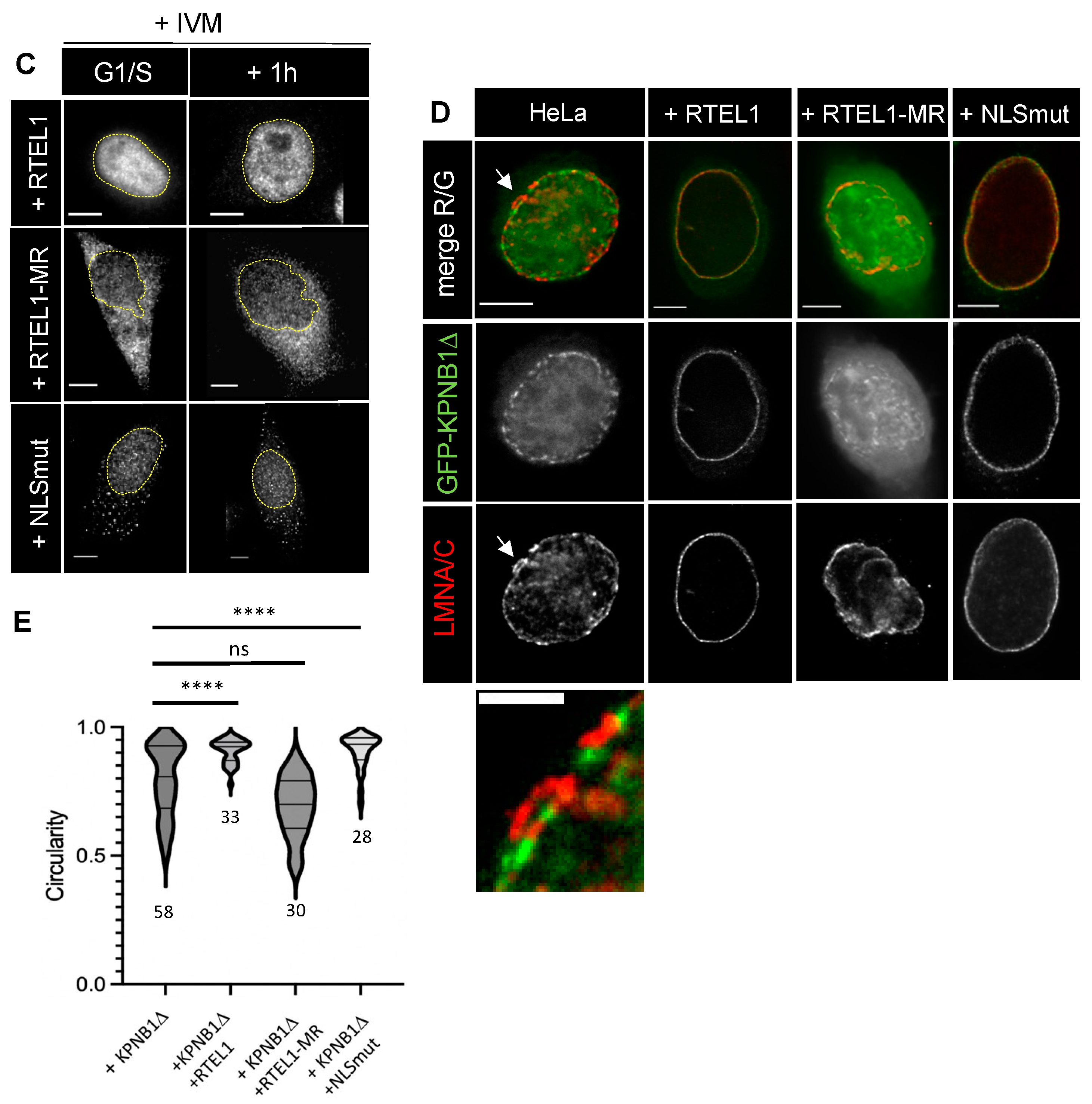
3.3. RTEL1 Overexpression Protects Cells from NE Defects Induced by Inhibition of KPNB1
3.4. RTEL1 Interacts, Albeit Weakly, with the N-Terminal Domain of KPNB1
3.5. The C-Terminus of RTEL1 Lacking the Helicase Domain Is Sufficient to Protect Cells from Importazole-Induced NE Defects
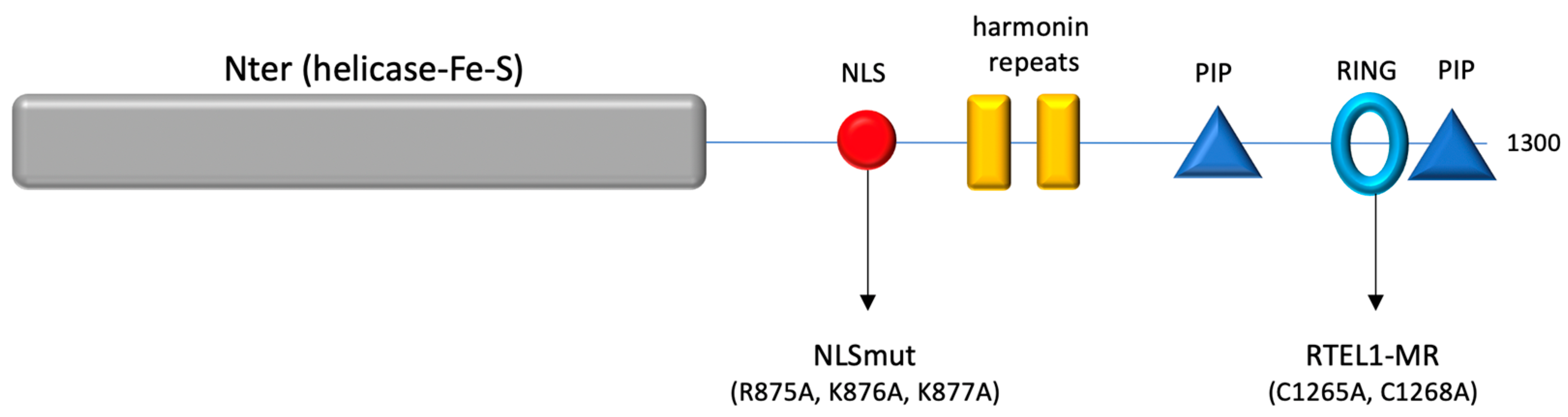
3.6. The 683-762 Domain in RTEL1 Is Required for the Induction of NE Deformation Phenotypes Caused by Mutations in the RING Domain
3.7. A 683-808 Domain in RTEL1 Promotes Nuclear Import through Targeting to the NPC
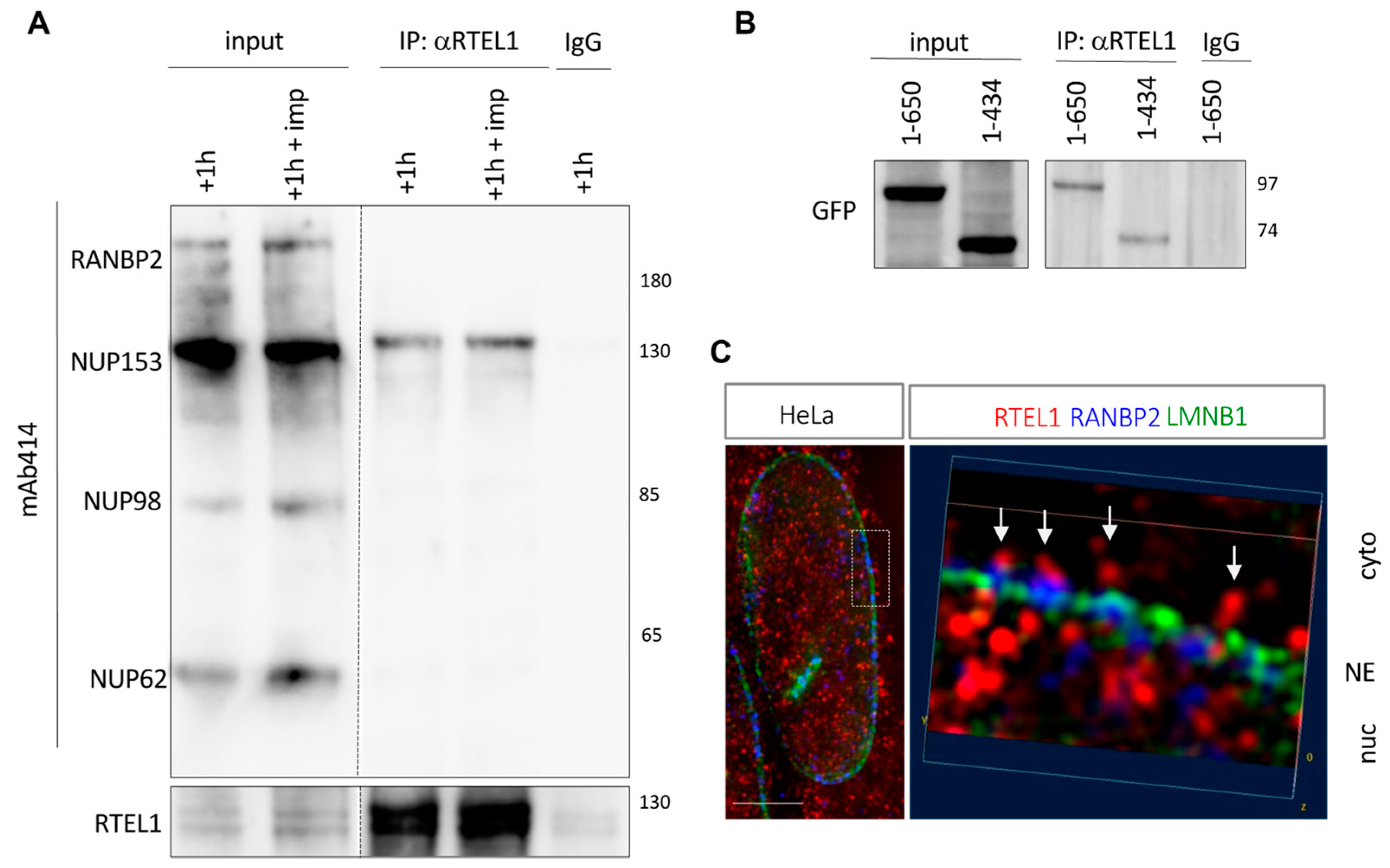
3.8. RTEL1 Interacts with NUP153
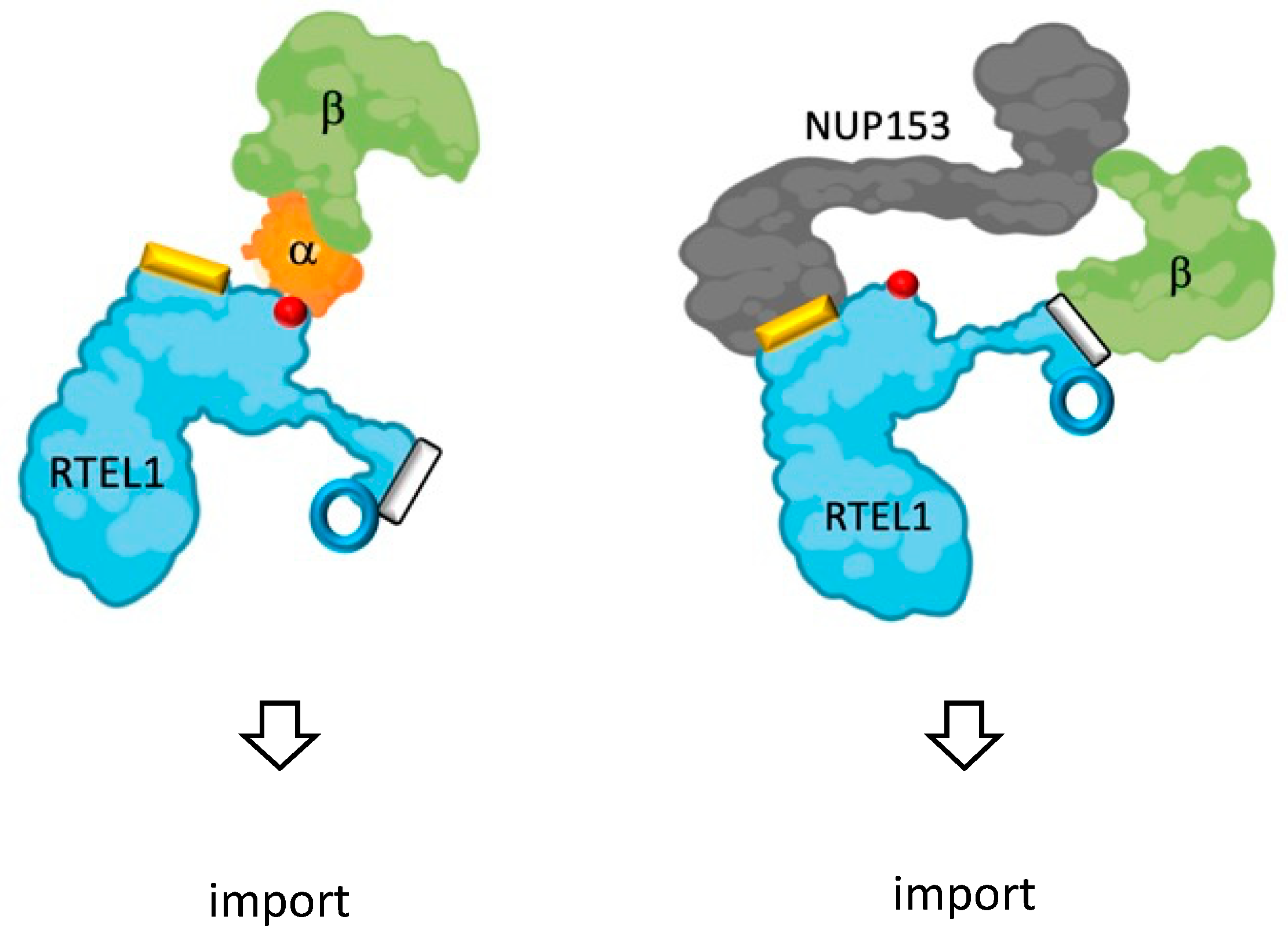
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ding, H.; Schertzer, M.; Wu, X.; Gertsenstein, M.; Selig, S.; Kammori, M.; Pourvali, R.; Poon, S.; Vulto, I.; Chavez, E.; et al. Regulation of murine telomere length by Rtel: An essential gene encoding a helicase-like protein. Cell 2004, 117, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Porreca, R.M.; Glousker, G.; Awad, A.; Matilla Fernandez, M.I.; Gibaud, A.; Naucke, C.; Cohen, S.B.; Bryan, T.M.; Tzfati, Y.; Draskovic, I.; et al. Human RTEL1 stabilizes long G-overhangs allowing telomerase-dependent over-extension. Nucleic Acids Res. 2018, 46, 4533–4545. [Google Scholar] [CrossRef] [PubMed]
- Vannier, J.B.; Pavicic-Kaltenbrunner, V.; Petalcorin, M.I.; Ding, H.; Boulton, S.J. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 2012, 149, 795–806. [Google Scholar] [CrossRef]
- Sarek, G.; Vannier, J.B.; Panier, S.; Petrini, J.H.J.; Boulton, S.J. TRF2 Recruits RTEL1 to Telomeres in S Phase to Promote T-Loop Unwinding. Mol. Cell 2016, 61, 788–789. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Glousker, G.; Lamm, N.; Tawil, S.; Hourvitz, N.; Smoom, R.; Revy, P.; Tzfati, Y. Full length RTEL1 is required for the elongation of the single-stranded telomeric overhang by telomerase. Nucleic Acids Res. 2020, 48, 7239–7251. [Google Scholar] [CrossRef]
