
Characterization of Three Somatic Mutations in the 3′UTR of RRAS2 and Their Inverse Correlation with Lymphocytosis in Chronic Lymphocytic Leukemia

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Cells
2.2. Flow Cytometry
2.3. gDNA Extraction
2.4. Sequencing Strategy for Patients’ Samples
2.5. RRAS2 Expression Measurement
2.6. Fluorescent Probes Method to Sequence rs8570 SNP in CLL Patients’ Samples
2.7. Statistical Analysis
3. Results
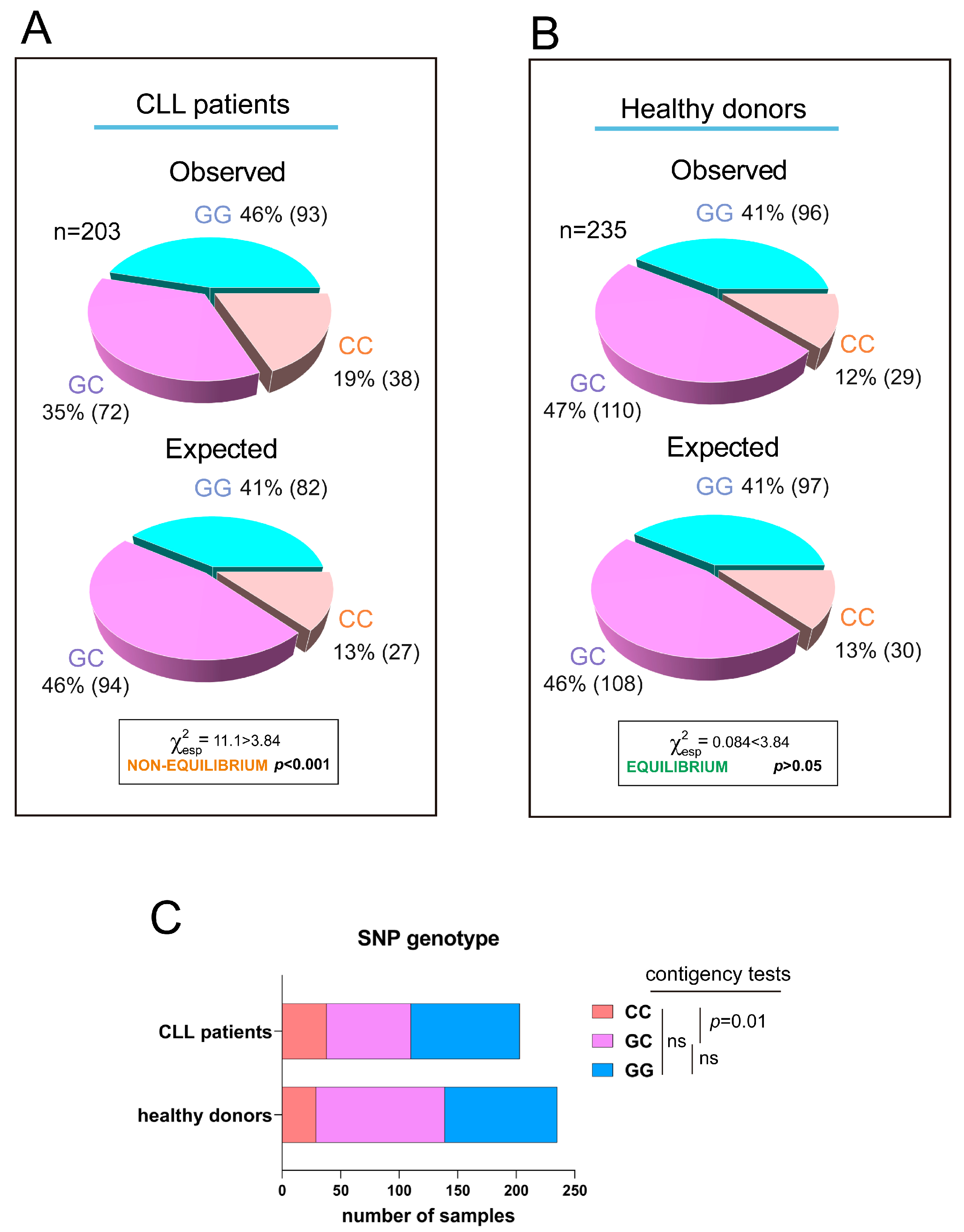
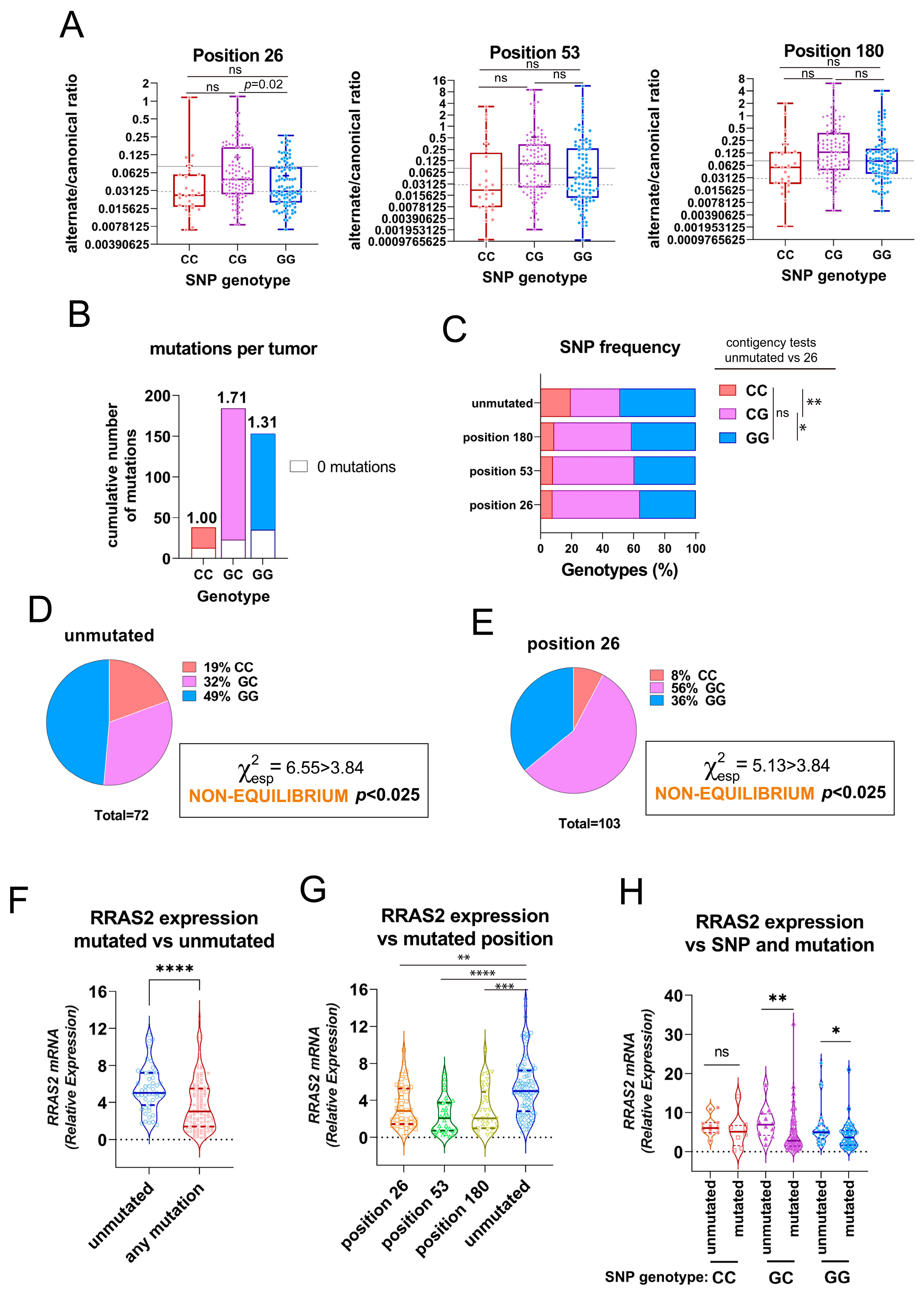
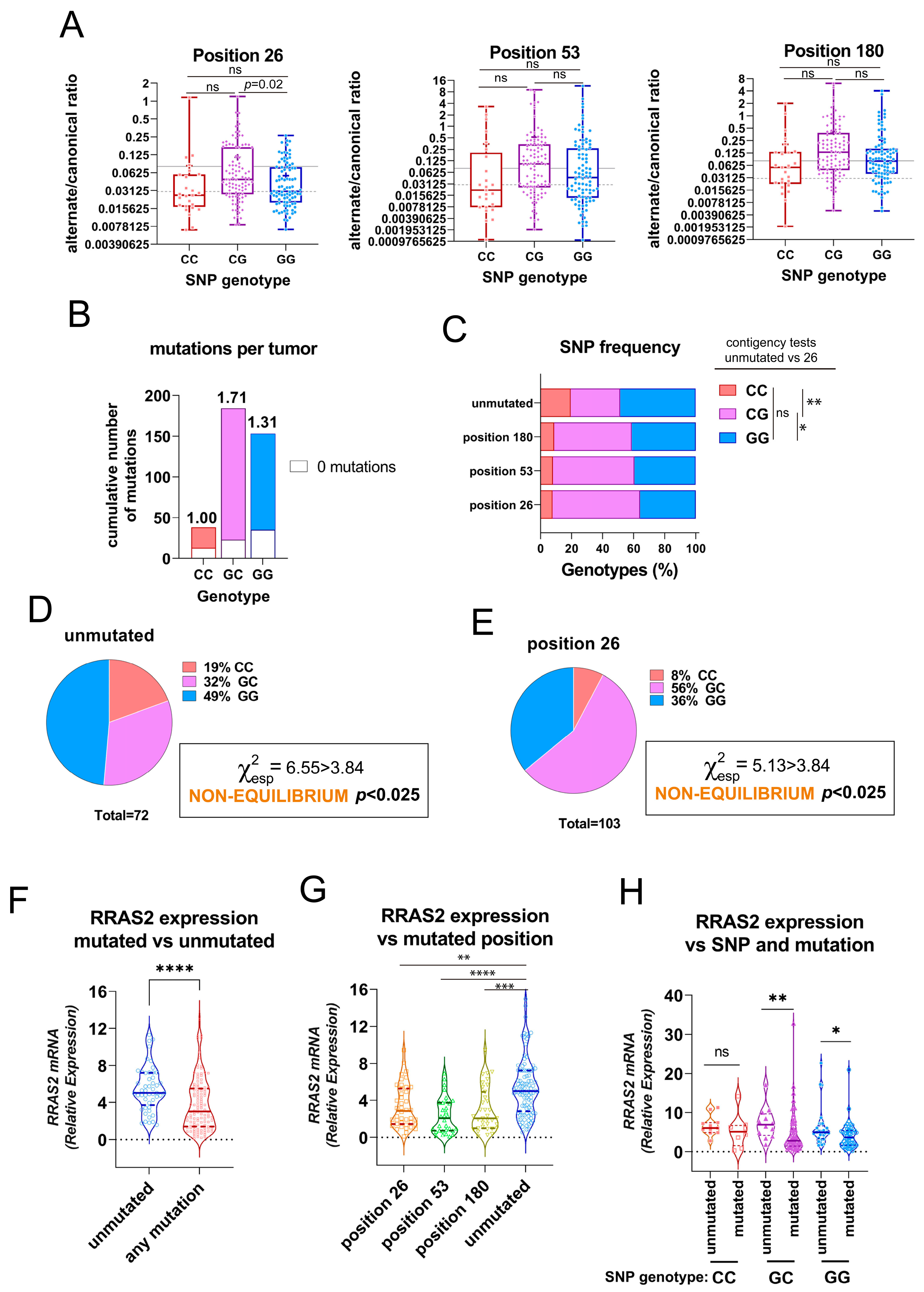
3.1. Presence of Somatic Mutations in the 3′UTR of RRAS2 in CLL Patients
3.2. Association of the Three 3′UTR Mutations with RRAS2 mRNA Abundance
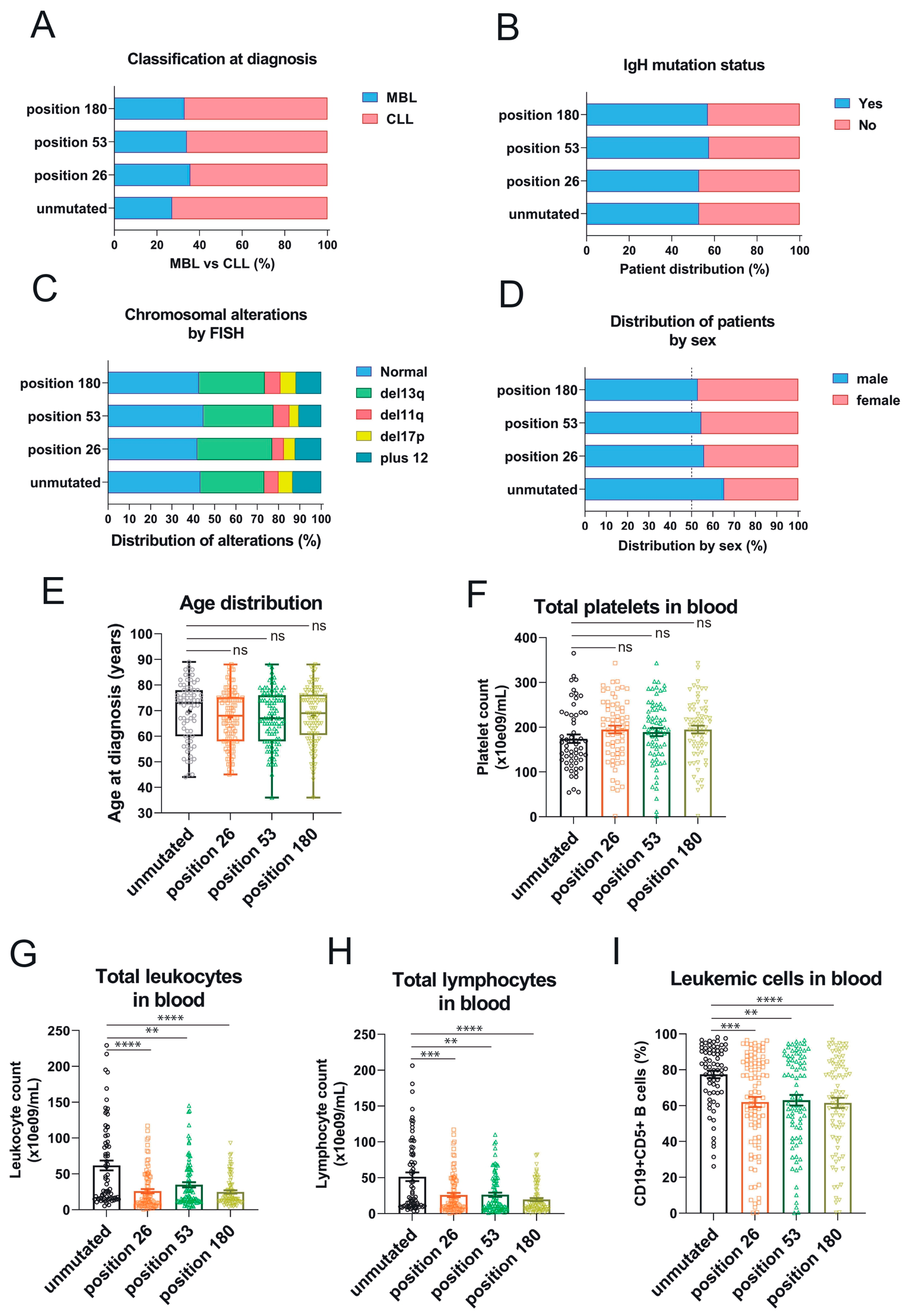
3.3. Association of the Three 3′UTR Mutations with Clinical Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ghia, P.; Ferreri, A.J.M.; Caligaris-Cappio, F. Chronic Lymphocytic Leukemia. Crit. Rev. Oncol. Hematol. 2007, 64, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, B.; Dreyling, M.; Robak, T.; Montserrat, E.; Hallek, M. Chronic Lymphocytic Leukemia: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2011, 22, vi50–vi54. [Google Scholar] [CrossRef] [PubMed]
