
Non-Intrinsic, Systemic Mechanisms of Cellular Senescence


{kind=link}
{kind=link}
{kind=link}
Abstract
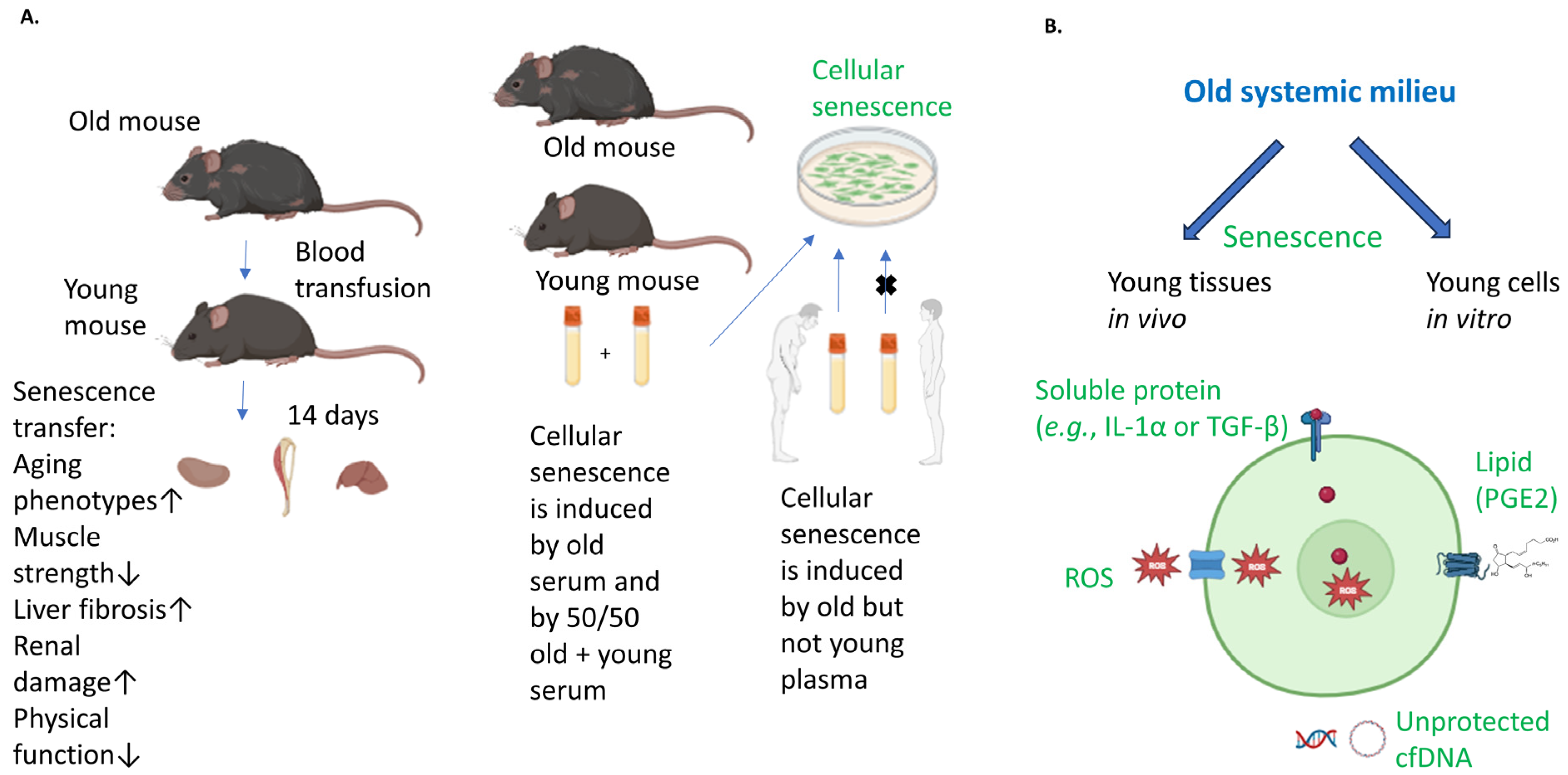
:1. Introduction
2. Single Component Soluble Secreted Factors
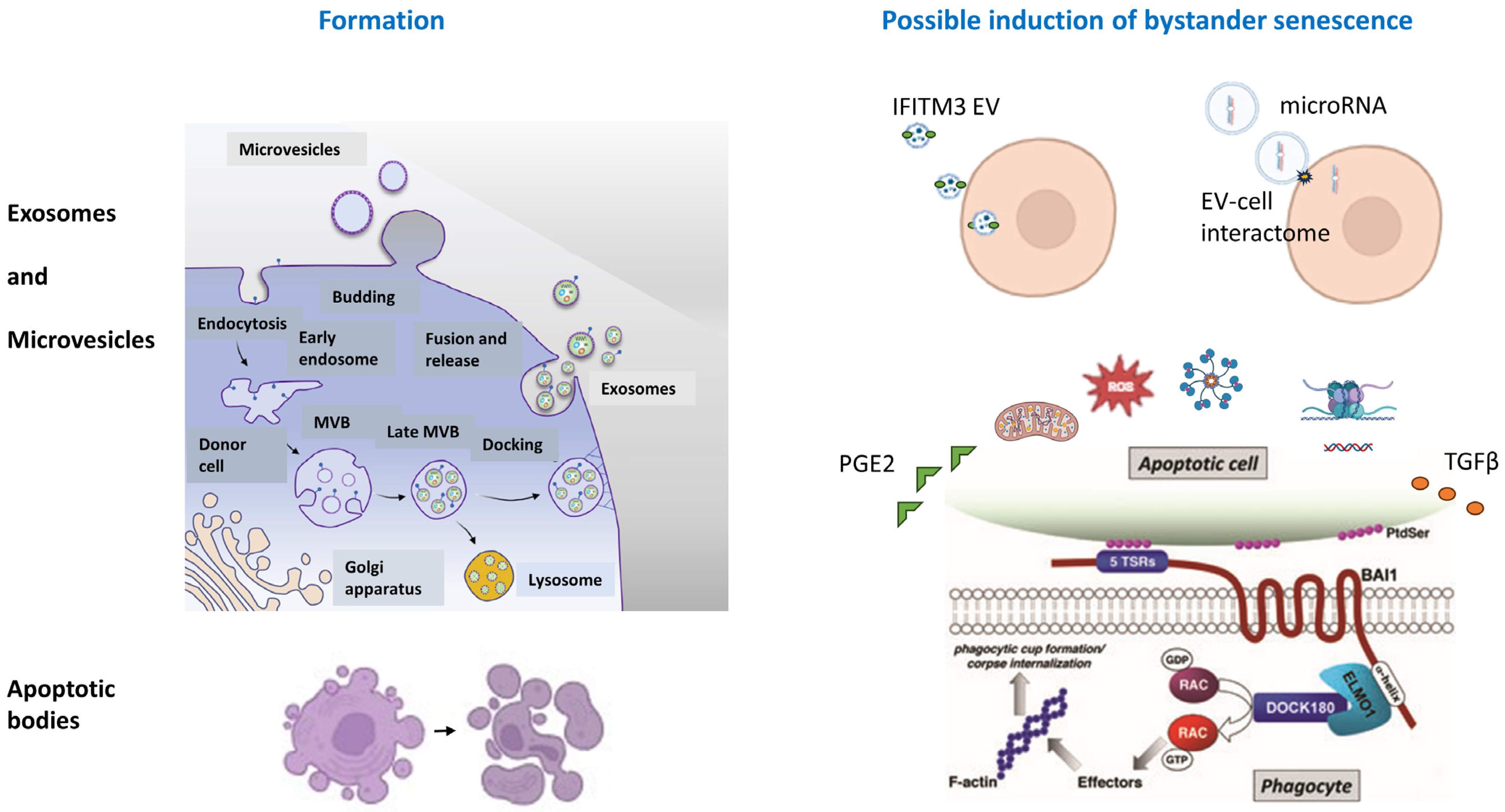
3. Membrane-Bound Bodies
4. Non-Vesicular Multi-Component Macromolecules
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roy, A.L.; Sierra, F.; Howcroft, K.; Singer, D.S.; Sharpless, N.; Hodes, R.J.; Wilder, E.L.; Anderson, J.M. A Blueprint for Characterizing Senescence. Cell 2020, 183, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gualda, E.; Baker, A.G.; Fruk, L.; Munoz-Espin, D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Singh, P.P.; Demmitt, B.A.; Nath, R.D.; Brunet, A. The Genetics of Aging: A Vertebrate Perspective. Cell 2019, 177, 200–220. [Google Scholar] [CrossRef]
- Jeon, O.H.; Mehdipour, M.; Gil, T.H.; Kang, M.; Aguirre, N.W.; Robinson, Z.R.; Kato, C.; Etienne, J.; Lee, H.G.; Alimirah, F.; et al. Systemic induction of senescence in young mice after single heterochronic blood exchange. Nat. Metab. 2022, 4, 995–1006. [Google Scholar] [CrossRef]
- Rebo, J.; Mehdipour, M.; Gathwala, R.; Causey, K.; Liu, Y.; Conboy, M.J.; Conboy, I.M. A single heterochronic blood exchange reveals rapid inhibition of multiple tissues by old blood. Nat. Commun. 2016, 7, 13363. [Google Scholar] [CrossRef]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef]
- da Silva, P.F.L.; Ogrodnik, M.; Kucheryavenko, O.; Glibert, J.; Miwa, S.; Cameron, K.; Ishaq, A.; Saretzki, G.; Nagaraja-Grellscheid, S.; Nelson, G.; et al. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 2019, 18, e12848. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFbeta-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ’bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Dagouassat, M.; Gagliolo, J.M.; Chrusciel, S.; Bourin, M.C.; Duprez, C.; Caramelle, P.; Boyer, L.; Hue, S.; Stern, J.B.; Validire, P.; et al. The cyclooxygenase-2-prostaglandin E2 pathway maintains senescence of chronic obstructive pulmonary disease fibroblasts. Am. J. Respir. Crit. Care Med. 2013, 187, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-kappaB signalling. Mech. Ageing Dev. 2018, 170, 30–36. [Google Scholar] [CrossRef]
