
Regulatory ILC2—Role of IL-10 Producing ILC2 in Asthma


Abstract
:1. Introduction
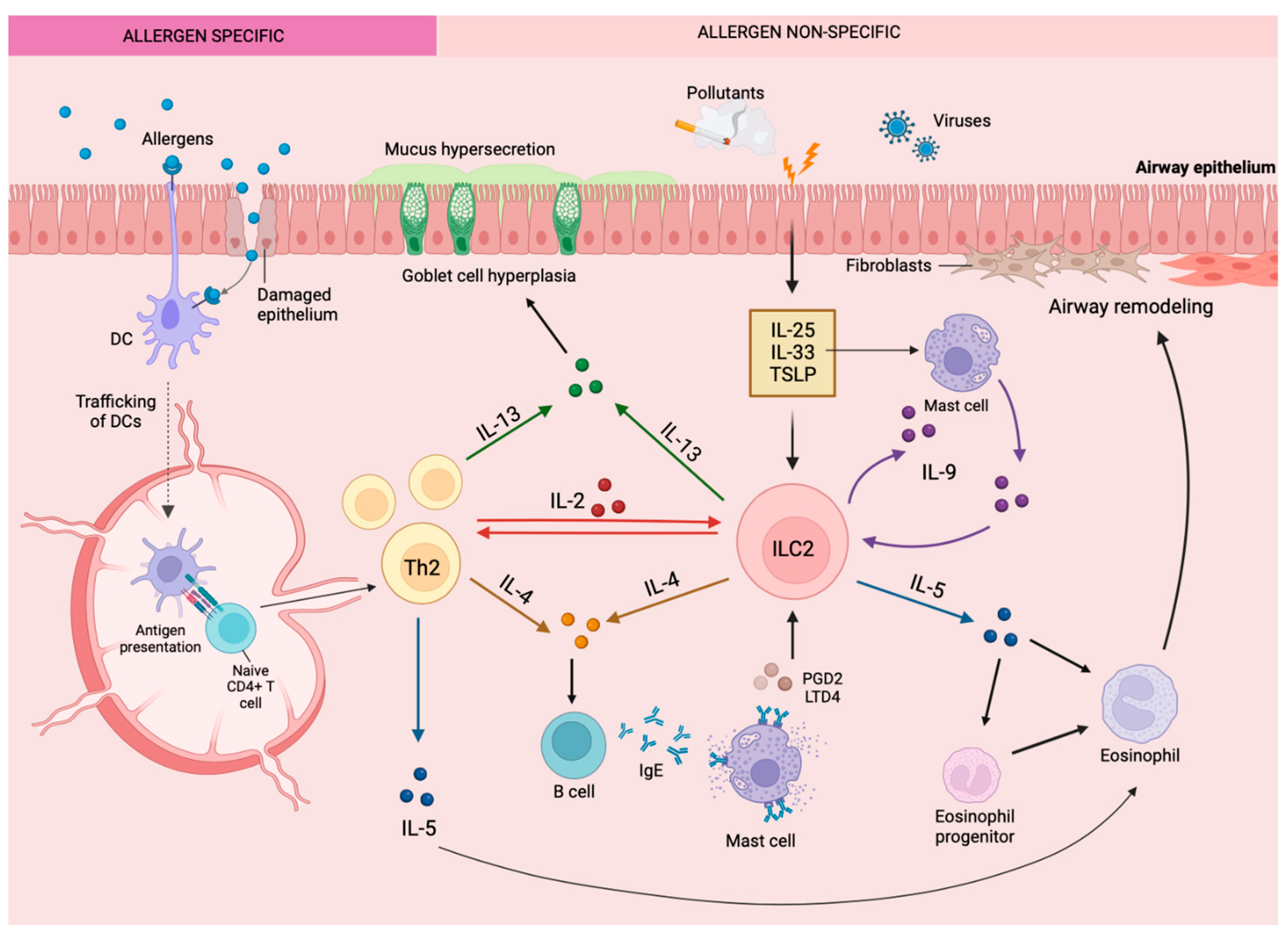
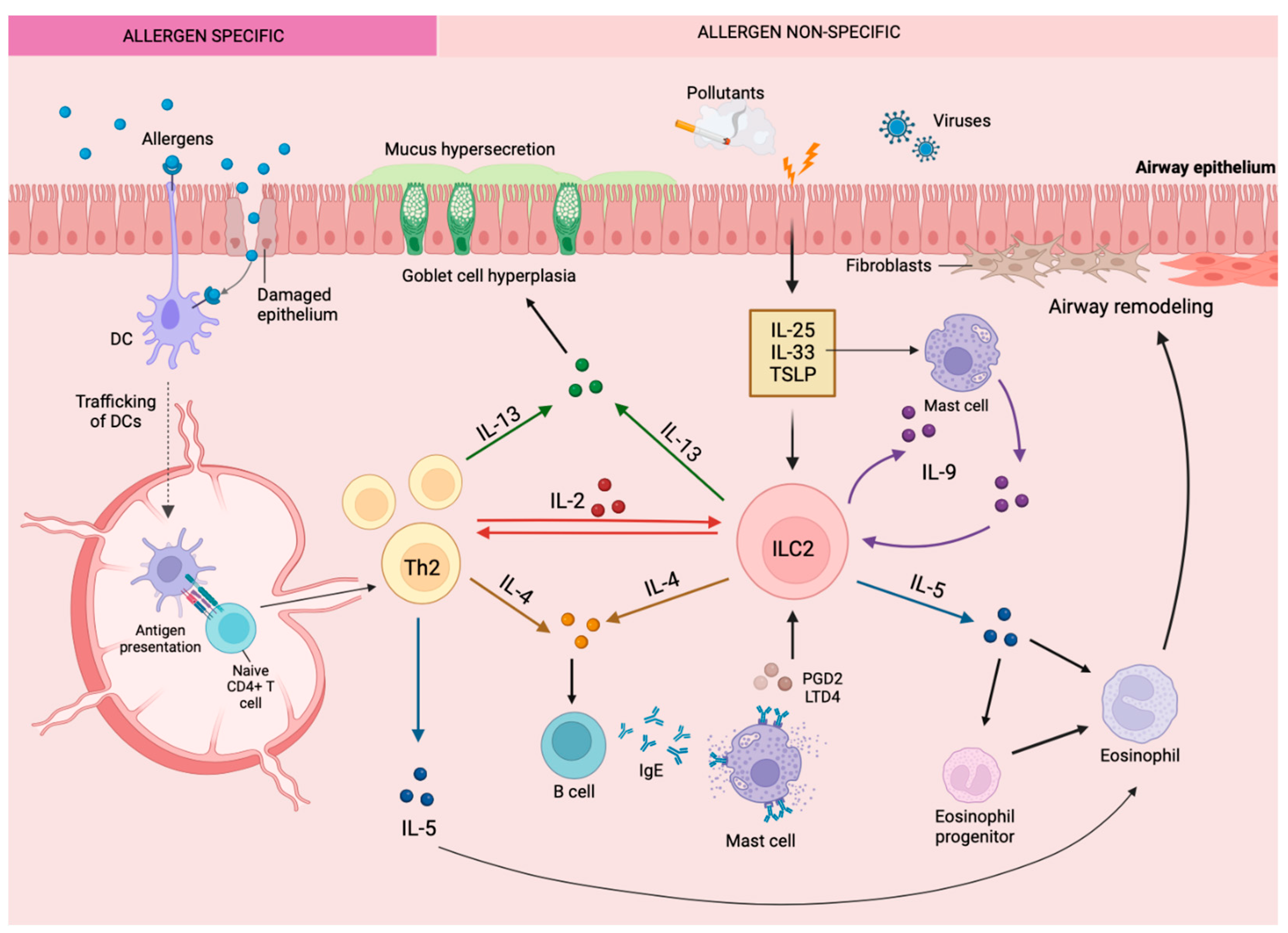
2. Asthma and Type 2 Inflammation
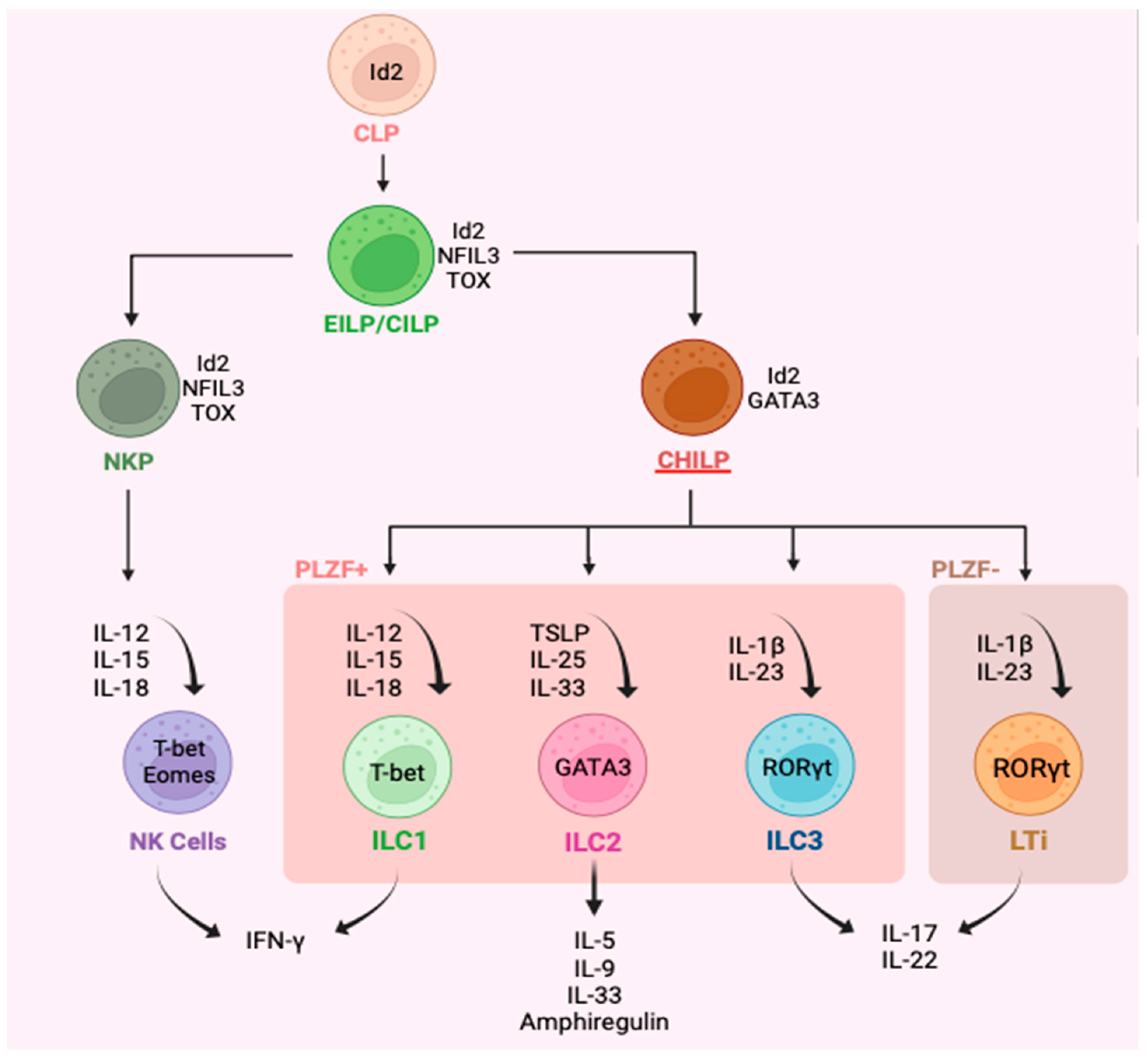
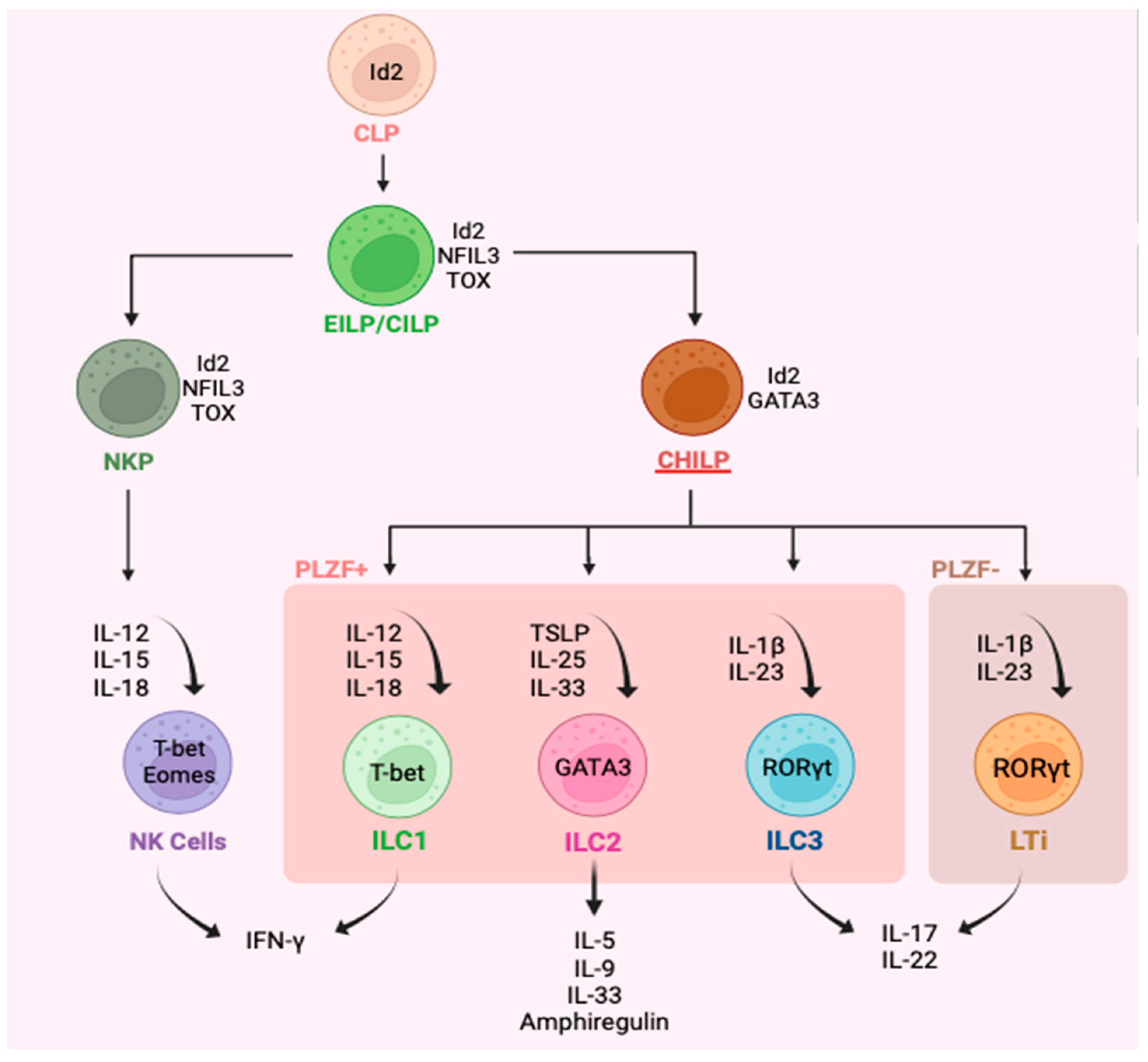
3. Plasticity of ILC
4. Interleukin-10
5. The Role of IL-10 Expressing ILC2
6. Positive Modulators of IL-10 Expression by ILC2
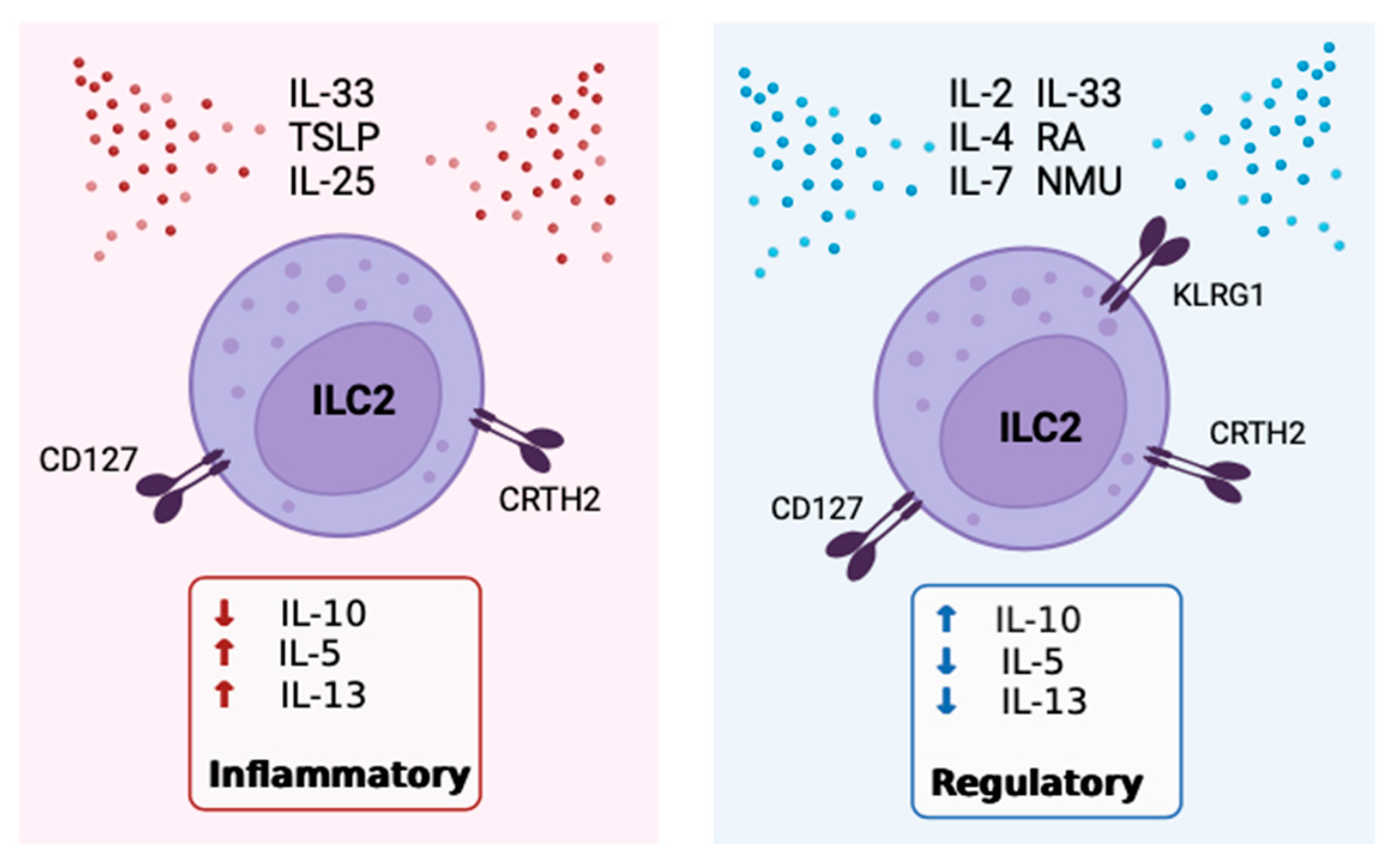
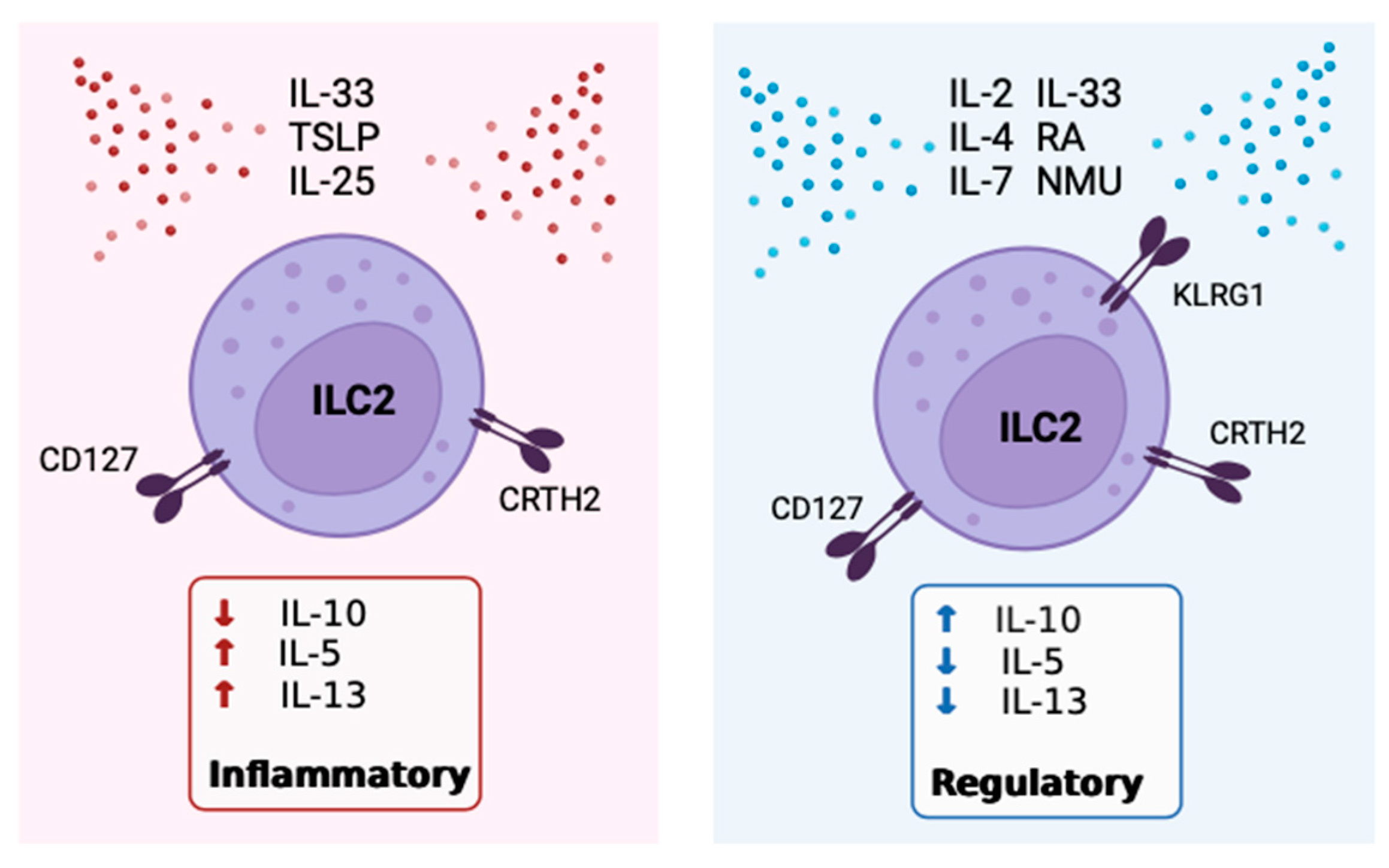
7. Inhibitors of IL-10 Expression by ILC2
8. Receptors, Sex Disparities and Neuroimmune Control of IL-10+ILC2

{kind=link}
{kind=link}
{kind=link}
| Study | Models and Methods | Inducers of IL-10 | Inhibitors of IL-10 |
|---|---|---|---|
| Bando J. et al. [21] | ILC2 from small intestine lamina propria of WT mice subjected to intestinal inflammation. | IL-2, IL-4, IL-10, IL-27, and NMU | TL1A |
| Seehus C. et al. [79] | WT murine blood and lung ILC2, pre-activated +/− intraperitoneal IL-33. | IL-2, IL-33, RA | TGF-β |
| Howard E. et al. [80] | Blood ILC2 from WT and RAG deficient (Rag2−/−γc−/−) in a mouse model of asthma. | IL-2, IL-4, cMaf, Blimp-1 | - |
| Golebski K. et al. [81] | Murine lung-tissue-extracted ILC2 using a model for grass pollen allergic rhinitis. Blood ILC2 from healthy controls and grass pollen allergic rhinitis. | IL-2, IL-33, RA, KLRG1 | - |
| Wang S. et al. [82] | Intestinal ILC2 from healthy and Rag1−/−Il10−/− mice and human intestinal tissue. | TGF-β | - |
| Boonpiyathad T. et al. [95] | PBMC from subjects with allergic rhinitis undergoing AIT. | RA, CD25, CTLA-4 | - |
| Baban B. et al. [101] | Examined CSF of traumatic brain injury patients and model mice and analyzed the effect of metformin. | KLRG1, IL-2, IL-33, AMPK | - |
| Laurent P. et al. [109] | Skin biopsies and peripheral blood of systemic sclerosis patients. | KLRG1 | TGF-β |
| Hurrell BP. et al. [118] | Asthma model mice and ICAM-1 deficient mice challenged intranasally with IL-33. | - | ICAM-1 |
| Naito M. et al. [120] | Isolated lung ILC2 from WT, allergic lung inflammation model mice, and Sema6D deficient mice. | - | Sema6D |
| Morita H. et al. [121] | Isolated ILC2 from the blood and nasal tissue of healthy and chronic rhinosinusitis subjects and HDM-sensitized mice. | IL-2, IL-33, RA, CTLA-4 | TGF-β |
9. Mechanisms Associated with IL-10 Expressing ILC2
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Komlósi, Z.I.; van de Veen, W.; Kovács, N.; Szűcs, G.; Sokolowska, M.; O’Mahony, L.; Akdis, M.; Akdis, C.A. Cellular and molecular mechanisms of allergic asthma. Mol. Asp. Med. 2022, 85, 100995. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wu, Y.; Zhang, Y.; Ni, B. IL-10-Producing ILCs: Molecular Mechanisms and Disease Relevance. Front. Immunol. 2021, 12, 650200. [Google Scholar] [CrossRef] [PubMed]
- Boos, M.D.; Yokota, Y.; Eberl, G.; Kee, B.L. Mature natural killer cell and lymphoid tissue–inducing cell development requires Id2-mediated suppression of E protein activity. J. Exp. Med. 2007, 204, 1119–1130. [Google Scholar] [CrossRef]
- Bartemes, K.R.; Kita, H. Roles of innate lymphoid cells (ILCs) in allergic diseases: The 10-year anniversary for ILC2s. J. Allergy Clin. Immunol. 2021, 147, 1531–1547. [Google Scholar] [CrossRef]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, G.; Fan, X.; Dikiy, S.; Lee, S.Y.; Rudensky, A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 2015, 350, 981–985. [Google Scholar] [CrossRef]
- Bonne-Année, S.; Bush, M.C.; Nutman, T.B. Differential Modulation of Human Innate Lymphoid Cell (ILC) Subsets by IL-10 and TGF-β. Sci. Rep. 2019, 9, 14305. [Google Scholar] [CrossRef]
- Smith, S.G.; Chen, R.; Kjarsgaard, M.; Huang, C.; Oliveria, J.-P.; O’Byrne, P.M.; Gauvreau, G.M.; Boulet, L.-P.; Lemiere, C.; Martin, J.; et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J. Allergy Clin. Immunol. 2016, 137, 75–86.e8. [Google Scholar] [CrossRef]
- Yang, Q.; Saenz, S.A.; Zlotoff, D.A.; Artis, D.; Bhandoola, A. Cutting Edge: Natural Helper Cells Derive from Lymphoid Progenitors. J. Immunol. 2011, 187, 5505–5509. [Google Scholar] [CrossRef]