- Vannier, J.B.; Sandhu, S.; Petalcorin, M.I.; Wu, X.; Nabi, Z.; Ding, H.; Boulton, S.J. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 2013, 342, 239–242. [Google Scholar] [CrossRef]
- Wu, W.; Bhowmick, R.; Vogel, I.; Ozer, O.; Ghisays, F.; Thakur, R.S.; Sanchez de Leon, E.; Richter, P.H.; Ren, L.; Petrini, J.H.; et al. RTEL1 suppresses G-quadruplex-associated R-loops at difficult-to-replicate loci in the human genome. Nat. Struct. Mol. Biol. 2020, 27, 424–437. [Google Scholar] [CrossRef]
- Uringa, E.J.; Lisaingo, K.; Pickett, H.A.; Brind’Amour, J.; Rohde, J.H.; Zelensky, A.; Essers, J.; Lansdorp, P.M. RTEL1 contributes to DNA replication and repair and telomere maintenance. Mol. Biol. Cell 2012, 23, 2782–2792. [Google Scholar] [CrossRef]
- Barber, L.J.; Youds, J.L.; Ward, J.D.; McIlwraith, M.J.; O’Neil, N.J.; Petalcorin, M.I.; Martin, J.S.; Collis, S.J.; Cantor, S.B.; Auclair, M.; et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 2008, 135, 261–271. [Google Scholar] [CrossRef]
- Campos, L.V.; Van Ravenstein, S.X.; Vontalge, E.J.; Greer, B.H.; Heintzman, D.R.; Kavlashvili, T.; McDonald, W.H.; Rose, K.L.; Eichman, B.F.; Dewar, J.M. RTEL1 and MCM10 overcome topological stress during vertebrate replication termination. Cell Rep. 2023, 42, 112109. [Google Scholar] [CrossRef]
- Takedachi, A.; Despras, E.; Scaglione, S.; Guerois, R.; Guervilly, J.H.; Blin, M.; Audebert, S.; Camoin, L.; Hasanova, Z.; Schertzer, M.; et al. SLX4 interacts with RTEL1 to prevent transcription-mediated DNA replication perturbations. Nat. Struct. Mol. Biol. 2020, 27, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, A.; Johansen, S.L.; Lin, L.; Schertzer, M.; Kanellis, D.C.; Katsori, A.M.; Christensen, S.T.; Luo, Y.; Andersen, J.S.; Elsasser, S.J.; et al. Human RTEL1 associates with Poldip3 to facilitate responses to replication stress and R-loop resolution. Genes. Dev. 2020, 34, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Ballew, B.J.; Joseph, V.; De, S.; Sarek, G.; Vannier, J.B.; Stracker, T.; Schrader, K.A.; Small, T.N.; O’Reilly, R.; Manschreck, C.; et al. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genet. 2013, 9, e1003695. [Google Scholar] [CrossRef] [PubMed]
