1. Introduction
Skeletal muscle is a highly plastic tissue, able to adapt to different events such as exercise or injury throughout life, which leads to a continuous recurrence of regeneration to maintain proper tissue homeostasis. Muscle regeneration is a fine-regulated process in which the main actors are the satellite cells (SCs), the adult muscle stem cells. They are located under the basal lamina in a quiescent state and, in response to injury, they exit this dormant state getting activated. This transition includes metabolic stimulation, cell-cycle entry, and migration [
1,
2,
3]. Once dividing, only a subset of SCs self-renew to restore the quiescent pool, while the majority of them differentiate and fuse with each other in order to complete the formation of skeletal muscle fibers. For this reason, SC fusion is a required step during the regeneration process [
4]. Although the transcriptional program governing skeletal muscle development and adult muscle myogenesis have been described in detail [
5,
6], the mechanisms that coordinate myoblast fusion remain partially understood. The conserved cell–cell fusion process so far described is composed of different steps: first, the recognition of two partner cells that have to migrate toward each other and bring themselves close enough (within 10 nm of one another) to allow the start of cells adhesion machinery; second, the induction of close membrane apposition through F-actin protrusions has to occur. The third step is the rearrangement of membrane lipid bilayer (hemifusion) and finally pore formation and cell cytoplasm fusion. In
Drosophila it has been demonstrated that mechanical forces play an essential role in cell–cell adhesion and cell–cell fusion, and are essential to overcome repulsive force among myoblasts [
7]. Due to hydration repulsion, the energy barrier is elevated and in this moment specialized proteins come in play; these proteins are called fusogenes and are required to initiate the fusion events [
4,
8]. In mammals, muscle-specific fusogenes, Myomaker and Myomerger, controlling different steps of the fusion process, hemifusion and pore formation respectively have been identified [
9,
10,
11,
12]. When Myomaker and Myomerger are co-expressed in fibroblasts, they are sufficient to induce fusion in these non-fusogenic cells [
11]. Moreover, numerous proteins are necessary for all the steps involved in the complex process of cell fusion [
13]. In particular, cell–cell adhesion molecules that are localized at the contact site such as M-cadherin, Integrin α9β1 and Adam12 [
14] are important not only to enhance the contact between two cells but also to start signaling transduction pathway. In addition, remodeling of the actin cytoskeleton and actin dynamics are essential to achieve a correct fusion between myoblasts [
15,
16,
17].
Using primary murine myoblasts, finger-like actin protrusions occurring at the site of fusion have been described for the first time by Randrianarison-Huetz and colleagues [
18]. Characterized as essential structures for the fusion process, the molecular mechanism underlying their formation has been partially unveiled. In this paper serum response factor (Srf) has been identified as a master regulator of SC fusion required in both fusion partners. In particular, it is crucial to control the organization of the actin cytoskeleton and actin-based protrusions for myoblast fusion in order to achieve efficient hypertrophic myofiber growth [
18]. Even if some downstream targets of Srf have been identified and could be implicated in the formation of actin-based protrusion regulating actin cytoskeleton organization, upstream regulators of Srf involved in this mechanism are still unknown. Interestingly Srf is one of the transcription factors activated by Ras homolog family member A (RhoA) [
19,
20] which translates physical forces into biochemical signalling and transmits this signal to the nucleus [
21].
RhoA is a small GTPase protein that oscillates between GTP-bound and GDP-bound states, regulating a wide spectrum of cellular functions. Indeed, Rho GTPases coordinate the differentiation of several cell types and are involved in the regulation of immunological responses, blood pressure levels, and glucose homeostasis. Recent works revealed that Rho GTPases play a critical role during muscle development, regeneration, and function, suggesting that this protein is an important player in the modulation of both embryonic and adult myogenesis [
22,
23]. RhoA is also necessary for the initial induction of the myogenic program via the stimulation of SRF-mediated gene expression programs and, subsequently, to maintain the myoblasts in a proliferative state [
24,
25]. Later on, during the myogenesis process, the activity of this GTPase must be shut down to prompt cell cycle withdrawal and the final myoblast fusion step. Cell culture experiments have shown that RhoA fluctuates between high and low activity states in proliferating and differentiating myoblasts, respectively [
26] underlying the complexity of its function. Thus, RhoA may control multiple aspects of muscle cell behavior. In this scenario, RhoA represents an interesting player to investigate during muscle adaptive responses. However, it remains to be determined whether such fluctuations also occur in vivo. Recently, it has been described that RhoA, activated in the whole muscle by Wnt4, maintains SCs in a quiescent state, promoting the inhibition of yes-associated protein (YAP) transcription factor in a Rho GTPase activates Rho kinase (ROCK) dependent manner [
27]. Moreover, other works investigated RhoA in the modulation of the myogenic potential of cells evidencing its role in promoting myogenesis during muscle regeneration [
28]. RhoA regulates muscle regeneration modulating autophagy flux and in consequence the switch from the quiescent to the activated state in satellite cells. Indeed, a recent study has shown that the inactivation of the RhoA–ROCK axis caused by the depletion of the upstream exchange factor ArhGEF3 promotes injury-induced muscle regeneration by increasing autophagy in mice [
29]. Another work showed that Vav2, a Rho GTPase activator, modulates the signaling output of the IGF1- and insulin-stimulated phosphatidylinositol 3-kinase pathway in muscle tissue, including RhoA in modulator of muscle mass [
30].
All these assertions show that Rho GTPases play important roles in muscle development, regeneration, and homeostasis. However, the redundant and/or compensatory effect of the other members of the Rho family represents a limitation in precisely defining the role of a single protein. In this paper we will focus on adult muscle stem cells. Moreover, the use of more sophisticated animal models (e.g., inducible, skeletal muscle-specific) may help to better define the role of Rho GTPases and identify their regulators and effector pathways in muscle homeostasis processes. Indeed, here we describe the role played by RhoA within SCs, taking advantage of conditional and inducible RhoA deletion, during skeletal muscle regeneration. We show that in the absence of RhoA, muscle regeneration and hypertrophy are compromised, mostly caused by impaired SC fusion. These defects are not correlated to actin disassembly or cytoskeleton shortfall. Furthermore, in differentiated primary muscle cells, we have evidence that RhoA ablation affects the expression of several genes, indicating novel functions of RhoA in adult skeletal muscle myogenesis and muscle plasticity.