- Borghesan, M.; Fafian-Labora, J.; Eleftheriadou, O.; Carpintero-Fernandez, P.; Paez-Ribes, M.; Vizcay-Barrena, G.; Swisa, A.; Kolodkin-Gal, D.; Ximenez-Embun, P.; Lowe, R.; et al. Small Extracellular Vesicles Are Key Regulators of Non-cell Autonomous Intercellular Communication in Senescence via the Interferon Protein IFITM3. Cell Rep. 2019, 27, 3956–3971. [Google Scholar] [CrossRef]
- Mas-Bargues, C.; Sanz-Ros, J.; Roman-Dominguez, A.; Gimeno-Mallench, L.; Ingles, M.; Vina, J.; Borras, C. Extracellular Vesicles from Healthy Cells Improves Cell Function and Stemness in Premature Senescent Stem Cells by miR-302b and HIF-1alpha Activation. Biomolecules 2020, 10, 957. [Google Scholar] [CrossRef]
- Mas-Bargues, C.; Sanz-Ros, J.; Romero-Garcia, N.; Huete-Acevedo, J.; Dromant, M.; Borras, C. Small extracellular vesicles from senescent stem cells trigger adaptive mechanisms in young stem cells by increasing antioxidant enzyme expression. Redox Biol. 2023, 62, 102668. [Google Scholar] [CrossRef]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles Derived from Indoxyl Sulfate Treated Endothelial Cells Induce Endothelial Progenitor Cells Dysfunction. Front Physiol. 2017, 8, 666. [Google Scholar] [CrossRef] [PubMed]
- Khayrullin, A.; Krishnan, P.; Martinez-Nater, L.; Mendhe, B.; Fulzele, S.; Liu, Y.; Mattison, J.A.; Hamrick, M.W. Very Long-Chain C24:1 Ceramide Is Increased in Serum Extracellular Vesicles with Aging and Can Induce Senescence in Bone-Derived Mesenchymal Stem Cells. Cells 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; Lin, C.C.; Eischen, C.M.; Bandyopadhyay, S.; Zhao, Z. Concordant dysregulation of miR-5p and miR-3p arms of the same precursor microRNA may be a mechanism in inducing cell proliferation and tumorigenesis: A lung cancer study. RNA 2015, 21, 1055–1065. [Google Scholar] [CrossRef]
- Weiner-Gorzel, K.; Dempsey, E.; Milewska, M.; McGoldrick, A.; Toh, V.; Walsh, A.; Lindsay, S.; Gubbins, L.; Cannon, A.; Sharpe, D.; et al. Overexpression of the microRNA miR-433 promotes resistance to paclitaxel through the induction of cellular senescence in ovarian cancer cells. Cancer Med. 2015, 4, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Moller, M.N.; Cuevasanta, E.; Orrico, F.; Lopez, A.C.; Thomson, L.; Denicola, A. Diffusion and Transport of Reactive Species Across Cell Membranes. Adv. Exp. Med. Biol. 2019, 1127, 3–19. [Google Scholar] [PubMed]