- Fürstenau, M.; Eichhorst, B. Novel Agents in Chronic Lymphocytic Leukemia: New Combination Therapies and Strategies to Overcome Resistance. Cancers 2021, 13, 1336. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.; Dalla-Favera, R. Chronic Lymphocytic Leukaemia: From Genetics to Treatment. Nat. Rev. Clin. Oncol. 2019, 16, 684–701. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Yao, Y.; Lin, X.; Li, F.; Jin, J.; Wang, H. The Global Burden and Attributable Risk Factors of Chronic Lymphocytic Leukemia in 204 Countries and Territories from 1990 to 2019: Analysis Based on the Global Burden of Disease Study 2019. Biomed. Eng. OnLine 2022, 21, 4. [Google Scholar] [CrossRef]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Non-Coding Recurrent Mutations in Chronic Lymphocytic Leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef]
- Graham, S.M.; Cox, A.D.; Drivas, G.; Rush, M.G.; D’Eustachio, P.; Der, C.J. Aberrant Function of the Ras-Related Protein TC21/R-Ras2 Triggers Malignant Transformation. Mol. Cell Biol. 1994, 14, 4108–4115. [Google Scholar]
- Graham, S.M.; Oldham, S.M.; Martin, C.B.; Drugan, J.K.; Zohn, I.E.; Campbell, S.; Der, C.J. TC21 and Ras Share Indistinguishable Transforming and Differentiating Activities. Oncogene 1999, 18, 2107–2116. [Google Scholar] [CrossRef]
