
Cell-Based Models of ‘Cytokine Release Syndrome’ Endorse CD40L and Granulocyte–Macrophage Colony-Stimulating Factor Knockout in Chimeric Antigen Receptor T Cells as Mitigation Strategy
 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cultivation of Cells
2.2. Production and Enrichment of CD19-Targeting CAR T cells
2.3. Genotyping and Assessment of Off-Target Effects
2.4. Flow Cytometry
2.5. Cytolytic Activity of CAR T Cells
2.6. CRS In Vitro Model
2.7. Cytokine Concentrations
2.8. Statistics
3. Results
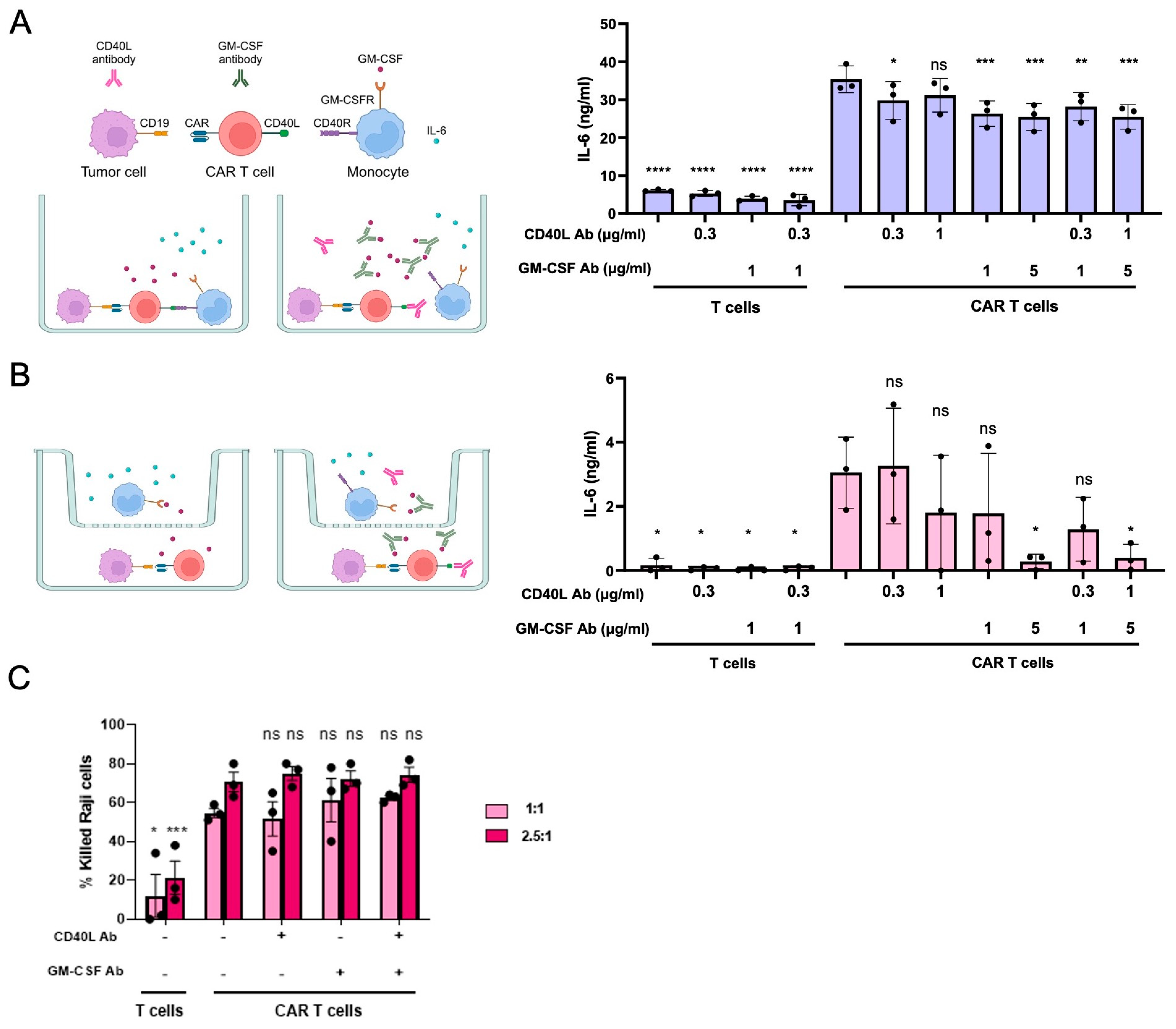
3.1. Modelling CRS In Vitro
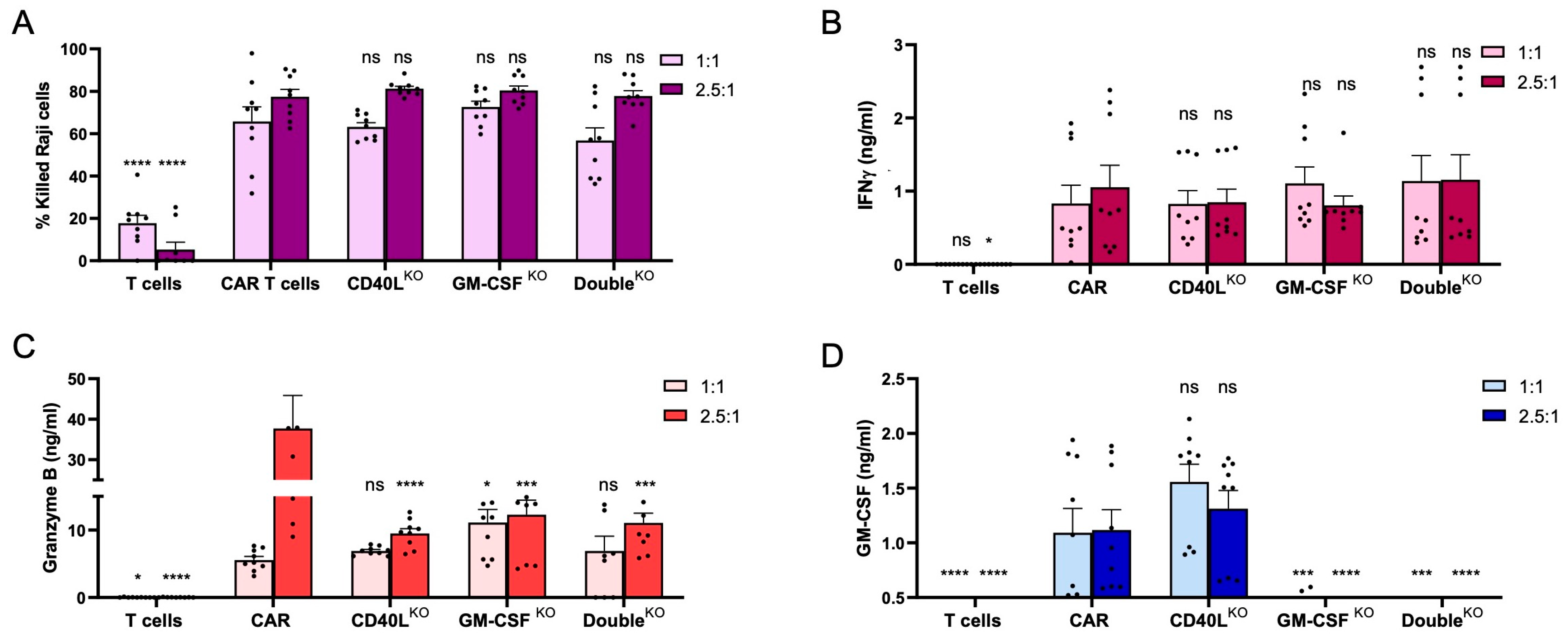
3.2. Disruption of CD40L and/or CSF2 in CAR T Cells
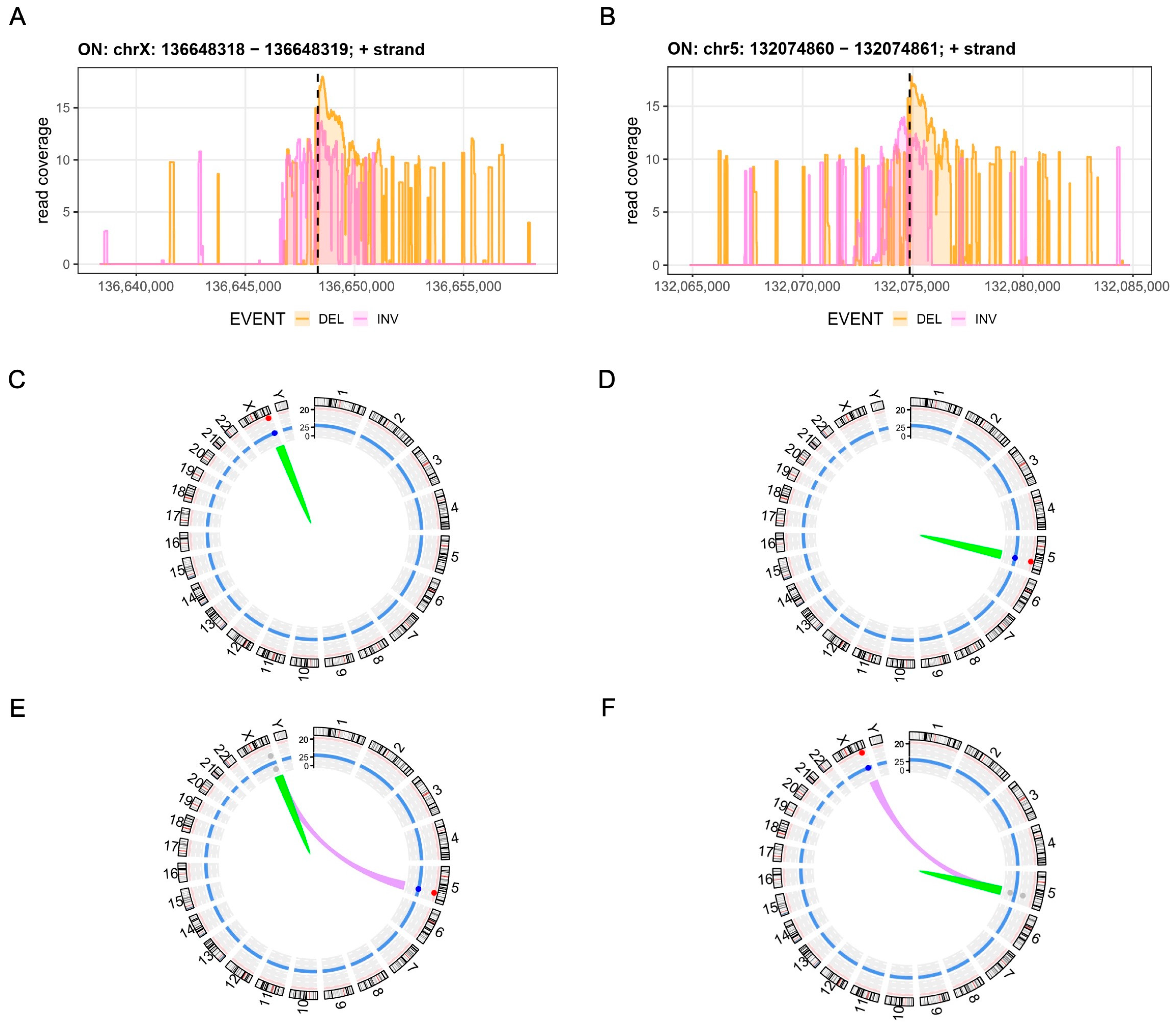
3.3. Assessment of Chromosomal Integrity
3.4. CD40LKO and GM-CSFKO CAR T Cells Eliminate CD19+ Tumor Cells
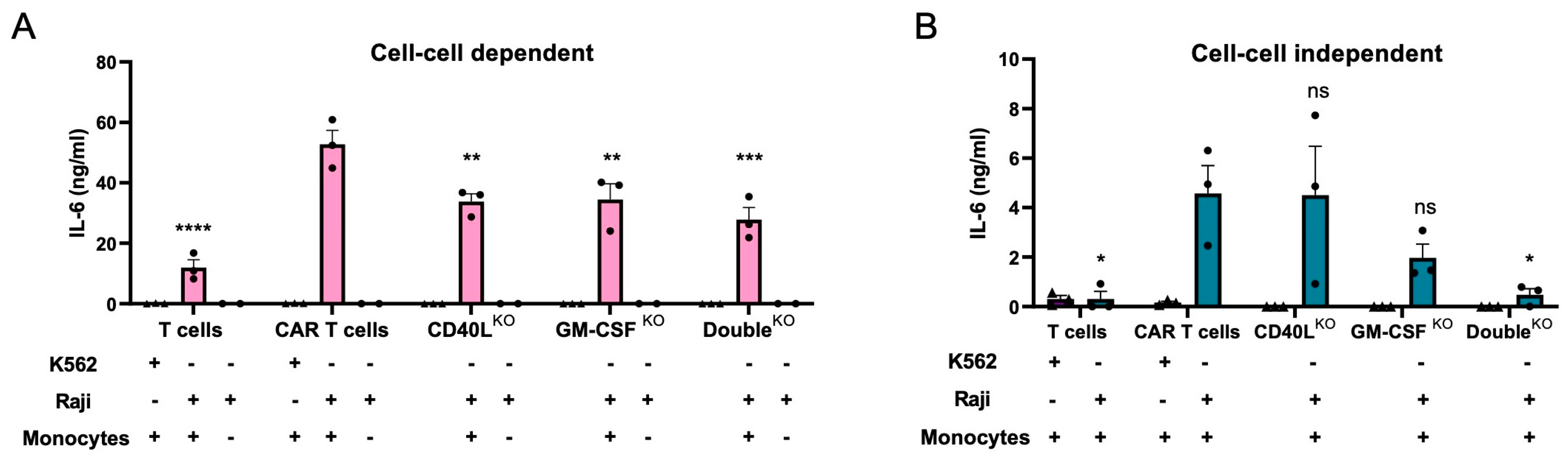
3.5. CD40LKO and/or GM-CSFKO Mitigates IL-6 Release
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abken, H. Building on Synthetic Immunology and T Cell Engineering: A Brief Journey through the History of Chimeric Antigen Receptors. Hum. Gene Ther. 2021, 32, 1011–1028. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Miklos, D.B.; Jacobson, C.A.; Perales, M.A.; Kersten, M.J.; Oluwole, O.O.; Ghobadi, A.; Rapoport, A.P.; McGuirk, J.; Pagel, J.M.; et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 640–654. