
Nanoscale CAR Organization at the Immune Synapse Correlates with CAR-T Effector Functions

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primary Cells and Cell Lines
- Untreated (UT) lymphocyte primary cells and CD138-CAR-T primary cells;
- FaDu cell lines—Human pharynx, squamous epithelial carcinoma.
- SKOV-3 cell lines—Human ovary epithelial adenocarcinoma, were a kind gift from the Dan Peer lab at Tel Aviv University;
- CAG—Human multiple myeloma cell line;
- OVCAR-3—Human ovarian adenocarcinoma.
2.2. Cell Growth and Treatment
2.3. Membrane Staining
2.4. Confocal Imaging
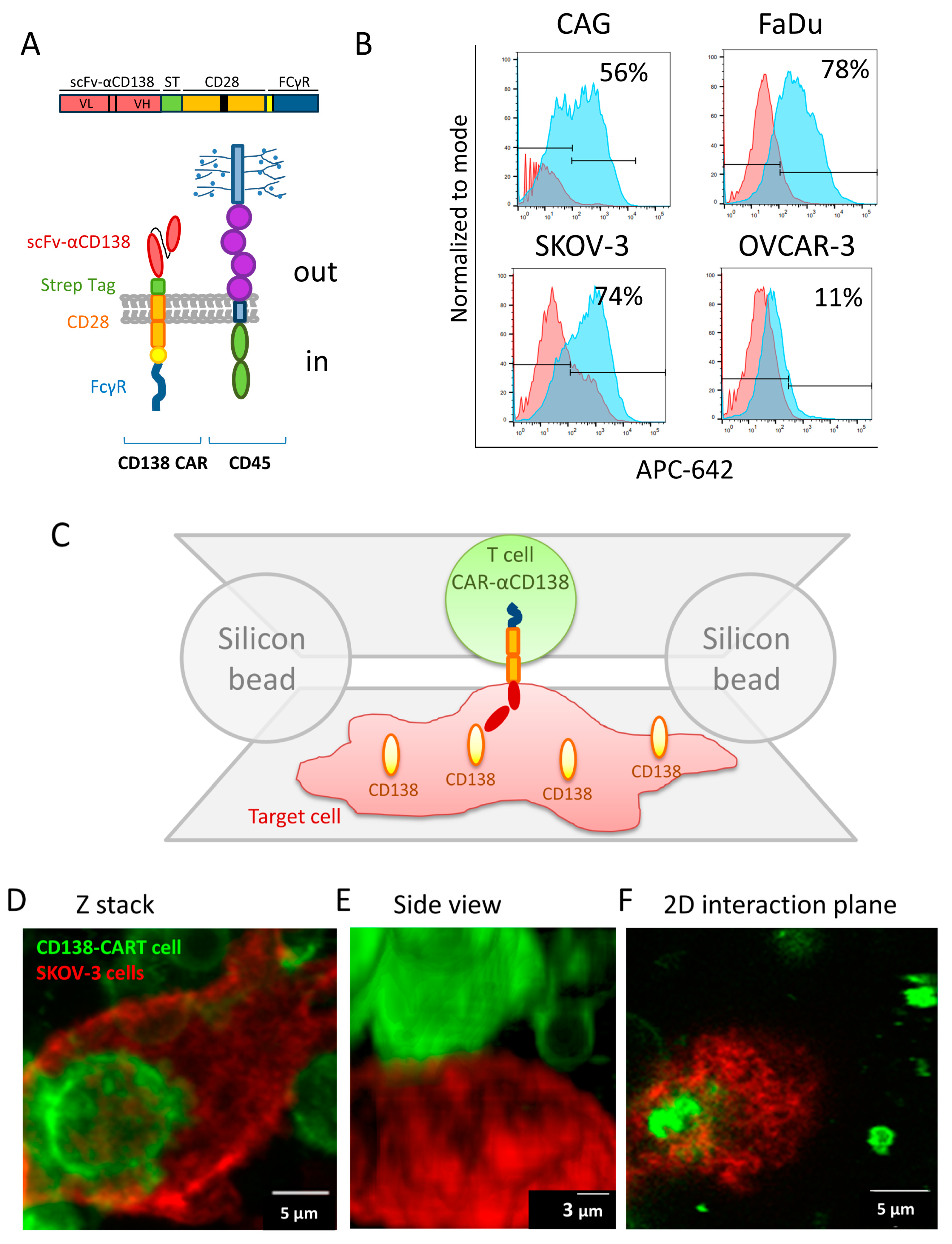
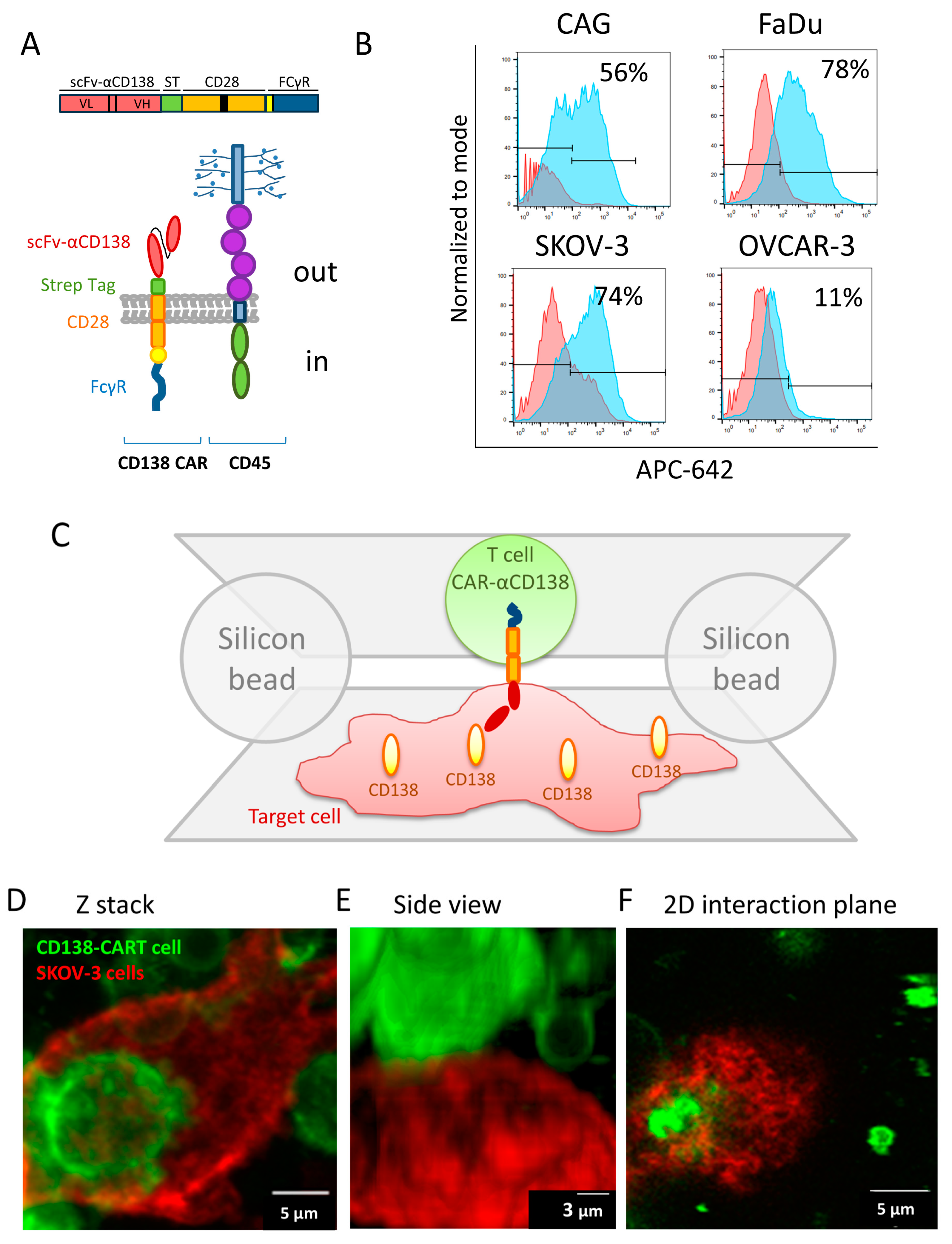
2.5. Cell Immuno-Staining
- Mouse Monoclonal Strep Tag II Antibody-FITC conjugated (clone 5A9F9) LS-C203631;
- Mouse monoclonal IgG1 α-Human CD45-Alexa647 conjugated (BioLegend, San Diego, CA, USA, 304056);
- Mouse monoclonal IgG1 α-Human CD138—APC conjugated (BioLegend, 352308).