- Le Guen, T.; Jullien, L.; Touzot, F.; Schertzer, M.; Gaillard, L.; Perderiset, M.; Carpentier, W.; Nitschke, P.; Picard, C.; Couillault, G.; et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum. Mol. Genet. 2013, 22, 3239–3249. [Google Scholar] [CrossRef] [PubMed]
- Walne, A.J.; Vulliamy, T.; Kirwan, M.; Plagnol, V.; Dokal, I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am. J. Hum. Genet. 2013, 92, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Revy, P.; Kannengiesser, C.; Bertuch, A.A. Genetics of human telomere biology disorders. Nat. Rev. Genet. 2023, 24, 86–108. [Google Scholar] [CrossRef]
- Cogan, J.D.; Kropski, J.A.; Zhao, M.; Mitchell, D.B.; Rives, L.; Markin, C.; Garnett, E.T.; Montgomery, K.H.; Mason, W.R.; McKean, D.F.; et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2015, 191, 646–655. [Google Scholar] [CrossRef]
- Kannengiesser, C.; Borie, R.; Menard, C.; Reocreux, M.; Nitschke, P.; Gazal, S.; Mal, H.; Taille, C.; Cadranel, J.; Nunes, H.; et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur. Respir. J. 2015, 46, 474–485. [Google Scholar] [CrossRef]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef]
- Borie, R.; Bouvry, D.; Cottin, V.; Gauvain, C.; Cazes, A.; Debray, M.P.; Cadranel, J.; Dieude, P.; Degot, T.; Dominique, S.; et al. Regulator of telomere length 1 (RTEL1) mutations are associated with heterogeneous pulmonary and extra-pulmonary phenotypes. Eur. Respir. J. 2019, 53, 1800508. [Google Scholar] [CrossRef]