- Zook, E.C.; Kee, B.L. Development of innate lymphoid cells. Nat. Immunol. 2016, 17, 775–782. [Google Scholar] [CrossRef]
- Seillet, C.; Rankin, L.C.; Groom, J.R.; Mielke, L.A.; Tellier, J.; Chopin, M.; Huntington, N.D.; Belz, G.T.; Carotta, S. Nfil3 is required for the development of all innate lymphoid cell subsets. J. Exp. Med. 2014, 211, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, F.; Harly, C.; Xing, S.; Ye, L.; Xia, X.; Wang, H.; Wang, X.; Yu, S.; Zhou, X.; et al. TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat. Immunol. 2015, 16, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Seehus, C.R.; Aliahmad, P.; De La Torre, B.; Iliev, I.D.; Spurka, L.; Funari, V.A.; Kaye, J. The development of innate lymphoid cells requires TOX-dependent generation of a common innate lymphoid cell progenitor. Nat. Immunol. 2015, 16, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Flach, M.; Möhle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of Type 1 ILCs from a Common Progenitor to All Helper-like Innate Lymphoid Cell Lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef]
- Ramirez, K.; Chandler, K.J.; Spaulding, C.; Zandi, S.; Sigvardsson, M.; Graves, B.J.; Kee, B.L. Gene Deregulation and Chronic Activation in Natural Killer Cells Deficient in the Transcription Factor ETS1. Immunity 2012, 36, 921–932. [Google Scholar] [CrossRef]
- Male, V.; Nisoli, I.; Kostrzewski, T.; Allan, D.S.J.; Carlyle, J.R.; Lord, G.M.; Wack, A.; Brady, H.J.M. The transcription factor E4bp4/Nfil3 controls commitment to the NK lineage and directly regulates Eomes and Id2 expression. J. Exp. Med. 2014, 211, 635–642. [Google Scholar] [CrossRef]
- Panda, S.K.; Colonna, M. Innate Lymphoid Cells in Mucosal Immunity. Front. Immunol. 2019, 10, 861. [Google Scholar] [CrossRef]
- Cope, A.; Le Friec, G.; Cardone, J.; Kemper, C. The Th1 life cycle: Molecular control of IFN-γ to IL-10 switching. Trends Immunol. 2011, 32, 278–286. [Google Scholar] [CrossRef]
- Hsieh, C.-S.; Macatonia, S.E.; Tripp, C.S.; Wolf, S.F.; O’Garra, A.; Murphy, K.M. Development of TH1 CD4+T Cells Through IL-12 Produced by Listeria-Induced Macrophages. Science 1993, 260, 547–549. [Google Scholar] [CrossRef]
- Chen, R.; Smith, S.G.; Salter, B.; El-Gammal, A.; Oliveria, J.P.; Obminski, C.; Watson, R.; O’byrne, P.M.; Gauvreau, G.M.; Sehmi, R. Allergen-induced Increases in Sputum Levels of Group 2 Innate Lymphoid Cells in Subjects with Asthma. Am. J. Respir. Crit. Care Med. 2017, 196, 700–712. [Google Scholar] [CrossRef]
- Bando, J.K.; Gilfillan, S.; Di Luccia, B.; Fachi, J.L.; Sécca, C.; Cella, M.; Colonna, M. ILC2s are the predominant source of intestinal ILC-derived IL-10. J. Exp. Med. 2019, 217, e20191520. [Google Scholar] [CrossRef] [PubMed]
- Klein Wolterink, R.G.J.; Serafini, N.; Van Nimwegen, M.; Vosshenrich, C.A.J.; De Bruijn, M.J.W.; Fonseca Pereira, D.; Veiga Fernandes, H.; Hendriks, R.W.; Di Santo, J.P. Essential, dose-dependent role for the transcription factor Gata3 in the development of IL-5+ and IL-13+ type 2 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10240–10245. [Google Scholar] [CrossRef] [PubMed]
- Hoyler, T.; Klose, C.S.N.; Souabni, A.; Turqueti-Neves, A.; Pfeifer, D.; Rawlins, E.L.; Voehringer, D.; Busslinger, M.; Diefenbach, A. The Transcription Factor GATA-3 Controls Cell Fate and Maintenance of Type 2 Innate Lymphoid Cells. Immunity 2012, 37, 634–648. [Google Scholar] [CrossRef] [PubMed]
- Mjösberg, J.; Bernink, J.; Golebski, K.; Karrich, J.J.; Peters, C.P.; Blom, B.; te Velde, A.A.; Fokkens, W.J.; van Drunen, C.M.; Spits, H. The Transcription Factor GATA3 Is Essential for the Function of Human Type 2 Innate Lymphoid Cells. Immunity 2012, 37, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Kiss, E.A.; Schwierzeck, V.; Ebert, K.; Hoyler, T.; D’hargues, Y.; Göppert, N.; Croxford, A.L.; Waisman, A.; Tanriver, Y.; et al. A T-bet gradient controls the fate and function of CCR6−RORγt+ innate lymphoid cells. Nature 2013, 494, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Salter, B.M.; Aw, M.; Sehmi, R. The role of type 2 innate lymphoid cells in eosinophilic asthma. J. Leukoc. Biol. 2019, 106, 889–901. [Google Scholar] [CrossRef]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef]