2. Materials and Methods
2.1. Mouse Protocol
RhoA
lx/lx mice are homozygous for RhoA floxed alleles harboring LoxP sites flanking exon 3 of the endogenous RhoA gene [
31]. Tg:Pax7-nGFP transgenic mice express nuclear localized EGFP under the Pax7 promoter [
32].
To investigate the effect of satellite cell-specific RhoA-deletion in adult muscle, the mouse strain following mice were generated: Pax7-Cre-ERT2:RhoAlx/lx. In all experiments, 3-month-old Pax7-Cre-ERT2:RhoAlx/lx mice (males and females) were given five intraperitoneal (i.p.) tamoxifen (Tam, 1 mg/day; MP Biomedicals, Santa Ana, CA, USA) injections to induce RhoA deletion (referred as Tam+). RhoAlx/lx mice not injected with Tam were used as control mice (referred to as Tam−).
Mice were genotyped by PCR using the following primers: Cre-F 5′-CCTGGAAAATGCTTCTGTCCG-3′; Cre-R 5′-CAGGGTGTTATAAGCAATCCC-3′; RhoAlx-F 5′-AGGGTTTCTCTGTACGGTAGTC-3′; RhoAlx-R 5′-GCAGCTAGTCTAACCCACTACA-3′. All animal experiments were conducted in accordance with the European guidelines for the care and use of laboratory animals and were approved by the institutional ethics committee and the French Ministry of Research (number A751402).
2.2. Compensatory Hypertrophy (CH) Protocol
Compensatory Hypertrophy (CH) of Plantaris muscles of control and RhoA-deleted mice was induced through the incapacitation of soleus and gastrocnemius muscles by sectioning their tendon, in both legs. During the process of CH, both mice groups were injected with Tam on days 2 and 4 after CH. 3 weeks after CH, Plantaris muscles were dissected and processed for histological analyses. When indicated, mice were administered 25 µg/g EdU (Life Technologies).
2.3. Cardiotoxin Protocol
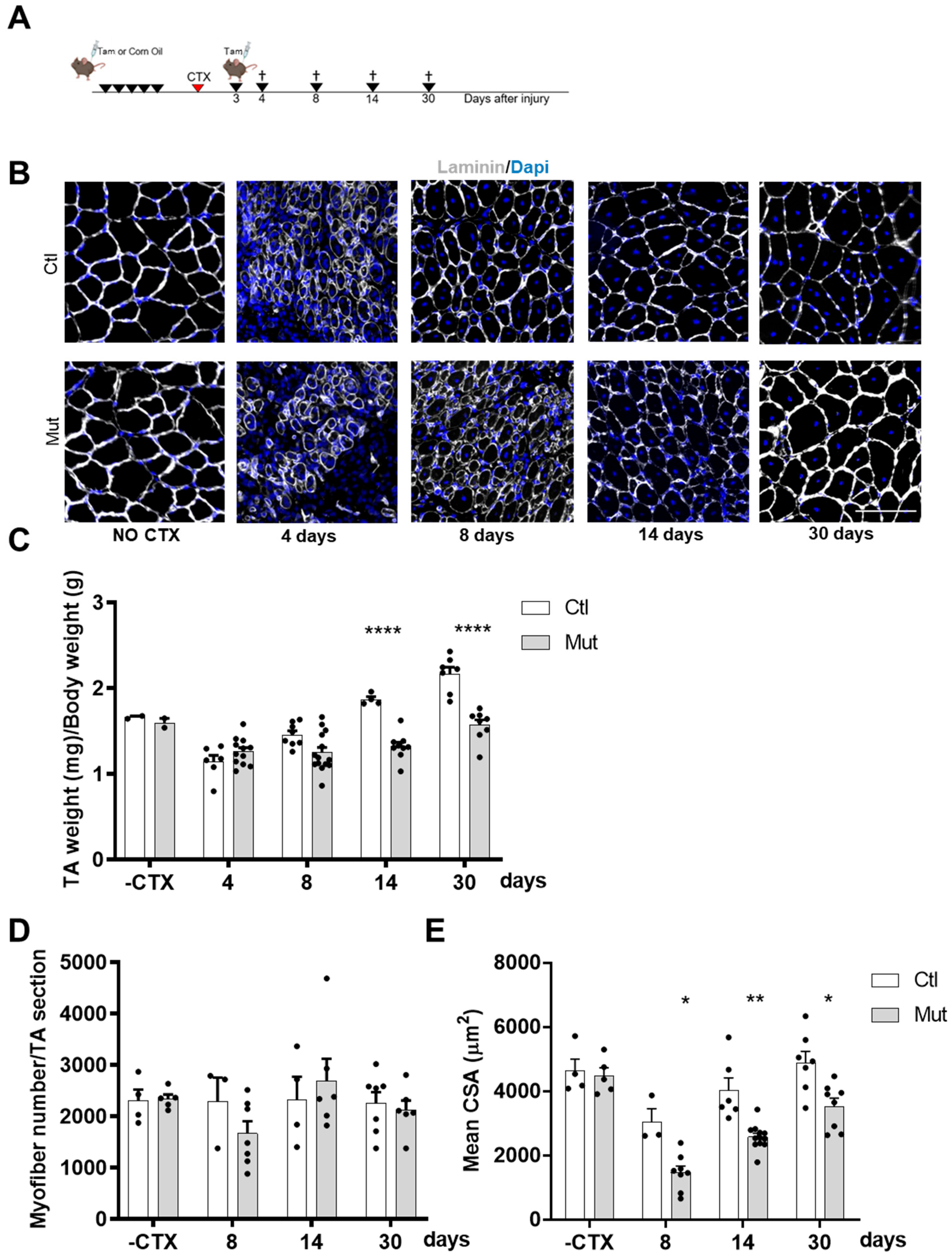
Muscle tissue injury in control and RhoA-deleted mice was achieved by a single intramuscular injection of 30 µL of 6 µM CTX (Latoxan, Valence, France) into TA muscle. During the process of regeneration, both mice groups were injected with Tam on days 3 after CTX. Mice were allowed to recover for 30 days and TA muscles were harvested.
