
Circulating Tumor Cell Subpopulations Predict Treatment Outcome in Pancreatic Ductal Adenocarcinoma (PDAC) Patients

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Blood Sample Processing
2.3. Polymerase Chain Reaction and Ligase Detection Reaction (PCR/LDR) Assay
2.4. Next-Generation Sequencing (NGS)
2.5. Statistical Tests
3. Results
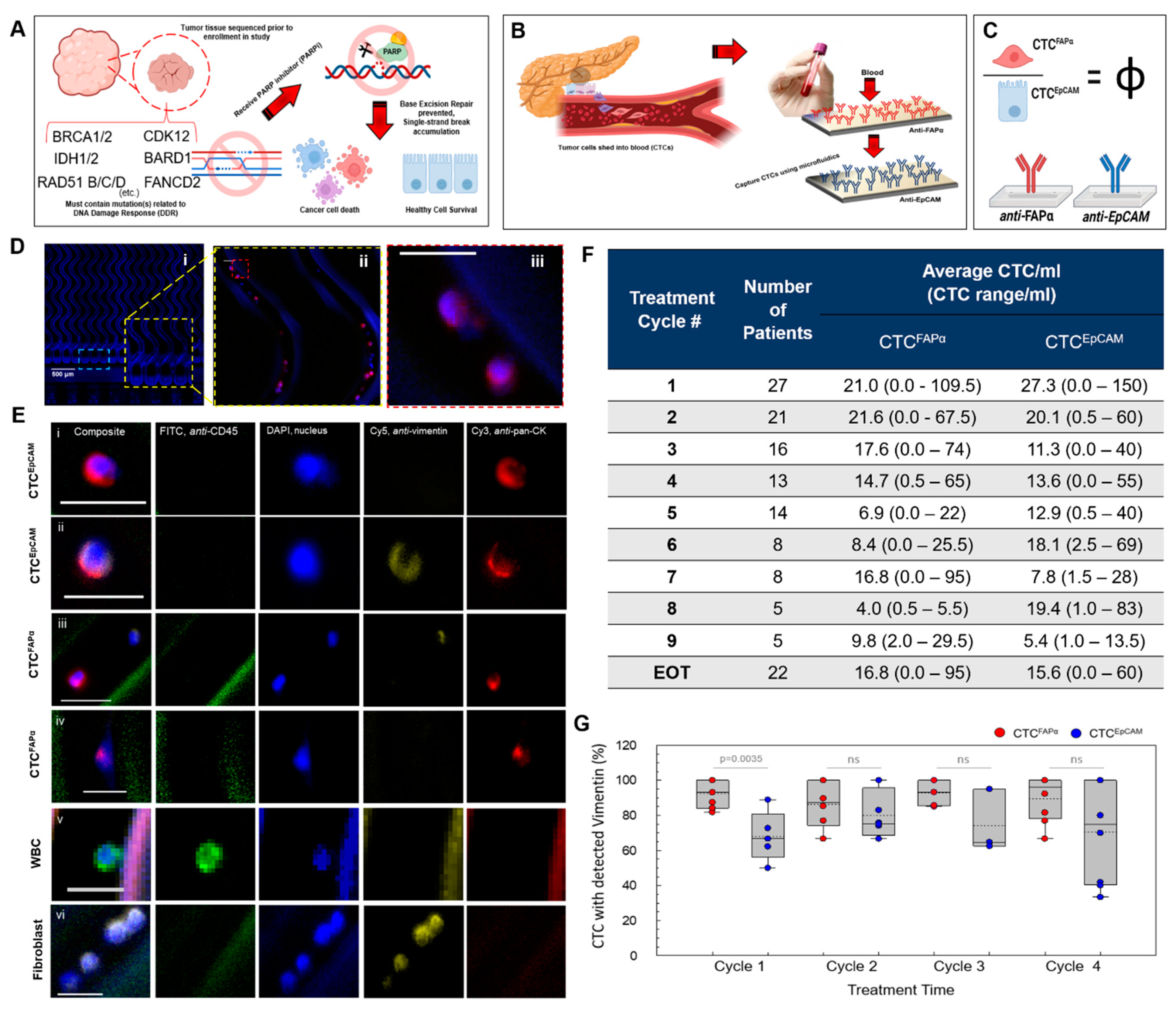
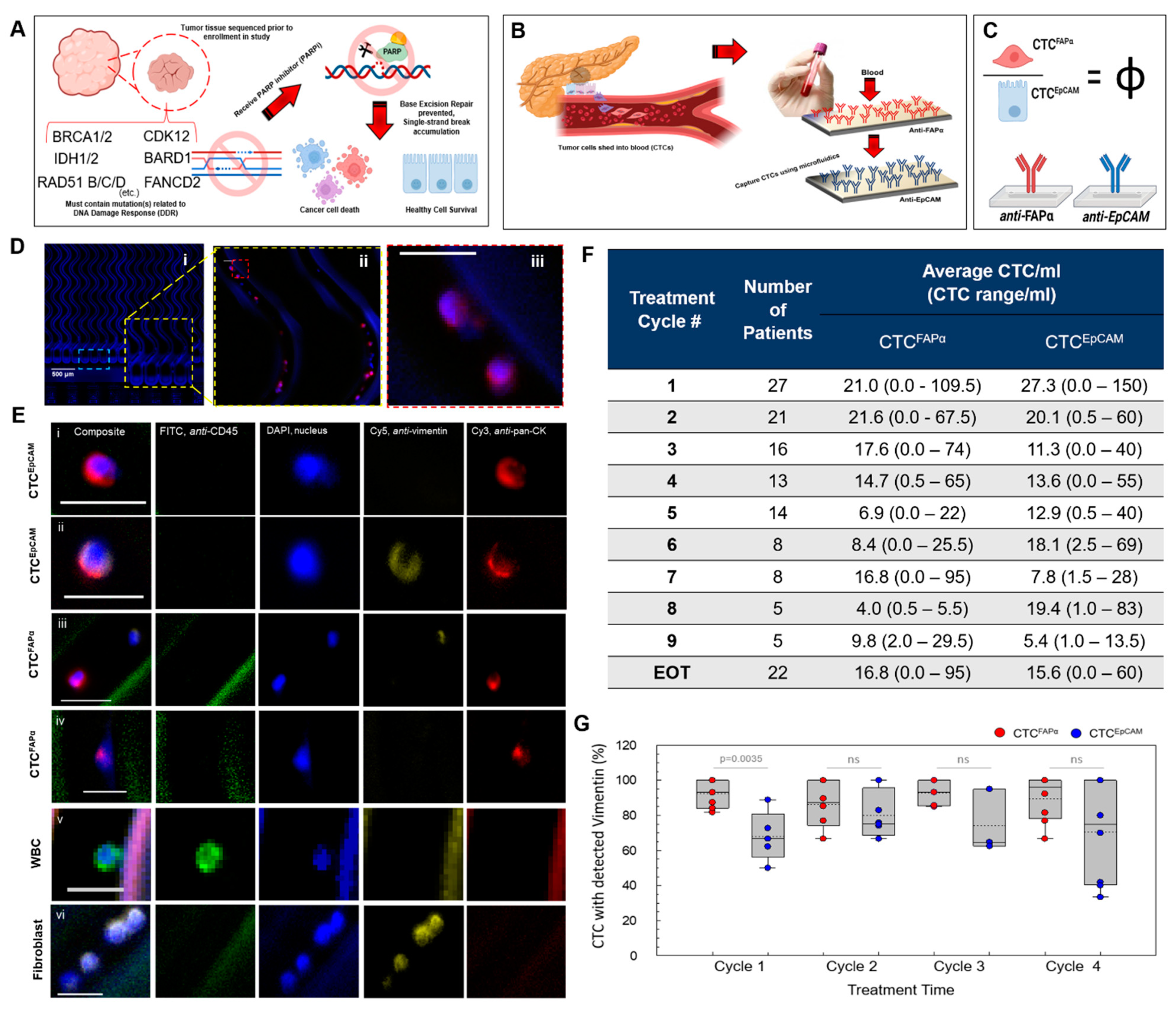
3.1. Automated Blood Sample Processing Using the Microfluidic CTC Isolation Device

3.2. Isolation of CTCs from Blood Samples
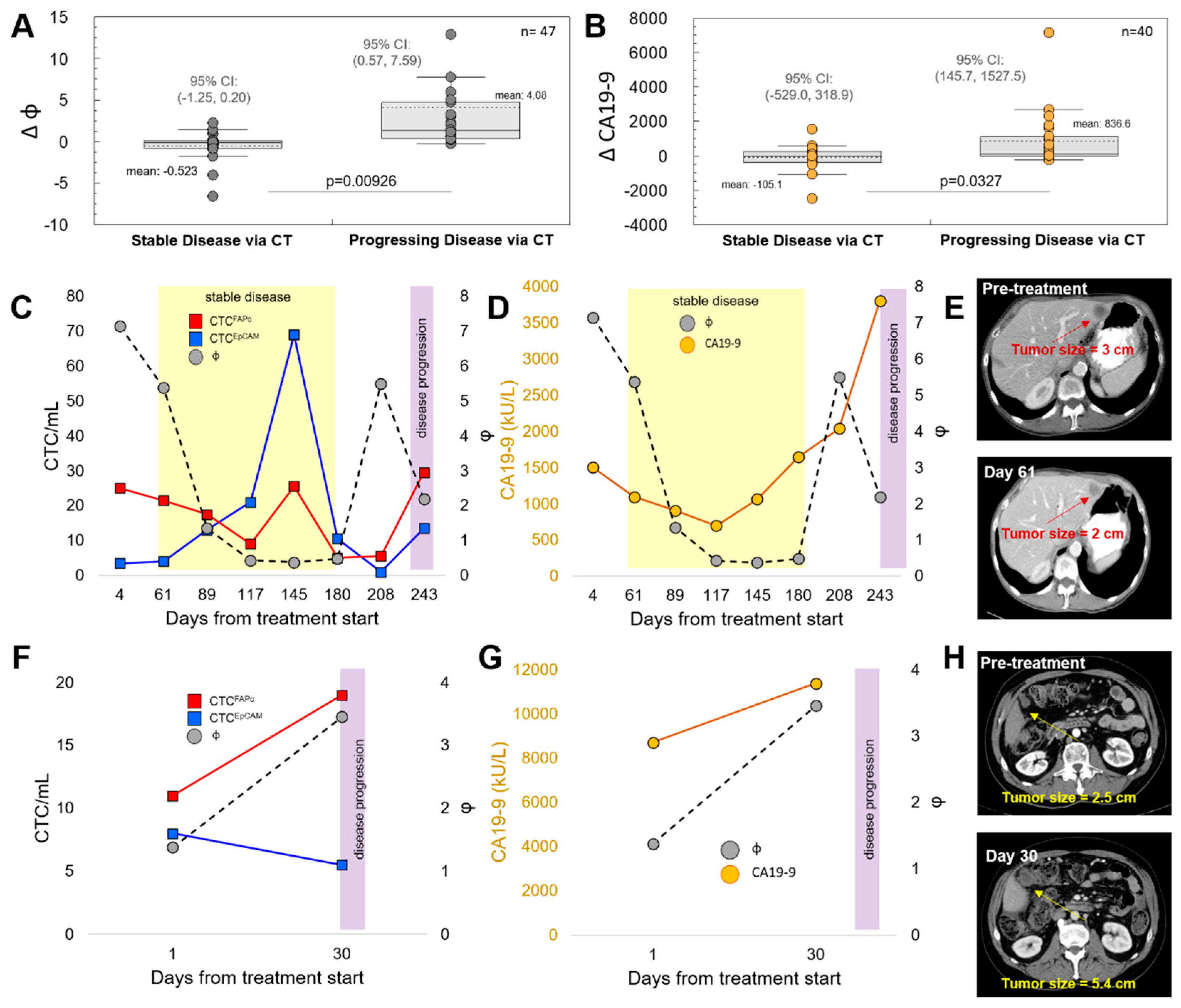
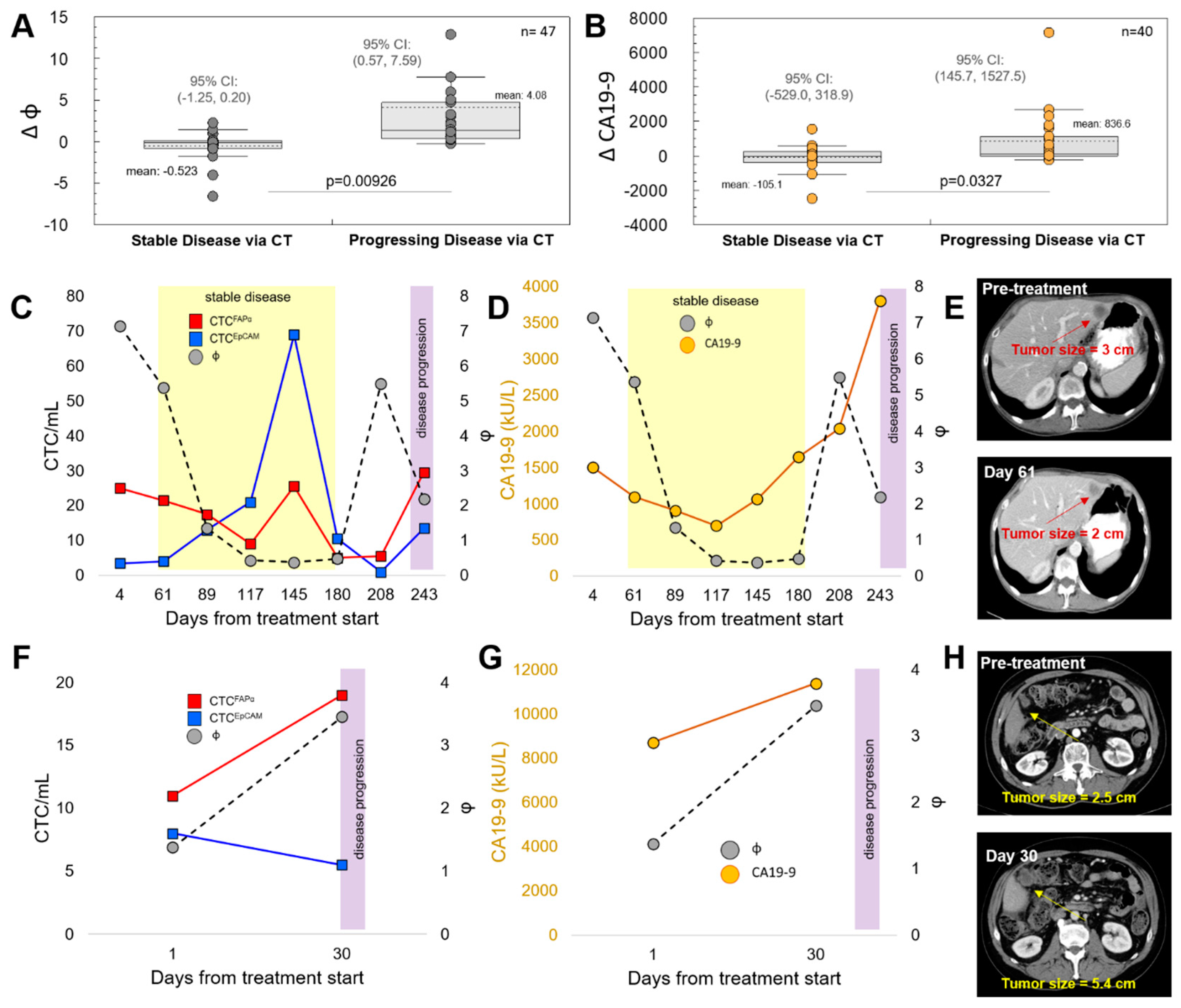
3.3. CTCFAPα to CTCEpCAM Ratio (Φ) and the Treatment Response
3.4. Progression-Free Survival (PFS) and Overall Survival (OS)
3.5. Next-Generation Sequencing (NGS) of CTCs to Search for DDR Mutations
3.6. KRAS Mutation Screening in CTCs Isolated from the Blood of PDAC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- De Dosso, S.; Siebenhüner, A.R.; Winder, T.; Meisel, A.; Fritsch, R.; Astaras, C.; Szturz, P.; Borner, M. Treatment landscape of metastatic pancreatic cancer. Cancer Treat. Rev. 2021, 96, 102180. [Google Scholar] [CrossRef] [PubMed]
- Zins, M.; Matos, C.; Cassinotto, C. Pancreatic adenocarcinoma staging in the era of preoperative chemotherapy and radiation therapy. Radiology 2018, 287, 374–390. [Google Scholar] [CrossRef] [PubMed]
- Koprowski, H.; Steplewski, Z.; Mitchell, K.; Herlyn, M.; Herlyn, D.; Fuhrer, P. Colorectal carcinoma antigens detected by hybridoma antibodies. Somat. Cell Genet. 1979, 5, 957–971. [Google Scholar] [CrossRef]