- Grenfell, S.; Smithers, N.; Miller, K.; Solari, R. Receptor-mediated endocytosis and nuclear transport of human interleukin 1 alpha. Biochem. J. 1989, 264, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Namba, T.; Takehara, M.; Nakaya, T.; Sugimoto, Y.; Araki, W.; Narumiya, S.; Suzuki, T.; Mizushima, T. Prostaglandin E2 stimulates the production of amyloid-beta peptides through internalization of the EP4 receptor. J. Biol. Chem. 2009, 284, 18493–18502. [Google Scholar] [CrossRef]
- Di Paolo, N.C.; Shayakhmetov, D.M. Interleukin 1alpha and the inflammatory process. Nat. Immunol. 2016, 17, 906–913. [Google Scholar] [CrossRef]
- Schwartz, R.E.; Shokhirev, M.N.; Andrade, L.R.; Gutkind, J.S.; Iglesias-Bartolome, R.; Shadel, G.S. Insights into epithelial cell senescence from transcriptome and secretome analysis of human oral keratinocytes. Aging 2021, 13, 4747–4777. [Google Scholar] [CrossRef]
- Mikula-Pietrasik, J.; Sosinska, P.; Janus, J.; Rubis, B.; Brewinska-Olchowik, M.; Piwocka, K.; Ksiazek, K. Bystander senescence in human peritoneal mesothelium and fibroblasts is related to thrombospondin-1-dependent activation of transforming growth factor-beta1. Int. J. Biochem. Cell Biol. 2013, 45, 2087–2096. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef] [PubMed]
- Vassilieva, I.; Kosheverova, V.; Vitte, M.; Kamentseva, R.; Shatrova, A.; Tsupkina, N.; Skvortsova, E.; Borodkina, A.; Tolkunova, E.; Nikolsky, N.; et al. Paracrine senescence of human endometrial mesenchymal stem cells: A role for the insulin-like growth factor binding protein 3. Aging 2020, 12, 1987–2004. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gan, Y.; Xu, Y.; Song, L.; Wang, L.; Ouyang, B.; Zhang, C.; Zhou, Q. The inflammatory cytokine TNF-alpha promotes the premature senescence of rat nucleus pulposus cells via the PI3K/Akt signaling pathway. Sci. Rep. 2017, 7, 42938. [Google Scholar] [CrossRef] [PubMed]
- Rentschler, M.; Chen, Y.; Pahl, J.; Soria-Martinez, L.; Braumuller, H.; Brenner, E.; Bischof, O.; Rocken, M.; Wieder, T. Nuclear Translocation of Argonaute 2 in Cytokine-Induced Senescence. Cell Physiol. Biochem. 2018, 51, 1103–1118. [Google Scholar] [CrossRef]
- Shin, J.W.; Lee, E.; Han, S.; Choe, S.A.; Jeon, O.H. Plasma Proteomic Signature of Cellular Senescence and Markers of Biological Aging among Postmenopausal Women. Rejuvenation Res. 2022, 25, 141–148. [Google Scholar] [CrossRef]
- Sharma, V.; Collins, L.B.; Chen, T.H.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget 2016, 7, 25377–25390. [Google Scholar] [CrossRef]