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Huang, S.; Chen, S.; Wang, Y.; Sun, Q.; Xu, X.; Li, Y. Mechanisms of cytokine release syndrome and neurotoxicity of CAR T-cell therapy and associated prevention and management strategies. J. Exp. Clin. Cancer Res. 2021, 40, 367. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Szymański, P.; Markowicz, M.; Mikiciuk-Olasik, E. Adaptation of high-throughput screening in drug discovery-toxicological screening tests. Int. J. Mol. Sci. 2012, 13, 427–452. [Google Scholar] [CrossRef] [PubMed]
- Alzubi, J.; Lock, D.; Rhiel, M.; Schmitz, S.; Wild, S.; Mussolino, C.; Hildenbeutel, M.; Brandes, C.; Rositzka, J.; Lennartz, S.; et al. Automated generation of gene-edited CAR T cells at clinical scale. Mol. Ther. Methods Clin. Dev. 2021, 20, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Bloh, K.; Kanchana, R.; Bialk, P.; Banas, K.; Zhang, Z.; Yoo, B.C.; Kmiec, E.B. Deconvolution of Complex DNA Repair (DECODR): Establishing a Novel Deconvolution Algorithm for Comprehensive Analysis of CRISPR-Edited Sanger Sequencing Data. CRISPR J. 2021, 4, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Turchiano, G.; Andrieux, G.; Klermund, J.; Blattner, G.; Pennucci, V.; El Gaz, M.; Monaco, G.; Poddar, S.; Mussolino, C.; Cornu, T.I.; et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell 2021, 28, 1136–1147.e1135. [Google Scholar] [CrossRef]