- Azizian, A.; Rühlmann, F.; Krause, T.; Bernhardt, M.; Jo, P.; König, A.; Kleiß, M.; Leha, A.; Ghadimi, M.; Gaedcke, J. CA19-9 for detecting recurrence of pancreatic cancer. Sci. Rep. 2020, 10, 1332. [Google Scholar] [CrossRef]
- Kim, S.; Park, B.K.; Seo, J.H.; Choi, J.; Choi, J.W.; Lee, C.K.; Chung, J.B.; Park, Y.; Kim, D.W. Carbohydrate antigen 19-9 elevation without evidence of malignant or pancreatobiliary diseases. Sci. Rep. 2020, 10, 8820. [Google Scholar] [CrossRef]
- Poruk, K.E.; Gay, D.Z.; Brown, K.; Mulvihill, J.D.; Boucher, K.M.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. The clinical utility of CA 19-9 in pancreatic adenocarcinoma: Diagnostic and prognostic updates. Curr. Mol. Med. 2013, 13, 340–351. [Google Scholar] [CrossRef]
- Martini, V.; Timme-Bronsert, S.; Fichtner-Feigl, S.; Hoeppner, J.; Kulemann, B. Circulating Tumor Cells in Pancreatic Cancer: Current Perspectives. Cancers 2019, 11, 1659. [Google Scholar] [CrossRef]
- Witek, M.A.; Aufforth, R.D.; Wang, H.; Kamande, J.W.; Jackson, J.M.; Carey, L.A.; Gehrig, P.A.; Milowsky, M.I.; Perou, C.M.; Soper, J.T.; et al. Discrete microfluidics for the isolation of circulating tumor cell subpopulations targeting fibroblast activation protein alpha and epithelial cell adhesion molecule. npj Precis. Oncol. 2017, 1, 24. [Google Scholar] [CrossRef]