2.4. Muscle Histology, Immunohistochemistry and Cell Staining
For assessment of tissue morphology, 8-μm-thick transverse sections were stained with hematoxylin and eosin (Hémalun de Mayer and Erythrosine 239 RAL Diagnostics) and examined under a light microscope.
TA muscles were collected and snap-frozen in liquid nitrogen-cooled isopentane. Eight μm-thick muscle sections were fixed in 4% PFA for 8 min at room temperature and blocked overnight at 4 °C in PBS 1×, 10% Horse serum, and 0.5% Triton X-100. Then, they were incubated with primary antibodies overnight at 4 °C in PBS 1×, 10% Horse serum, and 0.5% Triton X-100. The following primary antibodies were used: anti-eMHC (Alexis Biochemicals, San Diego, CA, USA, 805-504-L001, 1/50), anti-myogenin (sc-576 M-225, 1/100), anti-Laminin (Sigma, St. Louis, MO, USA, L9393, 1/200). After washes in PBS 1x, sections were incubated with secondary antibodies for 1 h at room temperature. The following secondary antibodies used were goat anti-mouse IgG1 Alexa 488 (ThermoFisher, Waltham, MA, USA, A21121, 1/1000) and donkey anti-rabbit Alexa 546 (Life Technologies, Carlsbad, CA, USA, A10040, 1/1000). Nuclei staining was performed using DAPI (Sigma, St. Louis, MO, USA, 14530, 1/10000). Muscles sections were then mounted in Dako Fluorescence Mounting Medium and kept at 4 °C until image acquisition.
For Pax7 staining, muscle sections were fixed in 4% PFA for 8 min at RT and permeabilized in ice-cold methanol for 6 min. Muscle sections were treated with Antigen Unmasking Solution pH6 (Vector, H-3300) for 15 min at 95 °C and cooled on ice for 30 min. Blocking and incubation with primary and secondary antibodies were conducted as described in the previous paragraph. Primary mouse anti-Pax7 antibody (Santa Cruz, sc-81648) was used at dilution 1/50.
EdU detection was performed using the Click-iT EdU Alexa Fluor 647 kit, according to the manufacturer’s instructions (Life Technologies).
Apoptotic cell detection was performed using ApopTag® Red Apoptosis Detection Kit (S7165), according to the manufacturer’s instructions (EMD Millipore, Chicago, IL, USA).
Muscle cells cultured in dishes were fixed for 8 min in 4% PFA and then permeabilized and blocked in PBS with 0.1% Triton X-100 and 5% horse serum for 1 h at RT. Cells were incubated overnight at 4 °C with the following primary antibodies: anti-MHC (DSHB,1/50) and anti-pMLC2 (cell signaling, 3671S, 1/1000) diluted in the same buffer. After incubation for 1 h at RT with fluorescent secondary antibody anti-mouse IgG1 Alexa 488 (1/1000; A21121), cells were stained with DAPI (for nuclei) and phalloidin Alexa Fluor 488 (1/500; Thermo Fisher Scientific; for F-actin) and mounted in Fluorescent Mounting Medium (Dako).
2.5. Primary Muscle Cell Culture
Primary cultures were derived from hindlimb muscles of control and RhoA-deleted of 6- to 8-week-old mice all harboring the Pax7-nGFP transgene that allowed prospective selection of SCs by FACS. The dissection of the muscles was performed with care to take off as much fat and connective tissue as possible. The muscles were minced in DMEM/F12 supplemented with 2% antibiotic/antimycotic (15240-062; Gibco, Waltham, MA, USA) in a sterile Petri dish on ice. The minced muscles were digested three times for 25 min at 37 °C with 1 mg/mL collagenase D (Roche) and 0.1% Trypsin (15090-046; Gibco, Waltham, MA, USA), and digestion was stopped by adding FCS (25% final). Cells were filtered through a 70-µm cell strainer and pelleted. Cells were then washed three times in DMEM/F12 and 2% antibiotic/antimycotic, resuspended in 1× PBS without Ca2+ and Mg2+, 2% FCS, and 2% antibiotic/antimycotic, and finally filtered with a 40-µm cell strainer. Pax7/GFP-positive SCs were sorted on FACS Aria III (BD) previously calibrated (fluorescence minus one and use of compensation beads) using the CYBIO Cochin Institute platform. Cells were collected in a FACS tube containing FCS and 2% antibiotic/antimycotic.
In standard conditions, myoblasts were grown in a growth medium (DMEM/F12, 2% Ultroser G [PALL Life Sciences, Portsmouth, United Kingdom], and 20% FCS) on plastic dishes coated with 0.02% Gelatin. For differentiation, myoblasts were seeded in Matrigel-coated dishes and cultured in a differentiation medium (DMEM/F12 and 2% horse serum).