- Rhiel, M.; Geiger, K.; Andrieux, G.; Rositzka, J.; Boerries, M.; Cathomen, T.; Cornu, T.I. T-CAST: An optimized CAST-Seq pipeline for TALEN confirms superior safety and efficacy of obligate-heterodimeric scaffolds. Front. Genome Ed. 2023, 5, 1130736. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Joung, J.K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet. 2016, 17, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Amit, I.; Iancu, O.; Levy-Jurgenson, A.; Kurgan, G.; McNeill, M.S.; Rettig, G.R.; Allen, D.; Breier, D.; Ben Haim, N.; Wang, Y.; et al. CRISPECTOR provides accurate estimation of genome editing translocation and off-target activity from comparative NGS data. Nat. Commun. 2021, 12, 3042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; Ma, Q.; Yang, H.; Signorovitch, J.; Wu, E. A Review of Two Regulatory Approved Anti-CD19 CAR T-Cell Therapies in Diffuse Large B-Cell Lymphoma: Why Are Indirect Treatment Comparisons Not Feasible? Adv. Ther. 2020, 37, 3040–3058. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.D.; Clark, C.R.; Chou, T.H. Granulocyte/macrophage colony-stimulating factor stimulates monocyte and tissue macrophage proliferation and enhances their responsiveness to macrophage colony-stimulating factor. Blood 1988, 71, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 2016, 45, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.; Duchateau, P.; Depil, S.; Poirot, L.; Valton, J. Granulocyte–macrophage