- Liu, F.; Qi, L.; Liu, B.; Liu, J.; Zhang, H.; Che, D.; Cao, J.; Shen, J.; Geng, J.; Bi, Y.; et al. Fibroblast activation protein overexpression and clinical implications in solid tumors: A meta-analysis. PLoS ONE 2015, 10, e0116683. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Jolly, M.K.; Mani, S.A.; Levine, H. Hybrid epithelial/mesenchymal phenotype(s): The ‘fittest’ for metastasis? Biochim. Biophys. Acta (BBA)—Rev. Cancer 2018, 1870, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef]
- Stott, S.L.; Hsu, C.-H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef]
- Curtin, N.J.; Mukhopadhyay, A.; Drew, Y.; Plummer, R. DNA Repair in Cancer Therapy; Kelley, M.R., Ed.; Academic Press: San Diego, CA, USA, 2012; pp. 55–73. [Google Scholar] [CrossRef]
- Wong, K.H.K.; Tessier, S.N.; Miyamoto, D.T.; Miller, K.L.; Bookstaver, L.D.; Carey, T.R.; Stannard, C.J.; Thapar, V.; Tai, E.C.; Vo, K.D.; et al. Whole blood stabilization for the microfluidic isolation and molecular characterization of circulating tumor cells. Nat. Commun. 2017, 8, 1733. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Adams, A.A.; Okagbare, P.; Feng, J.; McCarley, R.L.; Murphy, M.C.; Soper, S.A. Circulating tumor cell isolation and enumeration using polymer-based microfluidics with an integrated conductivity sensor. J. Am. Chem. Soc. 2008, 130, 8633–8641. [Google Scholar] [CrossRef]
- Phan, H.V.; van Gent, M.; Drayman, N.; Basu, A.; Gack, M.U.; Tay, S. High-throughput RNA sequencing of paraformaldehyde-fixed single cells. Nat. Commun. 2021, 12, 5636. [Google Scholar] [CrossRef] [PubMed]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Hsia, L.-T.; Ashley, N.; Ouaret, D.; Wang, L.M.; Wilding, J.; Bodmer, W.F. Myofibroblasts are distinguished from activated skin fibroblasts by the expression of AOC3 and other associated markers. Proc. Natl. Acad. Sci. USA 2016, 113, E2162–E2171. [Google Scholar] [CrossRef]