2.6. Microscope Slide Preparation for Cell-on-Cell
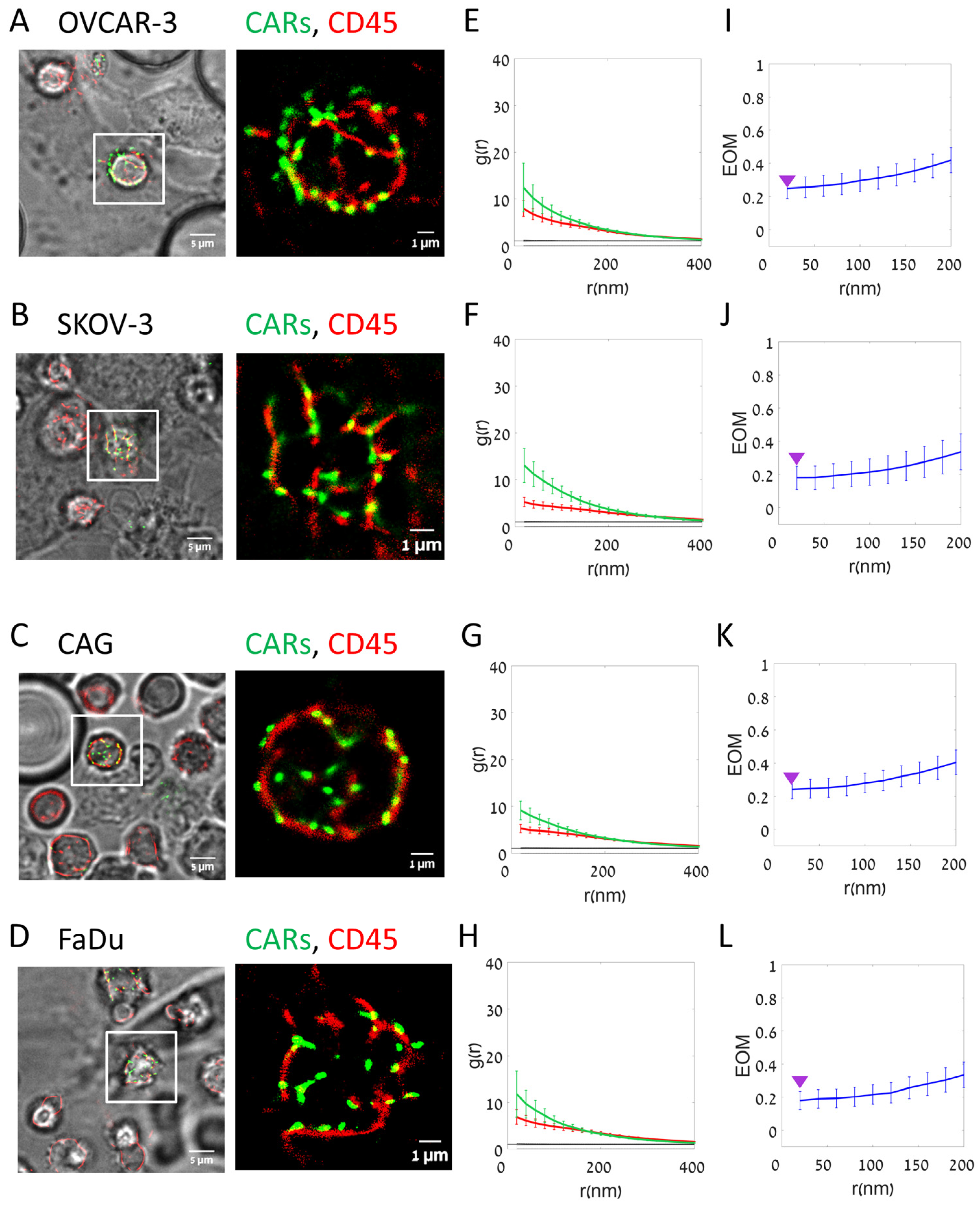
2.7. Single-Molecule Localization Microscopy
2.8. dSTORM Reconstruction
2.9. Three-Dimensional dSTORM
2.10. DBSCAN Cluster Size Analysis
2.11. Calcium Assay
2.12. FACS Analysis for CD138 Expression Level
2.13. Killing Analysis
2.14. MTT Assay
3. Results
3.1. High-Resolution Imaging of the Interface between CAR-T and Target Cells
3.2. Imaging and Analyses of Nanoscale Molecular Organization at the Interface
3.3. CARs Cluster and Segregate to a Higher Extent from CD45 in Interfaces with On-Target Cells
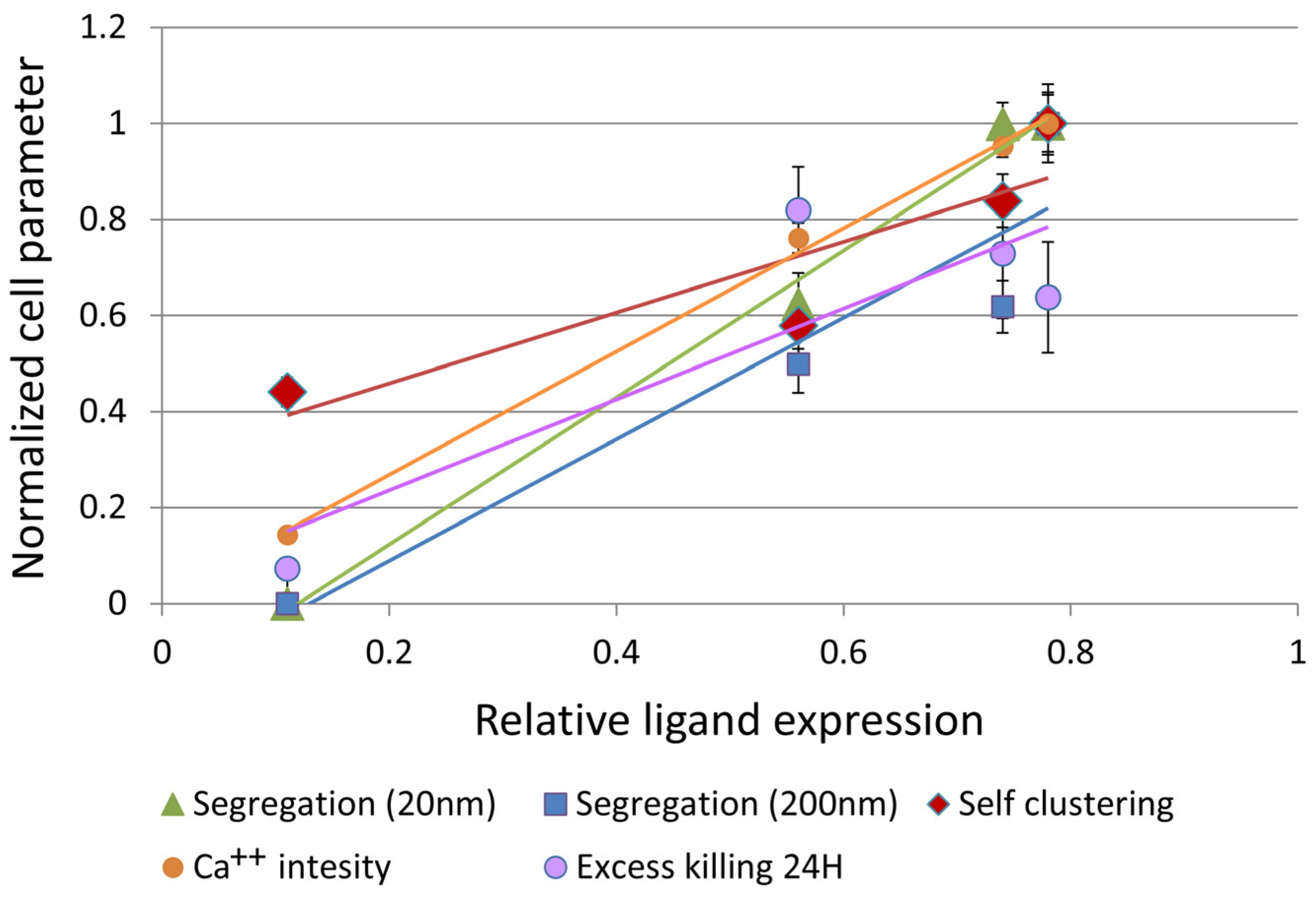
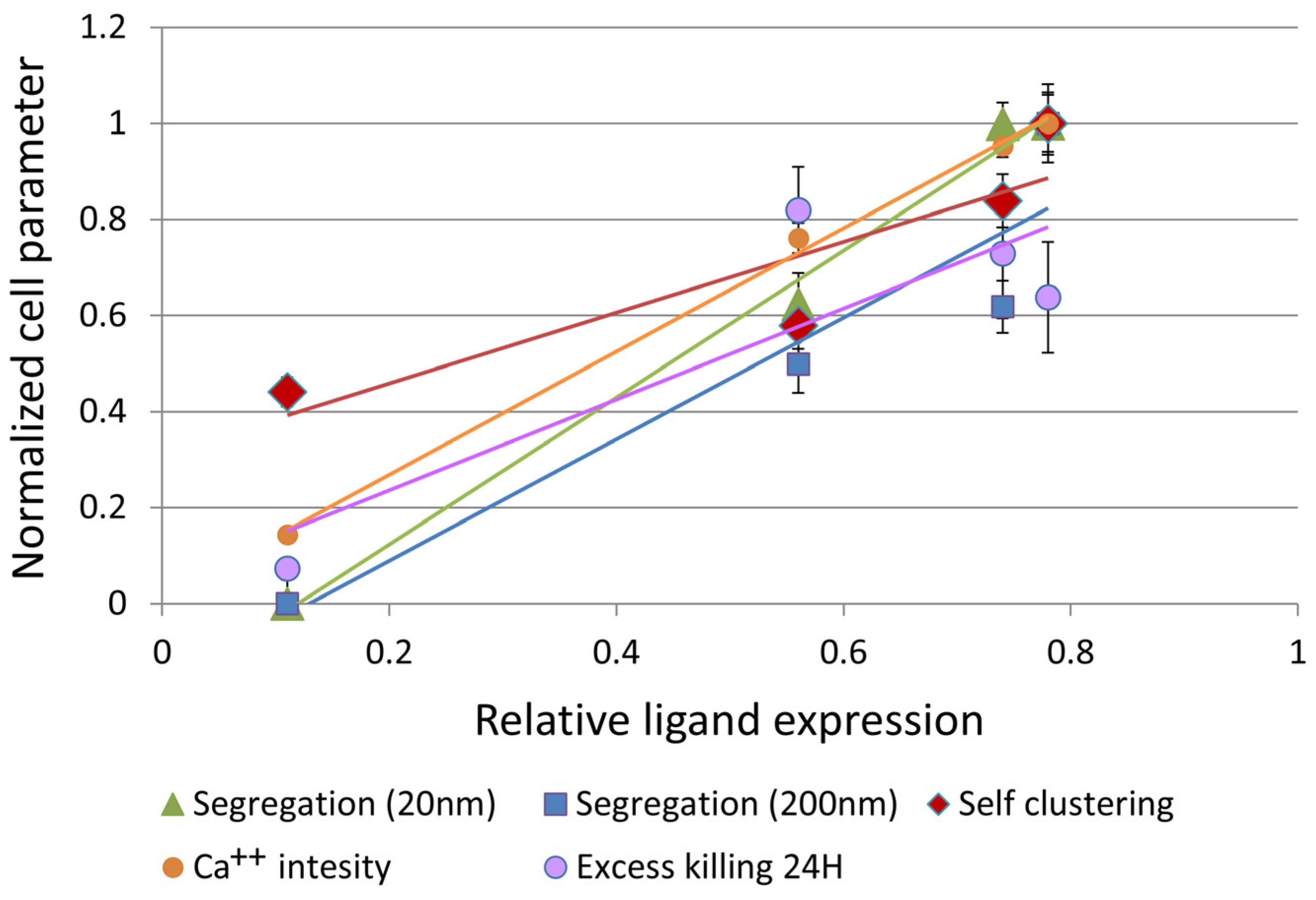
3.4. CAR–Glycoprotein Segregation Correlates with Key Effector Functions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monks, C.R.; Freiberg, B.A.; Kupfer, H.; Sciaky, N.; Kupfer, A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nat. Publ. Gr. 1998, 395, 82–86. [Google