- Han, J.; Song, J.W. Dyskeratosis congenita with heterozygous RTEL1 mutations presenting with fibrotic hypersensitivity pneumonitis. Respir. Med. Case Rep. 2023, 42, 101810. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.C.W.; Gutierrez-Rodrigues, F.; Cooper, J.; Jiang, J.; Gandhi, S.; Kajigaya, S.; Feng, X.; Ibanez, M.; Donaires, F.S.; Lopes da Silva, J.P.; et al. Heterozygous RTEL1 variants in bone marrow failure and myeloid neoplasms. Blood Adv. 2018, 2, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Schertzer, M.; Jouravleva, K.; Perderiset, M.; Dingli, F.; Loew, D.; Le Guen, T.; Bardoni, B.; de Villartay, J.P.; Revy, P.; Londono-Vallejo, A. Human regulator of telomere elongation helicase 1 (RTEL1) is required for the nuclear and cytoplasmic trafficking of pre-U2 RNA. Nucleic Acids Res. 2015, 43, 1834–1847. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical nuclear localization signals: Definition, function, and interaction with importin alpha. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Musser, S.M. Nuclear import time and transport efficiency depend on importin beta concentration. J. Cell Biol. 2006, 174, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Riddick, G.; Macara, I.G. A systems analysis of importin-alpha-beta mediated nuclear protein import. J. Cell Biol. 2005, 168, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Touzot, F.; Kermasson, L.; Jullien, L.; Moshous, D.; Menard, C.; Ikinciogullari, A.; Dogu, F.; Sari, S.; Giacobbi-Milet, V.; Etzioni, A.; et al. Extended clinical and genetic spectrum associated with biallelic RTEL1 mutations. Blood Adv. 2016, 1, 36–46. [Google Scholar] [CrossRef]
- Krokan, H.; Schaffer, P.; DePamphilis, M.L. Involvement of eucaryotic deoxyribonucleic acid polymerases alpha and gamma in the replication of cellular and viral deoxyribonucleic acid. Biochemistry 1979, 18, 4431–4443. [Google Scholar] [CrossRef]
- Bousset, K.; Diffley, J.F. The Cdc7 protein kinase is required for origin firing during S phase. Genes. Dev. 1998, 12, 480–490. [Google Scholar] [CrossRef]
- Soderholm, J.F.; Bird, S.L.; Kalab, P.; Sampathkumar, Y.; Hasegawa, K.; Uehara-Bingen, M.; Weis, K.; Heald, R. Importazole, a small molecule inhibitor of the transport receptor importin-beta. ACS Chem. Biol. 2011, 6, 700–708. [Google Scholar] [CrossRef]