- George, B.; Kent, M.; Surinach, A.; Lamarre, N.; Cockrum, P. The Association of Real-World CA 19-9 Level Monitoring Patterns and Clinical Outcomes Among Patients With Metastatic Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2021, 11, 754687. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing Oncology Panels. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef]
- Khanna, M.; Park, P.; Zirvi, M.; Cao, W.; Picon, A.; Day, J.; Paty, P.; Barany, F. Multiplex PCR/LDR for detection of K-ras mutations in primary colon tumors. Oncogene 1999, 18, 27–38. [Google Scholar] [CrossRef]
- ClinicalTrials.gov 2022; Volume 2022. Available online: ClinicalTrials.gov (accessed on 20 December 2022).
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef]
- Kawamura, Y.; Yamamoto, Y.; Sato, T.-A.; Ochiya, T. Extracellular vesicles as trans-genomic agents: Emerging roles in disease and evolution. Cancer Sci. 2017, 108, 824–830. [Google Scholar] [CrossRef]
- Wei, Z.; Batagov, A.O.; Schinelli, S.; Wang, J.; Wang, Y.; El Fatimy, R.; Rabinovsky, R.; Balaj, L.; Chen, C.C.; Hochberg, F.; et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat. Commun. 2017, 8, 1145. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: Biology and clinical significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef]