- Wang, W.; Mani, A.M.; Wu, Z.H. DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression. J. Cancer Metastasis Treat. 2017, 3, 45–59. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Iske, J.; Seyda, M.; Heinbokel, T.; Maenosono, R.; Minami, K.; Nian, Y.; Quante, M.; Falk, C.S.; Azuma, H.; Martin, F.; et al. Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs following transplantation. Nat. Commun. 2020, 11, 4289. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.N.; Victorelli, S.G.; Salmonowicz, H.; Dasgupta, N.; Liu, T.; Passos, J.F.; Adams, P.D. Cytoplasmic DNA: Sources, sensing, and role in aging and disease. Cell 2021, 184, 5506–5526. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [PubMed]
- Santa, P.; Garreau, A.; Serpas, L.; Ferriere, A.; Blanco, P.; Soni, C.; Sisirak, V. The Role of Nucleases and Nucleic Acid Editing Enzymes in the Regulation of Self-Nucleic Acid Sensing. Front. Immunol. 2021, 12, 629922. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, H.; Long, Y.; Li, P.; Gu, Y. The main sources of circulating cell-free DNA: Apoptosis, necrosis and active secretion. Crit. Rev. Oncol. Hematol. 2021, 157, 103166. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.; Koh, D.; Jeon, H.B.; Kim, K.M. The Role of Extracellular Vesicles in Senescence. Mol. Cells 2022, 45, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.; Mizen, H.; Bishop, C.L. The bright and dark side of extracellular vesicles in the senescence-associated secretory phenotype. Mech. Ageing Dev. 2020, 189, 111263. [Google Scholar] [CrossRef]
- Elliott, M.R.; Ravichandran, K.S. ELMO1 signaling in apoptotic germ cell clearance and spermatogenesis. Ann. N. Y. Acad. Sci. USA 2010, 1209, 30–36. [Google Scholar] [CrossRef]
- Lu, Z.; Elliott, M.R.; Chen, Y.; Walsh, J.T.; Klibanov, A.L.; Ravichandran, K.S.; Kipnis, J. Phagocytic activity of neuronal progenitors regulates adult neurogenesis. Nat. Cell Biol. 2011, 13, 1076–1083. [Google Scholar] [CrossRef]
- McKelvey, K.J.; Powell, K.L.; Ashton, A.W.; Morris, J.M.; McCracken, S.A. Exosomes: Mechanisms of Uptake. J. Circ. Biomark. 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Di, K.; Fan, B.; Wu, J.; Gu, X.; Sun, Y.; Khan, A.; Li, P.; Li, Z. MicroRNAs in extracellular vesicles: Sorting mechanisms, diagnostic value, isolation, and detection technology. Front. Bioeng. Biotechnol. 2022, 10, 948959. [Google Scholar] [CrossRef] [PubMed]
- Isaac, R.; Reis, F.C.G.; Ying, W.; Olefsky, J.M. Exosomes as mediators of intercellular crosstalk in metabolism. Cell Metab. 2021, 33, 1744–1762. [Google Scholar] [CrossRef] [PubMed]