- Delgado, P.; Cubelos, B.; Calleja, E.; Martinez-Martin, N.; Cipres, A.; Merida, I.; Bellas, C.; Bustelo, X.R.; Alarcon, B. Essential Function for the GTPase TC21 in Homeostatic Antigen Receptor Signaling. Nat. Immunol. 2009, 10, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, P.; Martinez-Martin, N.; Bovolenta, E.R.; Reyes-Garau, D.; Hernansanz-Agustin, P.; Delgado, P.; Diaz-Munoz, M.D.; Oeste, C.L.; Fernandez-Pisonero, I.; Castellano, E.; et al. R-Ras2 Is Required for Germinal Center Formation to Aid B Cells during Energetically Demanding Processes. Sci. Signal 2018, 11, eaal1506. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Pyon, J.K.; Lee, S.H.; Lee, Y.J.; Kang, S.G.; Kim, C.H.; Kim, D.W.; Nam, H.S.; Park, Y.H.; Jeong, D.J.; et al. Greater Expression of TC21/R-Ras2 in Highly Aggressive Malignant Skin Cancer. Int. J. Dermatol. 2011, 50, 956–960. [Google Scholar] [CrossRef]
- Macha, M.A.; Matta, A.; Sriram, U.; Thakkar, A.; Shukla, N.K.; Datta Gupta, S.; Ralhan, R. Clinical Significance of TC21 Overexpression in Oral Cancer. J. Oral. Pathol. Med. 2010, 39, 477–485. [Google Scholar] [CrossRef]
- Sharma, R.; Sud, N.; Chattopadhyay, T.K.; Ralhan, R. TC21/R-Ras2 Upregulation in Esophageal Tumorigenesis: Potential Diagnostic Implications. Oncology 2005, 69, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pisonero, I.; Clavaín, L.; Robles-Valero, J.; Lorenzo-Martín, L.F.; Caloto, R.; Nieto, B.; García-Macías, C.; Oeste, C.L.; Sánchez-Martín, M.; Abad, A.; et