- Allan, A.L.; Keeney, M. Circulating Tumor Cell Analysis: Technical and Statistical Considerations for Application to the Clinic. J. Oncol. 2010, 2010, 426218. [Google Scholar] [CrossRef] [PubMed]
- Smerage, J.B.; Barlow, W.E.; Hortobagyi, G.N.; Winer, E.P.; Leyland-Jones, B.; Srkalovic, G.; Tejwani, S.; Schott, A.F.; O’Rourke, M.A.; Lew, D.L.; et al. Circulating Tumor Cells and Response to Chemotherapy in Metastatic Breast Cancer: SWOG S0500. J. Clin. Oncol. 2014, 32, 3483–3489. [Google Scholar] [CrossRef] [PubMed]
- Elbanna, K.Y.; Jang, H.J.; Kim, T.K. Imaging diagnosis and staging of pancreatic ductal adenocarcinoma: A comprehensive review. Insights Imaging 2020, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Robert, M.; Jarlier, M.; Gourgou, S.; Desseigne, F.; Ychou, M.; Bouché, O.; Bennouna, J. Retrospective Analysis of CA19-9 Decrease in Patients with Metastatic Pancreatic Carcinoma Treated with FOLFIRINOX or Gemcitabine in a Randomized Phase III Study (ACCORD11/PRODIGE4). Oncology 2017, 93, 367–376. [Google Scholar] [CrossRef]
- Ziske, C.; Schlie, C.; Gorschlüter, M.; Glasmacher, A.; Mey, U.; Strehl, J.; Sauerbruch, T.; Schmidt-Wolf, I.G. Prognostic value of CA 19-9 levels in patients with inoperable adenocarcinoma of the pancreas treated with gemcitabine. Br. J. Cancer 2003, 89, 1413–1417. [Google Scholar] [CrossRef]
- Hess, V.; Glimelius, B.; Grawe, P.; Dietrich, D.; Bodoky, G.; Ruhstaller, T.; Bajetta, E.; Saletti, P.; Figer, A.; Scheithauer, W.; et al. CA 19-9 tumour-marker response to chemotherapy in patients with advanced pancreatic cancer enrolled in a randomised controlled trial. Lancet Oncol. 2008, 9, 132–138. [Google Scholar] [CrossRef]