- Kalderon, D.; Roberts, B.L.; Richardson, W.D.; Smith, A.E. A short amino acid sequence able to specify nuclear location. Cell 1984, 39, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, K.M.; Sivakumaran, H.; Heaton, S.M.; Harrich, D.; Jans, D.A. Ivermectin is a specific inhibitor of importin alpha/beta-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem. J. 2012, 443, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Biol. 2007, 8, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Gorlich, D.; Pante, N.; Kutay, U.; Aebi, U.; Bischoff, F.R. Identification of different roles for RanGDP and RanGTP in nuclear protein import. EMBO J. 1996, 15, 5584–5594. [Google Scholar] [CrossRef] [PubMed]
- Makise, M.; Mackay, D.R.; Elgort, S.; Shankaran, S.S.; Adam, S.A.; Ullman, K.S. The Nup153-Nup50 protein interface and its role in nuclear import. J. Biol. Chem. 2012, 287, 38515–38522. [Google Scholar] [CrossRef] [PubMed]
- Ben-Efraim, I.; Gerace, L. Gradient of increasing affinity of importin beta for nucleoporins along the pathway of nuclear import. J. Cell Biol. 2001, 152, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Burke, B.; May, D.G. BioID: A Screen for Protein-Protein Interactions. Curr. Protoc. Protein Sci. 2018, 91, 19–23. [Google Scholar] [CrossRef]
- Kimura, M.; Morinaka, Y.; Imai, K.; Kose, S.; Horton, P.; Imamoto, N. Extensive cargo identification reveals distinct biological roles of the 12 importin pathways. Elife 2017, 6, e21184. [Google Scholar] [CrossRef]
- Mackmull, M.T.; Klaus, B.; Heinze, I.; Chokkalingam, M.; Beyer, A.; Russell, R.B.; Ori, A.; Beck, M. Landscape of nuclear transport receptor cargo specificity. Mol. Syst. Biol. 2017, 13, 962. [Google Scholar] [CrossRef]
- Di Francesco, L.; Verrico, A.; Asteriti, I.A.; Rovella, P.; Cirigliano, P.; Guarguaglini, G.; Schinina, M.E.; Lavia, P. Visualization of human karyopherin beta-1/importin beta-1 interactions with protein partners in mitotic cells by co-immunoprecipitation and proximity ligation assays. Sci. Rep. 2018, 8, 1850. [Google Scholar] [CrossRef] [PubMed]