al. A Hotspot Mutation Targeting the R-RAS2 GTPase Acts as a Potent Oncogenic Driver in a Wide Spectrum of Tumors. Cell Rep. 2022, 38, 110522. [Google Scholar] [CrossRef] [PubMed]
- Hortal, A.M.; Oeste, C.L.; Cifuentes, C.; Alcoceba, M.; Fernández-Pisonero, I.; Clavaín, L.; Tercero, R.; Mendoza, P.; Domínguez, V.; García-Flores, M.; et al. Overexpression of Wild Type RRAS2, without Oncogenic Mutations, Drives Chronic Lymphocytic Leukemia. Mol. Cancer 2022, 21, 35. [Google Scholar] [CrossRef]
- Lefever, S.; Rihani, A.; Van der Meulen, J.; Pattyn, F.; Van Maerken, T.; Van Dorpe, J.; Hellemans, J.; Vandesompele, J. Cost-Effective and Robust Genotyping Using Double-Mismatch Allele-Specific Quantitative PCR. Sci. Rep. 2019, 9, 2150. [Google Scholar] [CrossRef]
- Hortal, A.; Lacuna, M.; Cifuentes, C.; Alcoceba, M.; Bustelo, X.R.; González, M.; Alarcón, B. An Optimized Single Nucleotide Polymorphism-Based Detection Method Suggests That Allelic Variants in the 3′ Untranslated Region of RRAS2 Correlate with Treatment Response in Chronic Lymphocytic Leukemia Patients. Cancers 2023, 15, 644. [Google Scholar] [CrossRef]
- Mayo, O. A Century of Hardy–Weinberg Equilibrium. Twin Res. Hum. Genet. 2008, 11, 249–256. [Google Scholar] [CrossRef]