- González-Martín, A.; Matulonis, U.A.; Korach, J.; Mirza, M.R.; Moore, K.N.; Wu, X.; York, W.; Gupta, D.; Lechpammer, S.; Monk, B.J. Niraparib treatment for patients with BRCA-mutated ovarian cancer: Review of clinical data and therapeutic context. Future Oncol. 2022, 18, 2505–2536. [Google Scholar] [CrossRef]
- Gemenetzis, G.; Groot, V.P.; Yu, J.; Ding, D.; Teinor, J.A.; Javed, A.A.; Wood, L.D.; Burkhart, R.A.; Cameron, J.L.; Makary, M.A.; et al. Circulating Tumor Cells Dynamics in Pancreatic Adenocarcinoma Correlate With Disease Status: Results of the Prospective CLUSTER Study. Ann. Surg. 2018, 268, 408–420. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Kulemann, B.; Rösch, S.; Seifert, S.; Timme, S.; Bronsert, P.; Seifert, G.; Martini, V.; Kuvendjiska, J.; Glatz, T.; Hussung, S.; et al. Pancreatic cancer: Circulating Tumor Cells and Primary Tumors show Heterogeneous KRAS Mutations. Sci. Rep. 2017, 7, 4510. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, F.; Suzuki, K.; Tamaki, S.; Abe, I.; Endo, Y.; Takayama, Y.; Ishikawa, H.; Kakizawa, N.; Saito, M.; Futsuhara, K.; et al. Longitudinal monitoring of KRAS-mutated circulating tumor DNA enables the prediction of prognosis and therapeutic responses in patients with pancreatic cancer. PLoS ONE 2019, 14, e0227366. [Google Scholar] [CrossRef] [PubMed]