2.6. Proliferation Assays
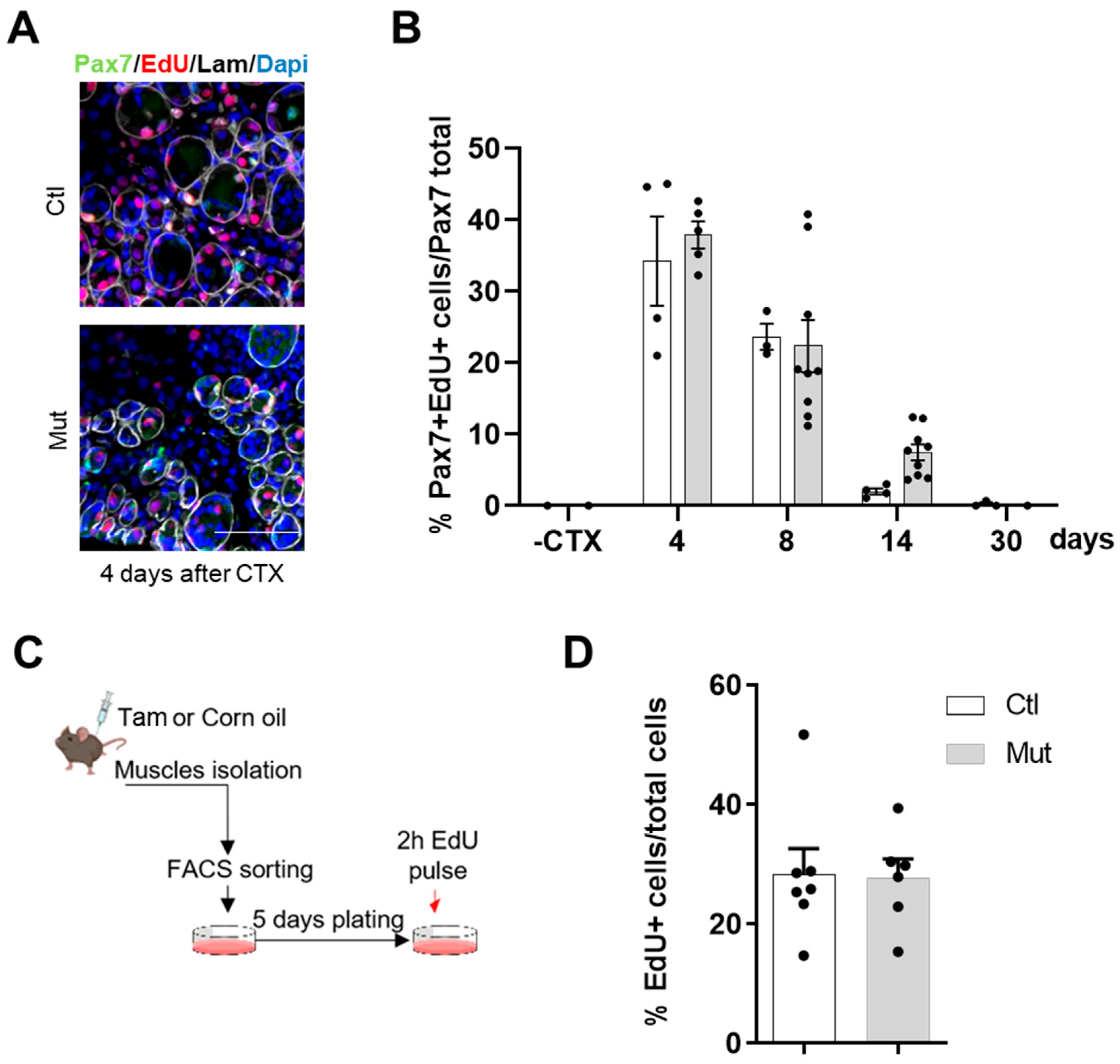
To detect S-phase entry, control and RhoA-deleted SCs were plated immediately after sorting, cultured for 5 days in a growth medium, and pulsed with EdU (10 µM; Life Technologies, Carlsbad, CA, USA) for 2 h before fixation with 4% PFA. EdU detection was performed using a Click-iT EdU Alexa Fluor 647 kit, according to the manufacturer’s instructions (Life Technologies, Carlsbad, CA, USA).
2.7. RNA Extraction and RT-qPCR
Total RNA was extracted using TRIzol reagent and reverse-transcribed with SuperScript III reverse transcriptase (Invitrogen). cDNA was synthesized from 1μg of RNA. Quantitative PCR analysis was performed using a Light Cycler (Roche) according to the manufacturer’s instructions using a SYBR Green I kit (Roche). Values were normalized using Hydroxymethylbilane synthetase (Hmbs). The following primers were used: RhoA-F 5′-AACCTGTGTGTTTTCAGCACC-3′; RhoA-R 5′-ACCTCTGGGAACTGGTCCTT-3′; Hmbs-F 5′-TGCACGATCCTGAAACTCTG-3′; Hmbs-R 5′-TGCATGCTATCTGAGCCATC-3′; MYH3-F 5′-GCAAAGACCCGTGACTTCACCTCTAG-3′; MYH3-R 5′-GCATGTGGAAAAGTGATACGTGG-3′; Myogenin-F 5′-GAAAGTGAATGAGGCCTTCG-3′; Myogenin-R 5′-ACGATGGACGTAAGGGAGTG-3′; MyoD-F 5′-GGCTCTCTCTGCTCCTTTGA-3′; MyoD-R 5′-AGTAGGGAAGTGTGCGTGCT-3′.
2.8. Cell Counting Analysis
To determine the average number of nuclei per fiber, we quantified the total number of DAPI+ nuclei contained within each muscle fiber or myotube.