- Chiaradia, E.; Tancini, B.; Emiliani, C.; Delo, F.; Pellegrino, R.M.; Tognoloni, A.; Urbanelli, L.; Buratta, S. Extracellular Vesicles under Oxidative Stress Conditions: Biological Properties and Physiological Roles. Cells 2021, 10, 1763. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Mancek-Keber, M. Half is enough: Oxidized lysophospholipids as novel bioactive molecules. Free Radic. Biol. Med. 2022, 188, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Shao, F.; Bai, X.Y.; Cai, G.; Lv, Y.; Chen, X. Changes in the expression of the Toll-like receptor system in the aging rat kidneys. PLoS ONE 2014, 9, e96351. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kiprov, D.D.; Luellen, C.; Lieb, M.; Liu, C.; Watanabe, E.; Mei, X.; Cassaleto, K.; Kramer, J.; Conboy, M.J.; et al. Old plasma dilution reduces human biological age: A clinical study. Geroscience 2022, 44, 2701–2720. [Google Scholar] [CrossRef]
- Qi, H.; Wang, Y.; Fa, S.; Yuan, C.; Yang, L. Extracellular Vesicles as Natural Delivery Carriers Regulate Oxidative Stress Under Pathological Conditions. Front. Bioeng. Biotechnol. 2021, 9, 752019. [Google Scholar] [CrossRef]
- Jeon, O.H.; Wilson, D.R.; Clement, C.C.; Rathod, S.; Cherry, C.; Powell, B.; Lee, Z.; Khalil, A.M.; Green, J.J.; Campisi, J.; et al. Senescence cell-associated extracellular vesicles serve as osteoarthritis disease and therapeutic markers. JCI Insight 2019, 4, e125019. [Google Scholar] [CrossRef]
- Alfonzo, M.C.; Al Saedi, A.; Fulzele, S.; Hamrick, M.W. Extracellular Vesicles as Communicators of Senescence in Musculoskeletal Aging. JBMR Plus 2022, 6, e10686. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Lowenstein, C.J. MiR-34, SIRT1 and p53: The feedback loop. Cell Cycle 2009, 8, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Vulpis, E.; Cuollo, L.; Borrelli, C.; Antonangeli, F.; Masuelli, L.; Cippitelli, M.; Fionda, C.; Caracciolo, G.; Petrucci, M.T.; Santoni, A.; et al. Doxorubicin-Mediated miR-433 Expression on Exosomes Promotes Bystander Senescence in Multiple Myeloma Cells in a DDR-Independent Manner. Int. J. Mol. Sci. 2023, 24, 6862. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, L.; Wu, J.; Qiu, Q.; Zhou, F.; Wu, H. The different expression profiles of microRNAs in elderly and young human dental pulp and the role of miR-433 in human dental pulp cells. Mech. Ageing Dev. 2015, 146–148, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mensa, E.; Guescini, M.; Giuliani, A.; Bacalini, M.G.; Ramini, D.; Corleone, G.; Ferracin, M.; Fulgenzi, G.; Graciotti, L.; Prattichizzo, F.; et al. Small extracellular vesicles deliver miR-21 and miR-217 as pro-senescence effectors to endothelial cells. J. Extracell. Vesicles 2020, 9, 1725285. [Google Scholar] [CrossRef]