- Mayr, C. What Are 3′ UTRs Doing? Cold Spring Harb. Perspect. Biol. 2019, 11, a034728. [Google Scholar] [CrossRef] [PubMed]
- Gaidano, G.; Rossi, D. The Mutational Landscape of Chronic Lymphocytic Leukemia and Its Impact on Prognosis and Treatment. Hematology 2017, 2017, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Gao, W.; Li, Q.; Liu, Y.; Chen, H.; Cui, Y.; Sun, Z.; Liu, Z. Comprehensive Characterization of Posttranscriptional Impairment-Related 3′-UTR Mutations in 2413 Whole Genomes of Cancer Patients. NPJ Genomic Med. 2022, 7, 34. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3′UTR Mutation | Allele | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|---|
| 26 | G (canonical) | CCCTTCAGTTTTAGCTACCCACG | GCTTCACAGAAAGGACTAGCC |
| T (alternative) | CCCTTCAGTTTTAGCTACCCACT | ||
| 53 | T (canonical) | GGAAAAGCCCTCATCTTCTATTT | AGCAGCCTTAGTGTTTCCTT |
| C (alternative) | GGAAAAGCCCTCATCTTCTATTC | ||
| 124 | G (canonical) | GATCACCATGTTAGCCTTATACC | AGCAGCCTTAGTGTTTCCTT |
| C (alternative) | GATCACCATGTTAGCCTTATACG | ||
| 180 | T (canonical) | CTACCAACGGCCAGGAAAAG | GCAGCCTTAGTGTTTCCTTGAAA |