- Del Re, M.; Vivaldi, C.; Rofi, E.; Vasile, E.; Miccoli, M.; Caparello, C.; D’Arienzo, P.D.; Fornaro, L.; Falcone, A.; Danesi, R. Early changes in plasma DNA levels of mutant KRAS as a sensitive marker of response to chemotherapy in pancreatic cancer. Sci. Rep. 2017, 7, 7931. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freed, I.M.; Kasi, A.; Fateru, O.; Hu, M.; Gonzalez, P.; Weatherington, N.; Pathak, H.; Hyter, S.; Sun, W.; Al-Rajabi, R.; et al. Circulating Tumor Cell Subpopulations Predict Treatment Outcome in Pancreatic Ductal Adenocarcinoma (PDAC) Patients. Cells 2023, 12, 2266. https://doi.org/10.3390/cells12182266
Freed IM, Kasi A, Fateru O, Hu M, Gonzalez P, Weatherington N, Pathak H, Hyter S, Sun W, Al-Rajabi R, et al. Circulating Tumor Cell Subpopulations Predict Treatment Outcome in Pancreatic Ductal Adenocarcinoma (PDAC) Patients. Cells. 2023; 12(18):2266. https://doi.org/10.3390/cells12182266
Chicago/Turabian StyleFreed, Ian M., Anup Kasi, Oluwadamilola Fateru, Mengjia Hu, Phasin Gonzalez, Nyla Weatherington, Harsh Pathak, Stephen Hyter, Weijing Sun, Raed Al-Rajabi, and et al. 2023. "Circulating Tumor Cell Subpopulations Predict Treatment Outcome in Pancreatic Ductal Adenocarcinoma (PDAC) Patients" Cells 12, no. 18: 2266. https://doi.org/10.3390/cells12182266
APA StyleFreed, I. M., Kasi, A., Fateru, O., Hu, M., Gonzalez, P., Weatherington, N., Pathak, H., Hyter, S., Sun, W., Al-Rajabi, R., Baranda, J., Hupert, M. L., Chalise, P., Godwin, A. K., A. Witek, M., & Soper, S. A. (2023). Circulating Tumor Cell Subpopulations Predict Treatment Outcome in Pancreatic Ductal Adenocarcinoma (PDAC) Patients. Cells, 12(18), 2266. https://doi.org/10.3390/cells12182266