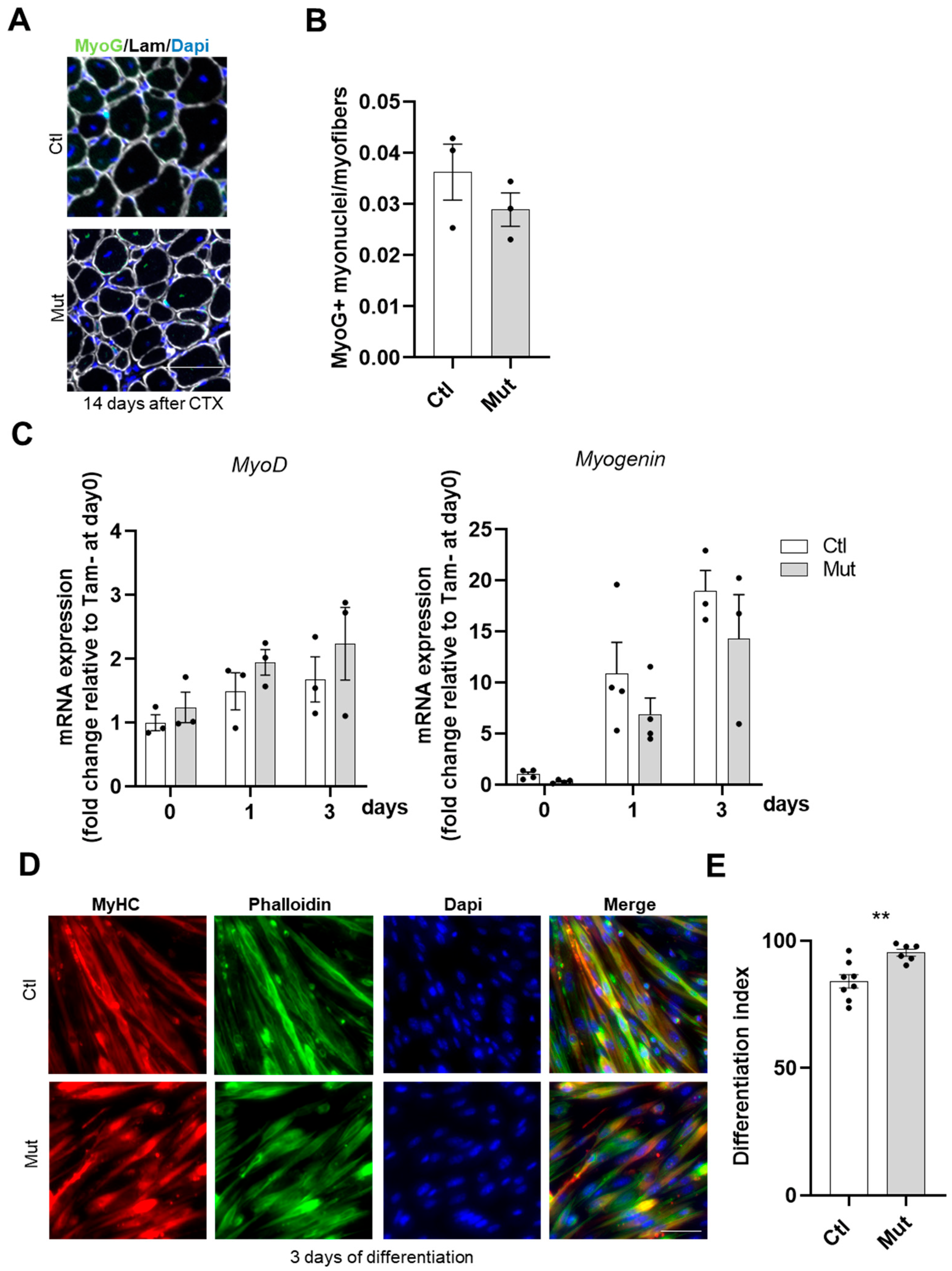
The fusion index was calculated as the fraction of nuclei contained within MyHC+ myotubes which had two or more nuclei, as compared to the number of total nuclei within differentiated cells (expressing MyHC).
In order to specify if fused cells are binucleated or bigger myotubes we determined the ratio between the number of nuclei contained within all MyHC+ cells and the number of cells MyHC+.
The differentiation index was calculated as the fraction of nuclei contained within all MyHC+ cells, including both mononuclear and multinuclear cells, as compared with the number of total nuclei within each image. Cells were considered positive for MyHC staining when the fluorescence intensity was clearly above background levels.
2.9. Cell Migration Assay
The migration of primary mouse muscle cells was quantified using time-lapse microscopy. Myoblasts were seeded in gelatin-coated eight-well Ibidi plates and maintained in a rich medium. The next day, cells were filmed using an inverted Axio Observer Z1 microscope (Zeiss) with an LCI PlN 20×/0.8 W DICII objective and an incubation chamber at 37 °C and 5% CO2. Live cells were monitored every 6 min for 4 h and 30 min with bright-field and Metamorph 7.7.5 software. Cell velocities were calculated in µm per minute using ImageJ (National Institutes of Health) by tracking the paths of cells. At least 100 cells were tracked for each group of two independent cell cultures.
2.10. Western Blot Analysis
Myoblast control and mutant were lysed in RIPA buffer (Sigma, St. Louis, MO, USA) and proteins were separated through denaturation SDS-PAGE electrophoresis using Mini-Protean TGX precast gels 4–15% (Biorad, Hercules, California, USA) and transferred on Nitrocellulose (0.2 micron, Biorad) membrane using the Trans-Blot turbo transfer system (Biorad). Membrane were blocked with 5% skinned milk in TBS-1% Tween (TBST) 1 h at room temperature and probed overnight at 4 °C with primary antibody. The following antibody were used: rabbit anti pMLC2 (1/50; #3674; Cell Signaling) and mouse anti α Tubulin (1/4000; T6064; Sigma, St. Louis, MO, USA)
Following washing in TBST, membranes were hybridized with secondary antibodies goat anti-mouse coupled to HRP (ThermoFisher, Waltham, MA, USA, 62-6520). Proteins were revealed using SuperSignal West Femto substrate (ThermoFisher, Waltham, MA, USA).