- Iyer-Bierhoff, A.; Grummt, I. Stop-and-Go: Dynamics of Nucleolar Transcription During the Cell Cycle. Epigenet Insights 2019, 12, 2516865719849090. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, C.M.; Lapointe, D.S.; Luong, M.X.; Peric-Hupkes, D.; Cho, B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. Gene profiling of cell cycle progression through S-phase reveals sequential expression of genes required for DNA replication and nucleosome assembly. Cancer Res. 2002, 62, 3233–3243. [Google Scholar] [PubMed]
- Ibarra, A.; Hetzer, M.W. Nuclear pore proteins and the control of genome functions. Genes. Dev. 2015, 29, 337–349. [Google Scholar] [CrossRef]
- Choudhry, S.K.; Neal, M.L.; Li, S.; Navare, A.T.; Van Eeuwen, T.; Wozniak, R.W.; Mast, F.D.; Rout, M.P.; Aitchison, J.D. Nuclear pore complexes mediate subtelomeric gene silencing by regulating PCNA levels on chromatin. J. Cell Biol. 2023, 222, e202207060. [Google Scholar] [CrossRef] [PubMed]
- Lowe, A.R.; Tang, J.H.; Yassif, J.; Graf, M.; Huang, W.Y.; Groves, J.T.; Weis, K.; Liphardt, J.T. Importin-beta modulates the permeability of the nuclear pore complex in a Ran-dependent manner. Elife 2015, 4, e04052. [Google Scholar] [CrossRef] [PubMed]
- Bertuch, A.A. The molecular genetics of the telomere biology disorders. RNA Biol. 2016, 13, 696–706. [Google Scholar] [CrossRef]
- Speckmann, C.; Sahoo, S.S.; Rizzi, M.; Hirabayashi, S.; Karow, A.; Serwas, N.K.; Hoemberg, M.; Damatova, N.; Schindler, D.; Vannier, J.B.; et al. Clinical and Molecular Heterogeneity of RTEL1 Deficiency. Front. Immunol. 2017, 8, 449. [Google Scholar] [CrossRef]
- Park, J.H.; Ryu, S.J.; Kim, B.J.; Cho, H.J.; Park, C.H.; Choi, H.J.C.; Jang, E.J.; Yang, E.J.; Hwang, J.A.; Woo, S.H.; et al. Disruption of nucleocytoplasmic trafficking as a cellular senescence driver. Exp. Mol. Med. 2021, 53, 1092–1108. [Google Scholar] [CrossRef]