- Mjösberg, J.M.; Trifari, S.; Crellin, N.K.; Peters, C.P.; van Drunen, C.M.; Piet, B.; Fokkens, W.J.; Cupedo, T.; Spits, H. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 2011, 12, 1055–1062. [Google Scholar] [CrossRef]
- Bennstein, S.B.; Uhrberg, M. Biology and therapeutic potential of human innate lymphoid cells. FEBS J. 2022, 289, 3967–3981. [Google Scholar] [CrossRef]
- Fallon, P.G.; Ballantyne, S.J.; Mangan, N.E.; Barlow, J.L.; Dasvarma, A.; Hewett, D.R.; McIlgorm, A.; Jolin, H.E.; McKenzie, A.N.J. Identification of an interleukin (IL)-25–dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J. Exp. Med. 2006, 203, 1105–1116. [Google Scholar] [CrossRef]
- Fonseca, W.; Lukacs, N.W.; Elesela, S.; Malinczak, C.-A. Role of ILC2 in Viral-Induced Lung Pathogenesis. Front. Immunol. 2021, 12, 675169. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.; Steer, C.A.; Mathä, L.; Gold, M.J.; Martinez-Gonzalez, I.; McNagny, K.M.; McKenzie, A.N.J.; Takei, F. Group 2 Innate Lymphoid Cells Are Critical for the Initiation of Adaptive T Helper 2 Cell-Mediated Allergic Lung Inflammation. Immunity 2014, 40, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Bartemes, K.R.; Iijima, K.; Kobayashi, T.; Kephart, G.M.; McKenzie, A.N.; Kita, H. IL-33–Responsive Lineage−CD25+CD44hi Lymphoid Cells Mediate Innate Type 2 Immunity and Allergic Inflammation in the Lungs. J. Immunol. 2012, 188, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Bailey, M.; Zaunders, J.; Mrad, N.; Sacks, R.; Sewell, W.; Harvey, R.J. Group 2 innate lymphoid cells (ILC2s) are increased in chronic rhinosinusitis with nasal polyps or eosinophilia. Clin. Exp. Allergy 2015, 45, 394–403. [Google Scholar] [CrossRef]
- Imai, Y.; Yasuda, K.; Sakaguchi, Y.; Haneda, T.; Mizutani, H.; Yoshimoto, T.; Nakanishi, K.; Yamanishi, K. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 13921–13926. [Google Scholar] [CrossRef]
- Kim, B.S.; Siracusa, M.C.; Saenz, S.A.; Noti, M.; Monticelli, L.A.; Sonnenberg, G.F.; Hepworth, M.R.; Van Voorhees, A.S.; Comeau, M.R.; Artis, D. TSLP Elicits IL-33–Independent Innate Lymphoid Cell Responses to Promote Skin Inflammation. Sci. Transl. Med. 2013, 5, 170ra16. [Google Scholar] [CrossRef]
- Shaw, J.L.; Fakhri, S.; Citardi, M.J.; Porter, P.C.; Corry, D.B.; Kheradmand, F.; Liu, Y.-J.; Luong, A. IL-33–Responsive Innate Lymphoid Cells Are an Important Source of IL-13 in Chronic Rhinosinusitis with Nasal Polyps. Am. J. Respir. Crit. Care Med. 2013, 188, 432–439. [Google Scholar] [CrossRef]
- Lee, J.-B.; Chen, C.-Y.; Liu, B.; Mugge, L.; Angkasekwinai, P.; Facchinetti, V.; Dong, C.; Liu, Y.-J.; Rothenberg, M.E.; Hogan, S.P.; et al. IL-25 and CD4+ TH2 cells enhance type 2 innate lymphoid cell–derived IL-13 production, which promotes IgE-mediated experimental food allergy. J. Allergy Clin. Immunol. 2016, 137, 1216–1225.e5. [Google Scholar] [CrossRef]
- Krempski, J.W.; Kobayashi, T.; Iijima, K.; McKenzie, A.N.; Kita, H. Group 2 Innate Lymphoid Cells Promote Development of T Follicular Helper Cells and Initiate Allergic Sensitization to Peanuts. J. Immunol. 2020, 204, 3086–3096. [Google Scholar] [CrossRef]
- Noval Rivas, M.; Burton, O.T.; Oettgen, H.C.; Chatila, T. IL-4 production by group 2 innate lymphoid cells promotes food allergy by blocking regulatory T-cell function. J. Allergy Clin. Immunol. 2016, 138, 801–811.e9. [Google Scholar] [CrossRef]
- Ju, X.; Miromoto, T.; Beaudin, S.; Salter, B.; Wattie, J.; Howie, K.J.; O’Byrne, P.M.; Gauvreau, G.M.; Sehmi, R. Enumeration of IL-17A/F Producing Innate Lymphoid Cells in Subjects with COPD. J. Allergy Clin. Immunol. 2019, 143, AB218. [Google Scholar] [CrossRef]
- Doherty, T.A.; Broide, D.H. Airway innate lymphoid cells in the induction and regulation of allergy. Allergol. Int. 2019, 68, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Lambrecht, B.N. The basic immunology of asthma. Cell 2021, 184, 1469–1485. [Google Scholar] [CrossRef] [PubMed]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.-I.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+Sca-1+ lymphoid cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.A.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef]