colony-stimulating factor inactivation in CAR T-cells prevents monocyte-dependent release of key cytokine release syndrome mediators. J. Biol. Chem. 2019, 294, 5430–5437. [Google Scholar] [CrossRef]
- Kuhn, N.F.; Purdon, T.J.; van Leeuwen, D.G.; Lopez, A.V.; Curran, K.J.; Daniyan, A.F.; Brentjens, R.J. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell 2019, 35, 473–488.e476. [Google Scholar] [CrossRef]
- Dettmer-Monaco, V.; Weißert, K.; Ammann, S.; Monaco, G.; Lei, L.; Gräßel, L.; Rhiel, M.; Rositzka, J.; Kaufmann, M.M.; Geiger, K.; et al. Gene editing of hematopoietic stem cells restores T cell response in familial hemophagocytic lymphohistiocytosis. J. Allergy Clin. Immunol. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef]
- AlJanahi, A.A.; Lazzarotto, C.R.; Chen, S.; Shin, T.H.; Cordes, S.; Fan, X.; Jabara, I.; Zhou, Y.; Young, D.J.; Lee, B.C.; et al. Prediction and validation of hematopoietic stem and progenitor cell off-target editing in transplanted rhesus macaques. Mol. Ther. 2022, 30, 209–222. [Google Scholar] [CrossRef]
- García, M.; Bonafont, J.; Martínez-Palacios, J.; Xu, R.; Turchiano, G.; Svensson, S.; Thrasher, A.J.; Larcher, F.; Del Rio, M.; Hernández-Alcoceba, R.; et al. Preclinical model for phenotypic correction of dystrophic epidermolysis bullosa by in vivo CRISPR-Cas9 delivery using adenoviral vectors. Mol. Ther. Methods Clin. Dev. 2022, 27, 96–108. [Google Scholar] [CrossRef]