| C (alternative) | GCAGCCTTAGTGTTTCCTTGAAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lacuna, M.; Hortal, A.M.; Cifuentes, C.; Gonzalo, T.; Alcoceba, M.; Bastos, M.; Bustelo, X.R.; González, M.; Alarcón, B. Characterization of Three Somatic Mutations in the 3′UTR of RRAS2 and Their Inverse Correlation with Lymphocytosis in Chronic Lymphocytic Leukemia. Cells 2023, 12, 2687. https://doi.org/10.3390/cells12232687
Lacuna M, Hortal AM, Cifuentes C, Gonzalo T, Alcoceba M, Bastos M, Bustelo XR, González M, Alarcón B. Characterization of Three Somatic Mutations in the 3′UTR of RRAS2 and Their Inverse Correlation with Lymphocytosis in Chronic Lymphocytic Leukemia. Cells. 2023; 12(23):2687. https://doi.org/10.3390/cells12232687
Chicago/Turabian StyleLacuna, Marta, Alejandro M. Hortal, Claudia Cifuentes, Tania Gonzalo, Miguel Alcoceba, Miguel Bastos, Xosé R. Bustelo, Marcos González, and Balbino Alarcón. 2023. "Characterization of Three Somatic Mutations in the 3′UTR of RRAS2 and Their Inverse Correlation with Lymphocytosis in Chronic Lymphocytic Leukemia" Cells 12, no. 23: 2687. https://doi.org/10.3390/cells12232687
APA StyleLacuna, M., Hortal, A. M., Cifuentes, C., Gonzalo, T., Alcoceba, M., Bastos, M., Bustelo, X. R., González, M., & Alarcón, B. (2023). Characterization of Three Somatic Mutations in the 3′UTR of RRAS2 and Their Inverse Correlation with Lymphocytosis in Chronic Lymphocytic Leukemia. Cells, 12(23), 2687. https://doi.org/10.3390/cells12232687