- D’Angelo, M.A.; Raices, M.; Panowski, S.H.; Hetzer, M.W. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 2009, 136, 284–295. [Google Scholar] [CrossRef]
- Larrieu, D.; Vire, E.; Robson, S.; Breusegem, S.Y.; Kouzarides, T.; Jackson, S.P. Inhibition of the acetyltransferase NAT10 normalizes progeric and aging cells by rebalancing the Transportin-1 nuclear import pathway. Sci. Signal 2018, 11, eaar5401. [Google Scholar] [CrossRef] [PubMed]
- Balmus, G.; Larrieu, D.; Barros, A.C.; Collins, C.; Abrudan, M.; Demir, M.; Geisler, N.J.; Lelliott, C.J.; White, J.K.; Karp, N.A.; et al. Targeting of NAT10 enhances healthspan in a mouse model of human accelerated aging syndrome. Nat. Commun. 2018, 9, 1700. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schertzer, M.; Jullien, L.; Pinto, A.L.; Calado, R.T.; Revy, P.; Londoño-Vallejo, A. Human RTEL1 Interacts with KPNB1 (Importin β) and NUP153 and Connects Nuclear Import to Nuclear Envelope Stability in S-Phase. Cells 2023, 12, 2798. https://doi.org/10.3390/cells12242798
Schertzer M, Jullien L, Pinto AL, Calado RT, Revy P, Londoño-Vallejo A. Human RTEL1 Interacts with KPNB1 (Importin β) and NUP153 and Connects Nuclear Import to Nuclear Envelope Stability in S-Phase. Cells. 2023; 12(24):2798. https://doi.org/10.3390/cells12242798
Chicago/Turabian StyleSchertzer, Michael, Laurent Jullien, André L. Pinto, Rodrigo T. Calado, Patrick Revy, and Arturo Londoño-Vallejo. 2023. "Human RTEL1 Interacts with KPNB1 (Importin β) and NUP153 and Connects Nuclear Import to Nuclear Envelope Stability in S-Phase" Cells 12, no. 24: 2798. https://doi.org/10.3390/cells12242798
APA StyleSchertzer, M., Jullien, L., Pinto, A. L., Calado, R. T., Revy, P., & Londoño-Vallejo, A. (2023). Human RTEL1 Interacts with KPNB1 (Importin β) and NUP153 and Connects Nuclear Import to Nuclear Envelope Stability in S-Phase. Cells, 12(24), 2798. https://doi.org/10.3390/cells12242798