- Price, A.E.; Liang, H.-E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically dispersed innate IL-13–expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494. [Google Scholar] [CrossRef]
- Christianson, C.A.; Goplen, N.P.; Zafar, I.; Irvin, C.; Good, J.T., Jr.; Rollins, D.R.; Gorentla, B.; Liu, W.; Gorska, M.M.; Chu, H.; et al. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J. Allergy Clin. Immunol. 2015, 136, 59–68.e14. [Google Scholar] [CrossRef]
- Fajt, M.L.; Gelhaus, S.L.; Freeman, B.; Uvalle, C.E.; Trudeau, J.B.; Holguin, F.; Wenzel, S.E. Prostaglandin D2 pathway upregulation: Relation to asthma severity, control, and TH2 inflammation. J. Allergy Clin. Immunol. 2013, 131, 1504–1512.e12. [Google Scholar] [CrossRef]
- Sehmi, R.; Smith, S.G.; Kjarsgaard, M.; Radford, K.; Boulet, L.-P.; Lemiere, C.; Prazma, C.M.; Ortega, H.; Martin, J.G.; Nair, P. Role of local eosinophilopoietic processes in the development of airway eosinophilia in prednisone-dependent severe asthma. Clin. Exp. Allergy 2016, 46, 793–802. [Google Scholar] [CrossRef]
- Busse, W.W.; Sedgwick, J.B. Eosinophils in Asthma. Ann. Allergy 1992, 68, 286–290. [Google Scholar]
- Nair, P.; Martin, J.G.; Cockcroft, D.C.; Dolovich, M.; Lemiere, C.; Boulet, L.-P.; O’Byrne, P.M. Airway Hyperresponsiveness in Asthma: Measurement and Clinical Relevance. J. Allergy Clin. Immunol. Pract. 2017, 5, 649–659.e2. [Google Scholar] [CrossRef] [PubMed]
- Nair, P. What is an “eosinophilic phenotype” of asthma? J. Allergy Clin. Immunol. 2013, 132, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Hargreave, F.E.; Nair, P. The definition and diagnosis of Asthma. Clin. Exp. Allergy 2009, 39, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, P.M.; Inman, M.D.; Parameswaran, K. The trials and tribulations of IL-5, eosinophils, and allergic asthma. J. Allergy Clin. Immunol. 2001, 108, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Fang, X.; Zhu, X.; Bai, C.; Zhu, L.; Jin, M.; Wang, X.; Hu, M.; Tang, R.; Chen, Z. IL-13+ Type 2 Innate Lymphoid Cells Correlate with Asthma Control Status and Treatment Response. Am. J. Respir. Cell Mol. Biol. 2016, 55, 675–683. [Google Scholar] [CrossRef]
- Winkler, C.; Hochdörfer, T.; Israelsson, E.; Hasselberg, A.; Cavallin, A.; Thörn, K.; Muthas, D.; Shojaee, S.; Lüer, K.; Müller, M.; et al. Activation of group 2 innate lymphoid cells after allergen challenge in asthmatic patients. J. Allergy Clin. Immunol. 2019, 144, 61–69.e7. [Google Scholar] [CrossRef]
- Klein Wolterink, R.G.J.; KleinJan, A.; van Nimwegen, M.; Bergen, I.; de Bruijn, M.; Levani, Y.; Hendriks, R.W. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur. J. Immunol. 2012, 42, 1106–1116. [Google Scholar] [CrossRef]
- Verma, M.; Liu, S.; Michalec, L.; Sripada, A.; Gorska, M.M.; Alam, R. Experimental asthma persists in IL-33 receptor knockout mice because of the emergence of thymic stromal lymphopoietin–driven IL-9+ and IL-13+ type 2 innate lymphoid cell subpopulations. J. Allergy Clin. Immunol. 2018, 142, 793–803.e8. [Google Scholar] [CrossRef]
- Bartemes, K.R.; Kephart, G.M.; Fox, S.J.; Kita, H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J. Allergy Clin. Immunol. 2014, 134, 671–678.e4. [Google Scholar] [CrossRef]
- Yu, Q.-N.; Tan, W.-P.; Fan, X.-L.; Guo, Y.-B.; Qin, Z.-L.; Li, C.-L.; Chen, D.; Lin, Z.B.; Wen, W.; Fu, Q.-L. Increased Group 2 Innate Lymphoid Cells Are Correlated with Eosinophilic Granulocytes in Patients with Allergic Airway Inflammation. Int. Arch. Allergy Immunol. 2018, 176, 124–132. [Google Scholar] [CrossRef]
- Liu, T.; Wu, J.; Zhao, J.; Wang, J.; Zhang, Y.; Liu, L.; Cao, L.; Liu, Y.; Dong, L. Type 2 innate lymphoid cells: A novel biomarker of eosinophilic airway inflammation in patients with mild to moderate asthma. Respir. Med. 2015, 109, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Nagakumar, P.; Denney, L.; Fleming, L.; Bush, A.; Lloyd, C.M.; Saglani, S. Type 2 innate lymphoid cells in induced sputum from children with severe asthma. J. Allergy Clin. Immunol. 2016, 137, 624–626.e6. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, C.J.; Barlow, J.L.; McKenzie, A.N.J. Insights into the initiation of type 2 immune responses. Immunology 2011, 134, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Surette, M.G.; Virchow, J.C. Neutrophilic asthma: Misconception or misnomer? Lancet Respir. Med. 2021, 9, 441–443. [Google Scholar] [CrossRef]