- Venkatesan, V.; Christopher, A.C.; Rhiel, M.; Azhagiri, M.K.K.; Babu, P.; Walavalkar, K.; Saravanan, B.; Andrieux, G.; Rangaraj, S.; Srinivasan, S.; et al. Editing the core region in HPFH deletions alters fetal and adult globin expression for treatment of β-hemoglobinopathies. Mol. Ther. Nucleic Acids 2023, 32, 671–688. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Target Site | Target Sequence (5′-3′) |
|---|---|---|
| CD40L-gRNA2 | CD40L exon 1 | AAAGTGCTGACCCAATCATC |
| CD40L-gRNA3 | CD40L exon 1 | AGTCCAGTGGCCGCAGATCG |
| CSF2-gRNA2 | CSF2 exon 3 | ACAGCTCCAGGCGGGTCTGT |
| CSF2-gRNA3 | CSF2 exon 3 | CAAGGGCCCCTTGACCATGA |
| Name | Sequence (5′-3′) |
|---|---|
| CD40L forward | GGAGAGAAGACTACGAAGCAC |
| CD40L reverse | GAGACTTCATTGACTAGGCAAC |
| CSF2 forward | TGACTACAGAGAGGCACAGA |
| CSF2 reverse | TCACCTCTGACCTCATTAACC |
| CD40L-decoy reverse | GAAGATACACAGCAAAAAGTGC |
| CD40L-decoy forward | ATAGAAGGTTGGACAAGGTAAGA |
| CD40L-bait | GTCTTCTCATGCTGCCTC |
| CD40L-bait nested | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTGCCACCTTCTCTGCCAGAAGATACC |
| CSF2-decoy reverse | GCAGTGCTGCTTGTAGTG |
| CSF2-decoy forward | CTCCAACCCCGGTGAGT |
| CSF2-bait | TGGTGGAGAGTTCTTGTAC |