2.11. Image Acquisition
Digital images were acquired using an Olympus BX63F microscope with 10× objective (UplanFL, numerical aperture 0.3) and 20× objective (UPLSAPO, 0.75), ORCA-Flash4.0 LT C11440-42U camera (Hamamatsu); an Axiovert 200 M microscope (Zeiss) with 5× objective (PLANFLUAR, 0.25) and 20× objective (LD PLANNEOFLUAR, 0.4), cooled CCD CoolSNAP-HQ2 camera (Photometrics); or a Spinning Disk Leica confocal microscope with a 100× oil-immersion objective (HCX PL APO, 1.47), cooled CCD CoolSNAP-HQ2 camera (Photometrics) and Metamorph v.7.7.5 (Molecular Devices). Images were composed and edited in ImageJ. The background was reduced using brightness and contrast adjustments applied to the whole image.
2.12. Affymetrix Microarrays
Microarray analysis was performed from three independent control and mutant cell cultures. Total RNAs were obtained from cells at day 0 (corresponding to myoblasts), day 1 (corresponding to myocytes), and day 3 (corresponding to myotubes) of differentiation, using RNeasy Mini kit (Qiagen, Les Ulis, France) and DNase treatment (Qiagen, Les Ulis, France). RNA integrity was certified on a bioanalyzer (Agilent, Santa Clara, CA, USA). Hybridization to Mouse Gene 2.0-ST arrays (Affymetrix) and scans (GCS3000 7G Expression Console software V1.4) were performed on the Genom’ic platform (Institut Cochin, Paris, France). Probe data normalization and gene expression levels were processed using the robust multiarray average (RMA) algorithm in the expression Console (Affymetrix, Santa Clara, CA, USA). Gene ontology analysis was performed using Ingenuity (IPA) software, version 51963813 (Release Date: 11 March 2020). Full data are available on Gene Expression Omnibus: GSE242637.
2.13. Morphometric Analysis
The Myofiber cross-section area (CSA) was analyzed by using immunostaining of Laminin, marking myofiber sarcolemma, and then using the MuscleJ tool [
33] or ImageJ macro previously developed in our laboratory [
18]. Between 1300 and 3000 myofibers were analyzed in regenerative areas. For the quantification of the number of nuclei per myofibers, ImageJ was used and at least 1300 myofibers were counted per muscle.
4. Discussion
Skeletal muscle regeneration and muscle hypertrophy rely on sequences of fine-correlated events, crucial for complete and functional muscle restoration, formation, and growth. In this study, we aimed to establish the role of RhoA, expressed specifically in SCs in this physiological process by using an inducible SC-targeted knock-down mouse model. We demonstrated that RhoA deletion in SCs negatively affected skeletal muscle regeneration and growth. RhoA-deleted SCs are less in number compared to control SCs after muscle injury, suggesting trouble in their activation, although their proliferation and their differentiation were not affected either in vivo or in vitro. Importantly we showed that RhoA is indispensable for satellite cell fusion both in vitro and in vivo upon regeneration and hypertrophy in a cell-autonomous manner by affecting the expression of genes not yet described as important for this function in mammals.