- Svenningsen, S.; Nair, P. Asthma Endotypes and an Overview of Targeted Therapy for Asthma. Front. Med. 2017, 4, 158. [Google Scholar] [CrossRef]
- Bal, S.M.; Golebski, K.; Spits, H. Plasticity of innate lymphoid cell subsets. Nat. Rev. Immunol. 2020, 20, 552–565. [Google Scholar] [CrossRef]
- Cai, T.; Qiu, J.; Ji, Y.; Li, W.; Ding, Z.; Suo, C.; Chang, J.; Wang, J.; He, R.; Qian, Y.; et al. IL-17–producing ST2+ group 2 innate lymphoid cells play a pathogenic role in lung inflammation. J. Allergy Clin. Immunol. 2019, 143, 229–244.e9. [Google Scholar] [CrossRef]
- Bernink, J.H.; Ohne, Y.; Teunissen, M.B.M.; Wang, J.; Wu, J.; Krabbendam, L.; Guntermann, C.; Volckmann, R.; Koster, J.; van Tol, S.; et al. c-Kit-positive ILC2s exhibit an ILC3-like signature that may contribute to IL-17-mediated pathologies. Nat. Immunol. 2019, 20, 992–1003. [Google Scholar] [CrossRef]
- Golebski, K.; Ros, X.R.; Nagasawa, M.; van Tol, S.; Heesters, B.A.; Aglmous, H.; Kradolfer, C.M.A.; Shikhagaie, M.M.; Seys, S.; Hellings, P.W.; et al. IL-1β, IL-23, and TGF-β drive plasticity of human ILC2s towards IL-17-producing ILCs in nasal inflammation. Nat. Commun. 2019, 10, 2162. [Google Scholar] [CrossRef]
- Chen, X.-J.; Liu, C.; Zhang, S.; Zhang, L.-F.; Meng, W.; Zhang, X.; Sun, M.; Zhang, Y.; Wang, R.-Z.; Yao, C.-F. ILC3-like ILC2 subset increases in minimal persistent inflammation after acute type II inflammation of allergic rhinitis and inhibited by Biminkang: Plasticity of ILC2 in minimal persistent inflammation. J. Leukoc. Biol. 2022, 112, 1445–1455. [Google Scholar] [CrossRef]
- Wen, Y.; Zeng, Q.; Luo, X.; Ma, R.; Tang, Y.; Liu, W. Leptin Promoted IL-17 Production from ILC2s in Allergic Rhinitis. Mediat. Inflamm. 2020, 2020, 9248479. [Google Scholar] [CrossRef] [PubMed]
- Simoni, Y.; Fehlings, M.; Kløverpris, H.N.; McGovern, N.; Koo, S.-L.; Loh, C.Y.; Lim, S.; Kurioka, A.; Fergusson, J.R.; Tang, C.-L.; et al. Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity 2017, 46, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Bernink, J.H.; Krabbendam, L.; Germar, K.; de Jong, E.; Gronke, K.; Kofoed-Nielsen, M.; Munneke, J.M.; Hazenberg, M.D.; Villaudy, J.; Buskens, C.J.; et al. Interleukin-12 and -23 Control Plasticity of CD127+ Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 2015, 43, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Doty, A.L.; Tang, Y.; Berrie, D.; Iqbal, A.; Tan, S.A.; Clare-Salzler, M.J.; Wallet, S.M.; Glover, S.C. Enrichment of IL-17A+IFN-γ+ and IL-22+IFN-γ+ T cell subsets is associated with reduction of NKp44+ILC3s in the terminal ileum of Crohn’s disease patients. Clin. Exp. Immunol. 2017, 190, 143–153. [Google Scholar] [CrossRef]
- Mazzurana, L.; Forkel, M.; Rao, A.; Van Acker, A.; Kokkinou, E.; Ichiya, T.; Almer, S.; Höög, C.; Friberg, D.; Mjösberg, J. Suppression of Aiolos and Ikaros expression by lenalidomide reduces human ILC3−ILC1/NK cell transdifferentiation. Eur. J. Immunol. 2019, 49, 1344–1355. [Google Scholar] [CrossRef]
- Silver, J.S.; Kearley, J.; Copenhaver, A.M.; Sanden, C.; Mori, M.; Yu, L.; Pritchard, G.H.; Berlin, A.A.; Hunter, C.A.; Bowler, R.; et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat. Immunol. 2016, 17, 626–635. [Google Scholar] [CrossRef]
- Bal, S.M.; Bernink, J.H.; Nagasawa, M.; Groot, J.; Shikhagaie, M.M.; Golebski, K.; van Drunen, C.M.; Lutter, R.; Jonkers, R.E.; Hombrink, P.; et al. IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat. Immunol. 2016, 17, 636–645. [Google Scholar] [CrossRef]
- Ogasawara, N.; Poposki, J.A.; Klingler, A.I.; Tan, B.K.; Weibman, A.R.; Hulse, K.E.; Stevens, W.W.; Peters, A.T.; Grammer, L.C.; Schleimer, R.P.; et al. IL-10, TGF-β, and glucocorticoid prevent the production of type 2 cytokines in human group 2 innate lymphoid cells. J. Allergy Clin. Immunol. 2018, 141, 1147–1151.e8. [Google Scholar] [CrossRef]