| CSF2-bait nested | GACTGGAGTTCAGACGTGTGCTCTTCCGATCTTGTGGGCACTTGGCCACTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dibas, A.; Rhiel, M.; Patel, V.B.; Andrieux, G.; Boerries, M.; Cornu, T.I.; Alzubi, J.; Cathomen, T. Cell-Based Models of ‘Cytokine Release Syndrome’ Endorse CD40L and Granulocyte–Macrophage Colony-Stimulating Factor Knockout in Chimeric Antigen Receptor T Cells as Mitigation Strategy. Cells 2023, 12, 2581. https://doi.org/10.3390/cells12212581
Dibas A, Rhiel M, Patel VB, Andrieux G, Boerries M, Cornu TI, Alzubi J, Cathomen T. Cell-Based Models of ‘Cytokine Release Syndrome’ Endorse CD40L and Granulocyte–Macrophage Colony-Stimulating Factor Knockout in Chimeric Antigen Receptor T Cells as Mitigation Strategy. Cells. 2023; 12(21):2581. https://doi.org/10.3390/cells12212581
Chicago/Turabian StyleDibas, Ala, Manuel Rhiel, Vidisha Bhavesh Patel, Geoffroy Andrieux, Melanie Boerries, Tatjana I. Cornu, Jamal Alzubi, and Toni Cathomen. 2023. "Cell-Based Models of ‘Cytokine Release Syndrome’ Endorse CD40L and Granulocyte–Macrophage Colony-Stimulating Factor Knockout in Chimeric Antigen Receptor T Cells as Mitigation Strategy" Cells 12, no. 21: 2581. https://doi.org/10.3390/cells12212581
APA StyleDibas, A., Rhiel, M., Patel, V. B., Andrieux, G., Boerries, M., Cornu, T. I., Alzubi, J., & Cathomen, T. (2023). Cell-Based Models of ‘Cytokine Release Syndrome’ Endorse CD40L and Granulocyte–Macrophage Colony-Stimulating Factor Knockout in Chimeric Antigen Receptor T Cells as Mitigation Strategy. Cells, 12(21), 2581. https://doi.org/10.3390/cells12212581