- Battistelli, M.; Falcieri, E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; d’Angelo, M. Neuronal Cells Rearrangement During Aging and Neurodegenerative Disease: Metabolism, Oxidative Stress and Organelles Dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef]
- Taglialatela, G.; Gegg, M.; Perez-Polo, J.R.; Williams, L.R.; Rose, G.M. Evidence for DNA fragmentation in the CNS of aged Fischer-344 rats. Neuroreport 1996, 7, 977–980. [Google Scholar] [CrossRef]
- Ali, S.; Garcia, J.M. Sarcopenia, cachexia and aging: Diagnosis, mechanisms and therapeutic options—A mini-review. Gerontology 2014, 60, 294–305. [Google Scholar] [CrossRef]
- Ribeiro, A.S.F.; Zerolo, B.E.; Lopez-Espuela, F.; Sanchez, R.; Fernandes, V.S. Cardiac System during the Aging Process. Aging Dis. 2023, 14, 1105–1122. [Google Scholar] [CrossRef]
- Kajstura, J.; Cheng, W.; Sarangarajan, R.; Li, P.; Li, B.; Nitahara, J.A.; Chapnick, S.; Reiss, K.; Olivetti, G.; Anversa, P. Necrotic and apoptotic myocyte cell death in the aging heart of Fischer 344 rats. Am. J. Physiol. 1996, 271 Pt 2, H1215–H1228. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Oka, T.; Son, H.G.; Oliver-Garcia, V.S.; Azin, M.; Eisenhaure, T.M.; Lieb, D.J.; Hacohen, N.; Demehri, S. Cytotoxic CD4(+) T cells eliminate senescent cells by targeting cytomegalovirus antigen. Cell 2023, 186, 1417–1431.E20. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, A.; Burton, D.G.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D ligands mediate immunosurveillance of senescent cells. Aging 2016, 8, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Thompson, P.J.; Wang, Y.; Bhattacharyya, A.; Apostolopoulou, H.; Hatano, R.; Naikawadi, R.P.; Shah, A.; Wolters, P.J.; Koliwad, S.; et al. Invariant Natural Killer T cells coordinate removal of senescent cells. Med. 2021, 2, 938–950. [Google Scholar] [CrossRef]