- Seehus, C.R.; Kadavallore, A.; de la Torre, B.; Yeckes, A.R.; Wang, Y.; Tang, J.; Kaye, J. Alternative activation generates IL-10 producing type 2 innate lymphoid cells. Nat. Commun. 2017, 8, 1900. [Google Scholar] [CrossRef]
- Howard, E.; Lewis, G.; Galle-Treger, L.; Hurrell, B.P.; Helou, D.G.; Shafiei-Jahani, P.; Painter, J.D.; Muench, G.A.; Soroosh, P.; Akbari, O. IL-10 production by ILC2s requires Blimp-1 and cMaf, modulates cellular metabolism, and ameliorates airway hyperreactivity. J. Allergy Clin. Immunol. 2021, 147, 1281–1295.e5. [Google Scholar] [CrossRef]
- Golebski, K.; Layhadi, J.A.; Sahiner, U.; Steveling-Klein, E.H.; Lenormand, M.M.; Li, R.C.Y.; Bal, S.M.; Heesters, B.A.; Vilà-Nadal, G.; Hunewald, O.; et al. Induction of IL-10-producing type 2 innate lymphoid cells by allergen immunotherapy is associated with clinical response. Immunity 2021, 54, 291–307.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xia, P.; Chen, Y.; Qu, Y.; Xiong, Z.; Ye, B.; Du, Y.; Tian, Y.; Yin, Z.; Xu, Z.; et al. Regulatory Innate Lymphoid Cells Control Innate Intestinal Inflammation. Cell 2017, 171, 201–216.e18. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; Vieira, P.; Fiorentino, D.F.; Trounstine, M.L.; Khan, T.A.; Mosmann, T.R. Homology of Cytokine Synthesis Inhibitory Factor (IL-10) to the Epstein-Barr Virus Gene BCRFI. Science 1990, 248, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the Interleukin-10 Receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc. Natl. Acad. Sci. USA 2005, 102, 8686–8691. [Google Scholar] [CrossRef]
- Iyer, S.S.; Cheng, G. Role of Interleukin 10 Transcriptional Regulation in Inflammation and Autoimmune Disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef]
- Glocker, E.-O.; Kotlarz, D.; Klein, C.; Shah, N.; Grimbacher, B. IL-10 and IL-10 receptor defects in humans. Ann. N. Y. Acad. Sci. 2011, 1246, 102–107. [Google Scholar] [CrossRef]
- Fiorentino, D.F.; Bond, M.W.; Mosmann, T.R. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J. Exp. Med. 1989, 170, 2081–2095. [Google Scholar] [CrossRef]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Abdulnour, R.-E.E.; Nyman, J.; Dionne, D.; Hofree, M.; Cuoco, M.S.; Rodman, C.; Farouq, D.; et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 2017, 549, 351–356. [Google Scholar] [CrossRef]
- Wang, X.; Wong, K.; Ouyang, W.; Rutz, S. Targeting IL-10 Family Cytokines for the Treatment of Human Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a028548. [Google Scholar] [CrossRef]
- Saraiva, M.; Vieira, P.; O’garra, A. Biology and therapeutic potential of interleukin-10. J. Exp. Med. 2019, 217, e20190418. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, C.; Kojo, S.; Yamashita, M.; Moro, K.; Lacaud, G.; Shiroguchi, K.; Taniuchi, I.; Ebihara, T. Runx/Cbfβ complexes protect group 2 innate lymphoid cells from exhausted-like hyporesponsiveness during allergic airway inflammation. Nat. Commun. 2019, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, W.; Zhu, R. The Role of Innate Immune Cells in Allergen Immunotherapy. Curr. Treat. Options Allergy 2023, 10, 148–165. [Google Scholar] [CrossRef]
- Boonpiyathad, T.; Tantilipikorn, P.; Ruxrungtham, K.; Pradubpongsa, P.; Mitthamsiri, W.; Piedvache, A.; Thantiworasit, P.; Sirivichayakul, S.; Jacquet, A.; Suratannon, N.; et al. IL-10–producing innate lymphoid cells increased in patients with house dust mite allergic rhinitis following immunotherapy. J. Allergy Clin. Immunol. 2021, 147, 1507–1510.e8. [Google Scholar] [CrossRef]
- Gupta, A.; Dimeloe, S.; Richards, D.F.; Chambers, E.S.; Black, C.; Urry, Z.; Ryanna, K.; Xystrakis, E.; Bush, A.; Saglani, S.; et al. Defective IL-10 expression and in vitro steroid-induced IL-17A in paediatric severe therapy-resistant asthma. Thorax 2014, 69, 508–515. [Google Scholar] [CrossRef]
- Nakagome, K.; Dohi, M.; Okunishi, K.; Tanaka, R.; Miyazaki, J.; Yamamoto, K. In vivo IL-10 gene delivery attenuates bleomycin induced pulmonary fibrosis by inhibiting the production and activation of TGF-β in the lung. Thorax 2006, 61, 886–894. [Google Scholar] [CrossRef]