RhoA has been described as an important regulator of cell cycle progression of some human cancers and cancer-associated mutations in Rho family regulators have been characterized [
46]. In contrast to these previous results, we demonstrated that the proliferation of RhoA-deleted SCs was not affected, in vivo and in vitro, excluding that RhoA could regulate muscle cell proliferation.
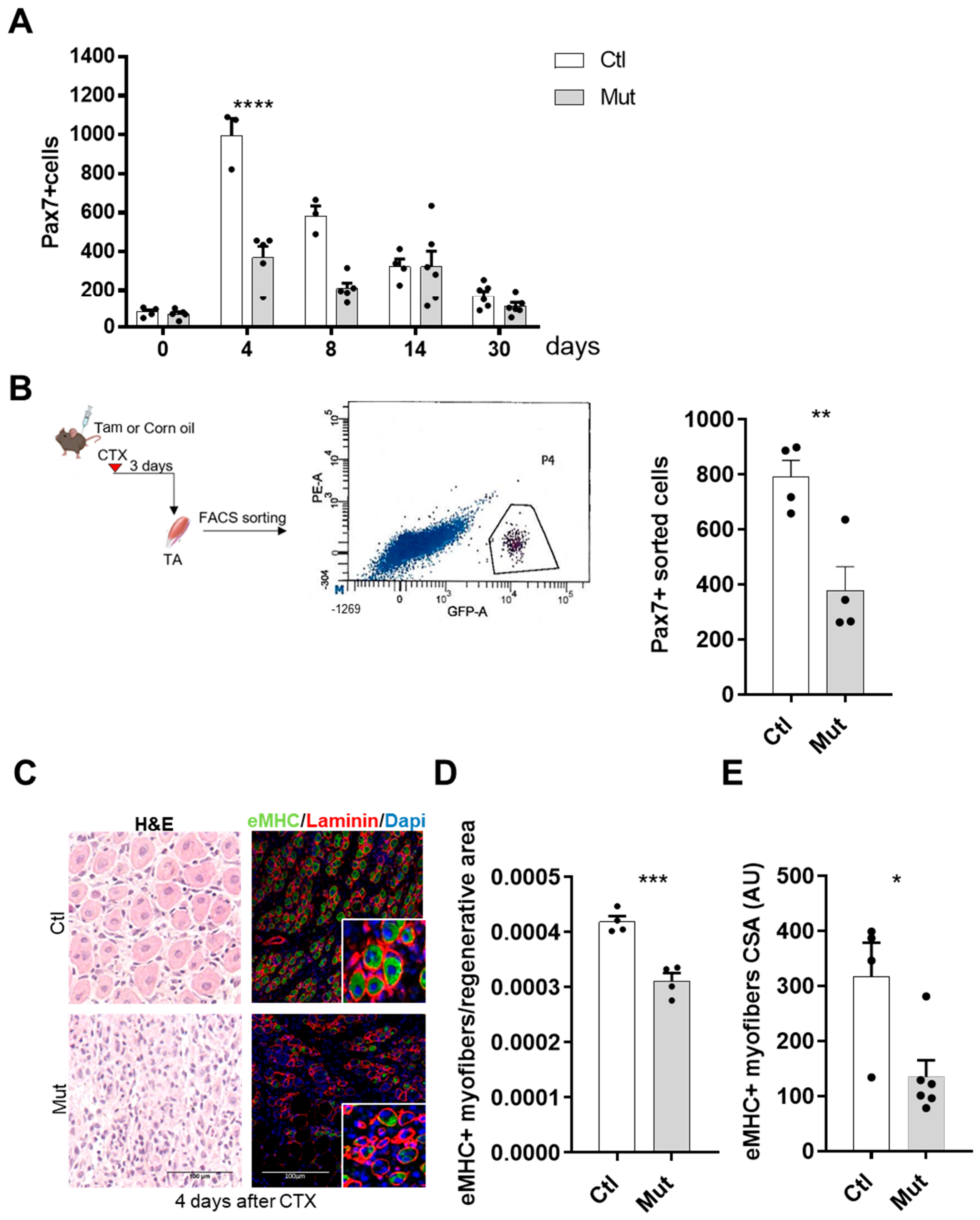
Despite a comparable number of RhoA-deleted SCs vs. control conditions, we found a reduced number of these cells in the early days of the regenerative process, suggesting an impairment in their ability to be activated. Nevertheless, Eliazer et al. showed that RhoA activation reinforces SC quiescence and when it was disrupted in SCs induces an abnormal activation of SCs in uninjured muscle [
27]. Our study did not confirm these data maybe because the mouse models used are different. In both cases, the deletion occurs specifically in the satellite cells but in the present study mice used are RhoA
lx/lx and the mice used by Eliazer et al. are RhoA
lx/+. In this later study, the authors reported a mild reduction of RhoA activity in SCs, but there is no report on residual RhoA expression. On the contrary in our case, we have a complete (almost 100%) reduction of RhoA expression.
At present, we cannot exclude the differences in the methods used to analyze SC proliferation. Moreover, this paper underlines the involvement of RhoA in cytoskeleton-related signaling (like YAP and actin) and whether we support the idea (based on array data) that RhoA could have alternative possible targets regulated in the regeneration context.
However, we cannot exclude a compensatory effect of other members of the Rho family in our model. Moreover, contrary to our results, they observed that after reduction in RhoA, SCs are located outside the niche, a finding that we did not observe in our model, indicating that the impaired muscle regeneration was independent of altered niching.
Anyway, further analyses are essential to elucidate the role of RhoA in SC activation.
We demonstrated that the differentiation of RhoA-deleted SCs was not affected in vivo or in vitro. This is in agreement with published data on the C2C12 cell line showing that both constitutive RhoA activation (RhoAV14) and RhoA activity inhibition (C3 transferase treatment) did not affect differentiation [
26]. It has been shown that RhoA activity must be tightly regulated in a finely coordinated time-dependent manner to ensure appropriate skeletal muscle formation. Indeed, in mouse C2C12 myoblast RhoA activity has been found to decrease in a biphasic manner during myogenic differentiation [
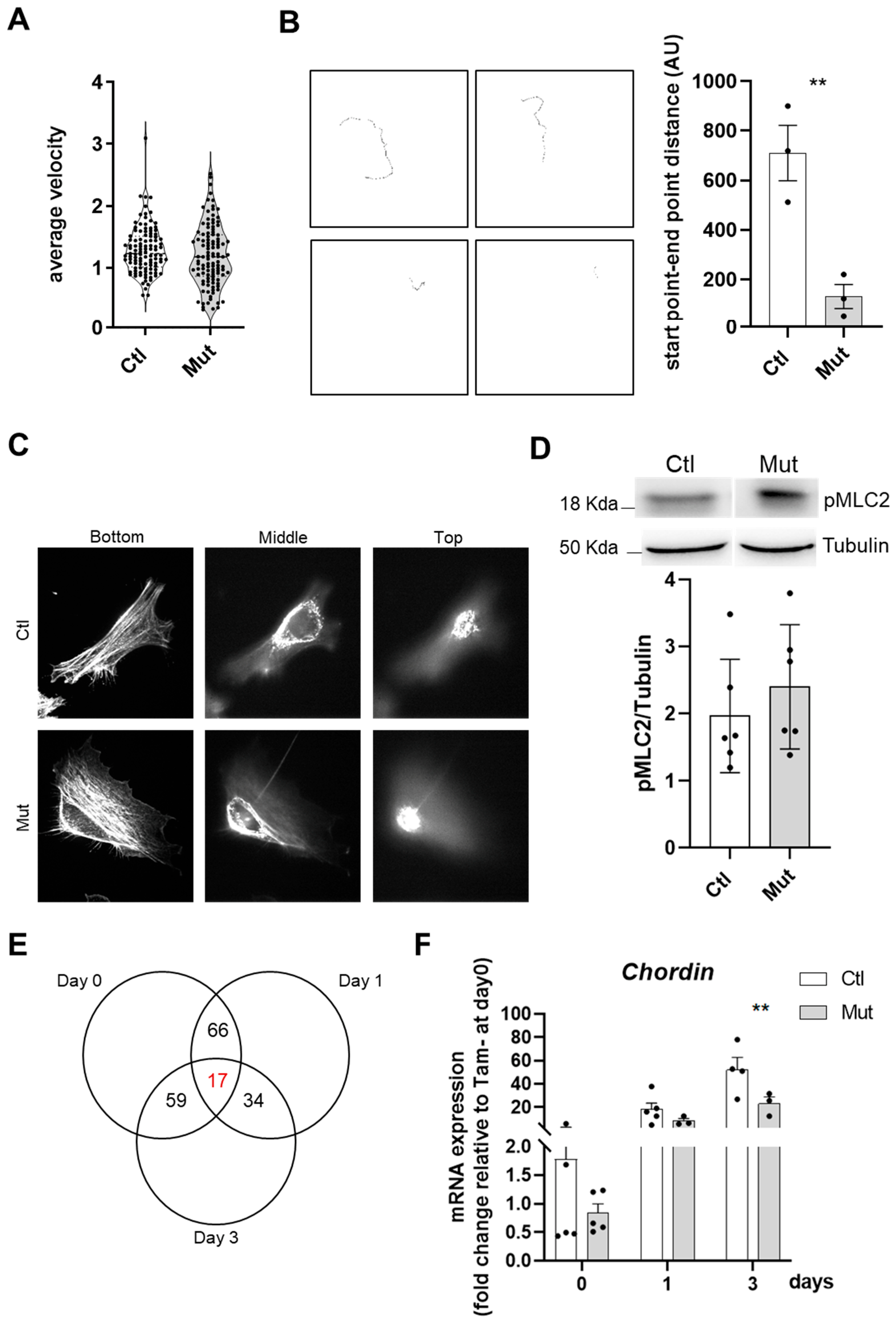
47]. Here, we showed for the first time in primary murine myoblasts that the complete deletion of RhoA does not affect differentiation. Moreover, mutant myotubes have a slightly increased differentiation index underlying the concept that the RhoA function should be finely modulated during all the steps to have a correct myogenesis.