- Kim, K.M.; Noh, J.H.; Bodogai, M.; Martindale, J.L.; Yang, X.; Indig, F.E.; Basu, S.K.; Ohnuma, K.; Morimoto, C.; Johnson, P.F.; et al. Identification of senescent cell surface targetable protein DPP4. Genes. Dev. 2017, 31, 1529–1534. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter-Hufford, A.; Ravichandran, K.S. Clearing the dead: Apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb. Perspect. Biol. 2013, 5, a008748. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.S.; Jiang, J.X.; Wu, J.; Halsted, C.; Friedman, S.L.; Zern, M.A.; Torok, N.J. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006, 43, 435–443. [Google Scholar] [CrossRef]
- Mu, M.; Gao, P.; He, J.; Tao, X.; Song, C. Vascular Endothelial Growth Factor Inhibits Phagocytosis of Apoptotic Cells by Airway Epithelial Cells. BioMed Res. Int. 2020, 2020, 5287131. [Google Scholar] [CrossRef]
- Szatmari-Toth, M.; Kristof, E.; Vereb, Z.; Akhtar, S.; Facsko, A.; Fesus, L.; Kauppinen, A.; Kaarniranta, K.; Petrovski, G. Clearance of autophagy-associated dying retinal pigment epithelial cells—A possible source for inflammation in age-related macular degeneration. Cell Death Dis. 2016, 7, e2367. [Google Scholar] [CrossRef]
- Tominaga, K.; Suzuki, H.I. TGF-beta Signaling in Cellular Senescence and Aging-Related Pathology. Int. J. Mol. Sci. 2019, 20, 5002. [Google Scholar] [CrossRef] [PubMed]
- Conboy, I.M.; Conboy, M.J.; Rebo, J. Systemic Problems: A perspective on stem cell aging and rejuvenation. Aging 2015, 7, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Walton, C.C.; Begelman, D.; Nguyen, W.; Andersen, J.K. Senescence as an Amyloid Cascade: The Amyloid Senescence Hypothesis. Front. Cell Neurosci. 2020, 14, 129. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.J.; Park, S.C. Targeting major vault protein in senescence-associated apoptosis resistance. Expert. Opin. Ther. Targets 2009, 13, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Kickhoefer, V.A.; Garcia, Y.; Mikyas, Y.; Johansson, E.; Zhou, J.C.; Raval-Fernandes, S.; Minoofar, P.; Zink, J.I.; Dunn, B.; Stewart, P.L.; et al. Engineering of vault nanocapsules with enzymatic and fluorescent properties. Proc. Natl. Acad. Sci. USA 2005, 102, 4348–4352. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Hulsmans, M.; Holvoet, P. MicroRNA-containing microvesicles regulating inflammation in association with atherosclerotic disease. Cardiovasc. Res. 2013, 100, 7–18. [Google Scholar] [CrossRef]
- Wu, J.; Yang, J.; Cho, W.C.; Zheng, Y. Argonaute proteins: Structural features, functions and emerging roles. J. Adv. Res. 2020, 24, 317–324. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef]