- Morita, H.; Arae, K.; Unno, H.; Miyauchi, K.; Toyama, S.; Nambu, A.; Oboki, K.; Ohno, T.; Motomura, K.; Matsuda, A.; et al. An Interleukin-33-Mast Cell-Interleukin-2 Axis Suppresses Papain-Induced Allergic Inflammation by Promoting Regulatory T Cell Numbers. Immunity 2015, 43, 175–186. [Google Scholar] [CrossRef]
- Ferreira, A.C.F.; Szeto, A.C.H.; Heycock, M.W.D.; Clark, P.A.; Walker, J.A.; Crisp, A.; Barlow, J.L.; Kitching, S.; Lim, A.; Gogoi, M.; et al. RORα is a critical checkpoint for T cell and ILC2 commitment in the embryonic thymus. Nat. Immunol. 2021, 22, 166–178. [Google Scholar] [CrossRef]
- Khare, A.; Krishnamoorthy, N.; Oriss, T.B.; Fei, M.; Ray, P.; Ray, A. Cutting Edge: Inhaled Antigen Upregulates Retinaldehyde Dehydrogenase in Lung CD103+ but Not Plasmacytoid Dendritic Cells To Induce Foxp3 De Novo in CD4+ T Cells and Promote Airway Tolerance. J. Immunol. 2013, 191, 25–29. [Google Scholar] [CrossRef]
- Baban, B.; Braun, M.; Khodadadi, H.; Ward, A.; Alverson, K.; Malik, A.; Nguyen, K.; Nazarian, S.; Hess, D.C.; Forseen, S.; et al. AMPK induces regulatory innate lymphoid cells after traumatic brain injury. JCI Insight 2021, 6, e126766. [Google Scholar] [CrossRef]
- Coomes, S.M.; Kannan, Y.; Pelly, V.S.; Entwistle, L.J.; Guidi, R.; Perez-Lloret, J.; Nikolov, N.; Müller, W.; Wilson, M.S. CD4+ Th2 cells are directly regulated by IL-10 during allergic airway inflammation. Mucosal Immunol. 2017, 10, 150–161. [Google Scholar] [CrossRef]
- Pelly, V.S.; Kannan, Y.; Coomes, S.M.; Entwistle, L.J.; Rückerl, D.; Seddon, B.; MacDonald, A.S.; McKenzie, A.; Wilson, M.S. IL-4-producing ILC2s are required for the differentiation of TH2 cells following Heligmosomoides polygyrus infection. Mucosal Immunol. 2016, 9, 1407–1417. [Google Scholar] [CrossRef]
- Symowski, C.; Voehringer, D. Th2 cell-derived IL-4/IL-13 promote ILC2 accumulation in the lung by ILC2-intrinsic STAT6 signaling in mice. Eur. J. Immunol. 2019, 49, 1421–1432. [Google Scholar] [CrossRef]
- Mitchell, R.E.; Hassan, M.; Burton, B.R.; Britton, G.; Hill, E.V.; Verhagen, J.; Wraith, D.C. IL-4 enhances IL-10 production in Th1 cells: Implications for Th1 and Th2 regulation. Sci. Rep. 2017, 7, 11315. [Google Scholar] [CrossRef]
- Zhu, B.; Buttrick, T.; Bassil, R.; Zhu, C.; Olah, M.; Wu, C.; Xiao, S.; Orent, W.; Elyaman, W.; Khoury, S.J. IL-4 and Retinoic Acid Synergistically Induce Regulatory Dendritic Cells Expressing Aldh1a2. J. Immunol. 2013, 191, 3139–3151. [Google Scholar] [CrossRef]
- Meylan, F.; Hawley, E.T.; Barron, L.; Barlow, J.L.; Penumetcha, P.; Pelletier, M.; Sciumè, G.; Richard, A.C.; Hayes, E.T.; Gomez-Rodriguez, J.; et al. The TNF-family cytokine TL1A promotes allergic immunopathology through group 2 innate lymphoid cells. Mucosal Immunol. 2014, 7, 958–968. [Google Scholar] [CrossRef]
- Machida, K.; Aw, M.; Salter, B.M.A.; Ju, X.; Mukherjee, M.; Gauvreau, G.M.; O’byrne, P.M.; Nair, P.; Sehmi, R. The Role of the TL1A/DR3 Axis in the Activation of Group 2 Innate Lymphoid Cells in Subjects with Eosinophilic Asthma. Am. J. Respir. Crit. Care Med. 2020, 202, 1105–1114. [Google Scholar] [CrossRef]
- Laurent, P.; Allard, B.; Manicki, P.; Jolivel, V.; Levionnois, E.; Jeljeli, M.; Henrot, P.; Izotte, J.; Leleu, D.; Groppi, A.; et al. TGFβ promotes low IL10-producing ILC2 with profibrotic ability involved in skin fibrosis in systemic sclerosis. Ann. Rheum. Dis. 2021, 80, 1594–1603. [Google Scholar] [CrossRef]
- Nakamura, S.; Kuroki, K.; Ohki, I.; Sasaki, K.; Kajikawa, M.; Maruyama, T.; Ito, M.; Kameda, Y.; Ikura, M.; Yamamoto, K.; et al. Molecular Basis for E-cadherin Recognition by Killer Cell Lectin-like Receptor G1 (KLRG1). J. Biol. Chem. 2009, 284, 27327–27335. [Google Scholar] [CrossRef]
- Henson, S.M.; Akbar, A.N. KLRG1—More than a marker for T cell senescence. Age 2009, 31, 285–291. [Google Scholar] [CrossRef]
- Laffont, S.; Blanquart, E.; Savignac, M.; Cénac, C.; Laverny, G.; Metzger, D.; Girard, J.-P.; Belz, G.T.; Pelletier, L.; Seillet, C.; et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1581–1592. [Google Scholar] [CrossRef]