RhoA has been described as an important regulator of actin polymerization and cytoskeleton assembly [
42]. However, in our hands, RhoA-deleted myoblasts did not present evident defects in F-actin cytoskeleton organization. Moreover, the formation of F-actin bundles did not appear affected by the absence of RhoA. However, we clearly showed that RhoA affected cell motility: not cell velocity but cell direction. Recent work demonstrated that syndecan-4 regulates the correct polarization of migrating mammalian myoblasts, in part in coordination with RhoA [
48,
49]. Our work and this last cited paper provide the first foundations to further elucidate the role of RhoA and its partners in SC motility orchestration.
In this study, we demonstrated that the main step impacted by the absence of RhoA is SC fusion. By CTX-induced regeneration we showed that primary (myoblast-myoblast) and secondary (myoblast-myotube) fusions were affected in RhoA-depleted SCs, considering the fact that the number of myonuclei per myofibers is reduced during all the regeneration process, impacting myofibers growth. This concept is also confirmed in vitro where not only the number of myonuclei per myofibers was reduced but also the fusion index, meaning that there is an impairment in the fusion of two mononucleated cells and of myotube and mononucleated cells. Moreover, during muscle overload-induced CH, we showed that the fusion of RhoA-deleted SCs to the growing myofibers is impaired as well, suggesting that even in heterotypic milieu (myofiber expressing RhoA/SC RhoA deleted), RhoA-deleted SCs are not able to fuse. These experiments suggest that the absence of RhoA in SCs renders them incapable of fusing either to myoblasts (regeneration situation) or to myofibers, RhoA-deleted (during muscle regeneration) or RhoA-expressing (during muscle hypertrophy).
Indeed, despite the reduced number of myonuclei per fiber 4 days after muscle damage, the number of satellite cells 14 and 30 days after ctx injury is comparable between control and mutant, suggesting that the decreased amount of myonuclei per myofiber might principally depend on fusion defect occurring in SCs lacking RhoA per se.
Furthermore, we showed that this defect was cell autonomous as murine primary SCs lacking RhoA were unable to fuse in vitro. Reports already published on C2C12 cells have asserted that RhoA must be deactivated to enable myoblast fusion [
50]. Somehow, our results confirm that RhoA expression/activity should be carefully balanced during myogenesis because of the deleterious effects of its chronic deletion on SC fusion.
Surprisingly, the absence of RhoA in proliferative myoblasts and differentiated myotubes did not affect the expression of genes already known to be involved in muscle cell fusion. Indeed, there was no difference in the expression of
Myomaker or
Myomixer in differentiated RhoA-deleted cells. Even the expression of
Srf, a master regulator of myoblast fusion, was not affected in RhoA-deleted cells, explaining in part why there is no actin assembly issue [
18]. Indeed there is no overlap with the genes deregulated in Srf deleted muscle cells [
18] suggesting that the impaired fusion occurring in the RhoA deleted model is independent of the cytoskeleton issue and of the RhoA/Srf axis. We can suppose that RhoA in SCs interacts with new partners not yet identified and that there are other RhoA downstream effectors controlling SCs fusion and muscle growth.
One gene we found downregulated on day 0, day 1, and day 3 of the differentiation process of RhoA deleted cells is the
Chordin. Chordin is an important negative regulator of BMP activity by inhibiting the binding of these ligands to their receptors [
51]. Previously, it has been shown that
Chordin expression increases during the differentiation process in C2C12 cells. This is in line with the fact that Chordin, as an intrinsic inhibitor of BMP signaling, supports myoblast differentiation and fusion [
52]. In our hands,
Chordin expression decreased in the absence of RhoA, suggesting that its role in BMP pathway inhibition was reduced and, in consequence, the fusion program was altered.
In our previous work, we described that among the most downregulated genes by the absence of RhoA in myofibers there were genes associated with extracellular matrix rearrangement [
40]. Noteworthy, among the Upstream Regulators significantly predicted inhibited (Z-score < 2) in RhoA-deleted three days differentiated myotubes, there was Adam12, a disintegrin and metalloprotease protein. We can speculate that Adam12, as an upstream regulator, controls matrix protein regulating adhesion and fusion of primary myoblast.
In conclusion, we described a novel role for RhoA within SCs during skeletal muscle regeneration. We propose that RhoA mainly affects SC fusion in part through a modification of SC movement and in the other part by an alteration of uncommon molecular mechanisms that are only partially described by this paper.
It will be important in future works to define more comprehensively the molecular pathway(s) involved and the specific RhoA cellular contributions. Clearing up signaling involved in the control of muscle regeneration, in particular, in response to muscle injury, may be essential to identify and design treatments for different pathological and traumatic conditions affecting skeletal muscle regeneration.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}