- Gallo, A.; Tandon, M.; Alevizos, I.; Illei, G.G. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS ONE 2012, 7, e30679. [Google Scholar] [CrossRef]
- Grabuschnig, S.; Bronkhorst, A.J.; Holdenrieder, S.; Rosales Rodriguez, I.; Schliep, K.P.; Schwendenwein, D.; Ungerer, V.; Sensen, C.W. Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms. Int. J. Mol. Sci. 2020, 21, 8062. [Google Scholar] [CrossRef] [PubMed]
- Gahan, P.B.; Stroun, M. The virtosome-a novel cytosolic informative entity and intercellular messenger. Cell Biochem. Funct. 2010, 28, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Olmo, D.C.; Dominguez, C.; Garcia-Arranz, M.; Anker, P.; Stroun, M.; Garcia-Verdugo, J.M.; Garcia-Olmo, D. Cell-free nucleic acids circulating in the plasma of colorectal cancer patients induce the oncogenic transformation of susceptible cultured cells. Cancer Res. 2010, 70, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Zhang, Q.; Franklin, J.L.; Coffey, R.J. Extracellular vesicles and nanoparticles: Emerging complexities. Trends Cell Biol. 2023, 33, 667–681. [Google Scholar] [CrossRef] [PubMed]
- Aarthy, R.; Mani, S.; Velusami, S.; Sundarsingh, S.; Rajkumar, T. Role of Circulating Cell-Free DNA in Cancers. Mol. Diagn. Ther. 2015, 19, 339–350. [Google Scholar] [CrossRef]
- Ventura, A.E.; Varela, A.R.P.; Dingjan, T.; Santos, T.C.B.; Fedorov, A.; Futerman, A.H.; Prieto, M.; Silva, L.C. Lipid domain formation and membrane shaping by C24-ceramide. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183400. [Google Scholar] [CrossRef]
- Chesarino, N.M.; McMichael, T.M.; Yount, J.S. Regulation of the trafficking and antiviral activity of IFITM3 by post-translational modifications. Future Microbiol. 2014, 9, 1151–1163. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwartz, R.E.; Conboy, I.M. Non-Intrinsic, Systemic Mechanisms of Cellular Senescence. Cells 2023, 12, 2769. https://doi.org/10.3390/cells12242769
Schwartz RE, Conboy IM. Non-Intrinsic, Systemic Mechanisms of Cellular Senescence. Cells. 2023; 12(24):2769. https://doi.org/10.3390/cells12242769
Chicago/Turabian StyleSchwartz, Rachael E., and Irina M. Conboy. 2023. "Non-Intrinsic, Systemic Mechanisms of Cellular Senescence" Cells 12, no. 24: 2769. https://doi.org/10.3390/cells12242769
APA StyleSchwartz, R. E., & Conboy, I. M. (2023). Non-Intrinsic, Systemic Mechanisms of Cellular Senescence. Cells, 12(24), 2769. https://doi.org/10.3390/cells12242769