- Loering, S.; Cameron, G.J.; Bhatt, N.P.; Belz, G.T.; Foster, P.S.; Hansbro, P.M.; Starkey, M.R. Differences in pulmonary group 2 innate lymphoid cells are dependent on mouse age, sex and strain. Immunol. Cell Biol. 2021, 99, 542–551. [Google Scholar] [CrossRef]
- Zhao, H.; Moarbes, V.; Gaudreault, V.; Shan, J.; Aldossary, H.; Cyr, L.; Fixman, E.D. Sex Differences in IL-33-Induced STAT6-Dependent Type 2 Airway Inflammation. Front. Immunol. 2019, 10, 859. [Google Scholar] [CrossRef]
- Kadel, S.; Ainsua-Enrich, E.; Hatipoglu, I.; Turner, S.; Singh, S.; Khan, S.; Kovats, S. A Major Population of Functional KLRG1– ILC2s in Female Lungs Contributes to a Sex Bias in ILC2 Numbers. ImmunoHorizons 2018, 2, 74–86. [Google Scholar] [CrossRef]
- Cephus, J.-Y.; Stier, M.T.; Fuseini, H.; Yung, J.A.; Toki, S.; Bloodworth, M.H.; Zhou, W.; Goleniewska, K.; Zhang, J.; Garon, S.L.; et al. Testosterone Attenuates Group 2 Innate Lymphoid Cell-Mediated Airway Inflammation. Cell Rep. 2017, 21, 2487–2499. [Google Scholar] [CrossRef]
- Lei, A.-H.; Xiao, Q.; Liu, G.-Y.; Shi, K.; Yang, Q.; Li, X.; Liu, Y.-F.; Wang, H.-K.; Cai, W.-P.; Guan, Y.-J.; et al. ICAM-1 controls development and function of ILC2. J. Exp. Med. 2018, 215, 2157–2174. [Google Scholar] [CrossRef]
- Hurrell, B.P.; Howard, E.; Galle-Treger, L.; Helou, D.G.; Shafiei-Jahani, P.; Painter, J.D.; Akbari, O. Distinct Roles of LFA-1 and ICAM-1 on ILC2s Control Lung Infiltration, Effector Functions, and Development of Airway Hyperreactivity. Front. Immunol. 2020, 11, 542818. [Google Scholar] [CrossRef]
- Cardoso, V.; Chesné, J.; Ribeiro, H.; García-Cassani, B.; Carvalho, T.; Bouchery, T.; Shah, K.; Barbosa-Morais, N.L.; Harris, N.; Veiga-Fernandes, H. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 2017, 549, 277–281. [Google Scholar] [CrossRef]
- Naito, M.; Nakanishi, Y.; Motomura, Y.; Takamatsu, H.; Koyama, S.; Nishide, M.; Naito, Y.; Izumi, M.; Mizuno, Y.; Yamaguchi, Y.; et al. Semaphorin 6D–expressing mesenchymal cells regulate IL-10 production by ILC2s in the lung. Life Sci. Alliance 2022, 5, e202201486. [Google Scholar] [CrossRef]
- Morita, H.; Kubo, T.; Rückert, B.; Ravindran, A.; Soyka, M.B.; Rinaldi, A.O.; Sugita, K.; Wawrzyniak, M.; Wawrzyniak, P.; Motomura, K.; et al. Induction of human regulatory innate lymphoid cells from group 2 innate lymphoid cells by retinoic acid. J. Allergy Clin. Immunol. 2019, 143, 2190–2201.e9. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.; Heinrich, F.; Neumann, K.; Junghans, V.; Mashreghi, M.-F.; Ahlers, J.; Janke, M.; Rudolph, C.; Mockel-Tenbrinck, N.; Kühl, A.A.; et al. Role of Blimp-1 in programing Th effector cells into IL-10 producers. J. Exp. Med. 2014, 211, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Dodd, H.; Moser, E.K.; Sharma, R.; Braciale, T.J. CD4+ T cell help and innate-derived IL-27 induce Blimp-1-dependent IL-10 production by antiviral CTLs. Nat. Immunol. 2011, 12, 327–334. [Google Scholar] [CrossRef]
- Xu, M.; Pokrovskii, M.; Ding, Y.; Yi, R.; Au, C.; Harrison, O.J.; Galan, C.; Belkaid, Y.; Bonneau, R.; Littman, D.R. c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 2018, 554, 373–377. [Google Scholar] [CrossRef]
- Rutz, S.; Ouyang, W. Regulation of Interleukin-10 Expression. Regulation of Cytokine Gene Expression in Immunity and Diseases. In Advances in Experimental Medicine and Biology; Ma, X., Ed.; Springer: Dordrecht, The Netherlands, 2016; pp. 89–116. ISBN 978-94-024-0921-5. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emami Fard, N.; Xiao, M.; Sehmi, R. Regulatory ILC2—Role of IL-10 Producing ILC2 in Asthma. Cells 2023, 12, 2556. https://doi.org/10.3390/cells12212556
Emami Fard N, Xiao M, Sehmi R. Regulatory ILC2—Role of IL-10 Producing ILC2 in Asthma. Cells. 2023; 12(21):2556. https://doi.org/10.3390/cells12212556
Chicago/Turabian StyleEmami Fard, Nahal, Maria Xiao, and Roma Sehmi. 2023. "Regulatory ILC2—Role of IL-10 Producing ILC2 in Asthma" Cells 12, no. 21: 2556. https://doi.org/10.3390/cells12212556
APA StyleEmami Fard, N., Xiao, M., & Sehmi, R. (2023). Regulatory ILC2—Role of IL-10 Producing ILC2 in Asthma. Cells, 12(21), 2556. https://doi.org/10.3390/cells12212556