Scholar] [CrossRef]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse: A molecular machine controlling T cell activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef]
- Rossy, J.; Owen, D.M.; Williamson, D.J.; Yang, Z.; Gaus, K. Conformational states of the kinase Lck regulate clustering in early T cell signaling. Nat Immunol. Nat. Immunol. 2013, 14, 82–89. [Google Scholar] [CrossRef]
- Sherman, E.; Barr, V.; Manley, S.; Patterson, G.; Balagopalan, L.; Akpan, I.; Regan, C.K.; Merrill, R.K.; Sommers, C.L.; Lippincott-Schwartz, J.; et al. Functional Nanoscale Organization of Signaling Molecules Downstream of the T Cell Antigen Receptor. Immunity 2011, 35, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L.; Bf, L.; Ma, M.; Mb, F.; Jb, H.; Jt, G.; Mm, D. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat. Immunol. 2010, 11, 90–96. [Google Scholar]
- Yokosuka, T.; Sakata-Sogawa, K.; Kobayashi, W.; Hiroshima, M.; Hashimoto-Tane, A.; Tokunaga, M.; Dustin, M.L.; Saito, T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 2005, 6, 1253. [Google Scholar] [CrossRef] [PubMed]
- Campi, G.; Varma, R.; Dustin, M.L. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J. Exp. Med. 2005, 202, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Schamel, W.W.; Arechaga, I.; Risueño, R.M.; van Santen, H.M.; Cabezas, P.; Risco, C.; Valpuesta, J.M.; Alarcón, B. Coexistence of multivalent and monovalent TCRs explains high sensitivity and wide range of response. J. Exp. Med. 2005, 202, 493–503. [Google Scholar] [CrossRef]
- Daviskineti, S.J.; van der Merwe, P.A. The c-segregation model: TCR triggering and beyond. Nat. Immunol. 2006, 7, 803–809. [Google Scholar]
- Chang, V.T.; Fernandes, R.A.; Ganzinger, K.A.; Lee, S.F.; Siebold, C.; McColl, J.; Jönsson, P.; Palayret, M.; Harlos, K.; Coles, C.H.; et al. Initiation of T cell signaling by CD45 segregation at ‘close contacts’. Nat. Immunol. 2016, 17, 574–582. [Google Scholar] [CrossRef]
- Razvag, Y.; Neve-Oz, Y.; Sajman, J.; Reches, M.; Sherman, E. Nanoscale kinetic segregation of TCR and CD45 in engaged microvilli facilitates early T cell activation. Nat. Commun. 2018, 9, 732. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D., Jr.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [PubMed]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef]
- Sajman, J.; Razvag, Y.; Schidorsky, S.; Kinrot, S.; Hermon, K.; Yakovian, O.; Sherman, E. Adhering interacting cells to two opposing coverslips allows super-resolution imaging of cell-cell interfaces. Commun. Biol. 2021, 4, 439. [Google Scholar] [CrossRef]
- Globerson Levin, A.; Rawet Slobodkin, M.; Waks, T.; Horn, G.; Ninio-Many, L.; Deshet Unger, N.; Ohayon, Y.; Suliman, S.; Cohen, Y.; Tartakovsky, B.; et al. Treatment of Multiple Myeloma Using Chimeric Antigen Receptor T Cells with Dual Specificity. Cancer Immunol. Res. 2020, 8, 1485–1495. [Google Scholar] [CrossRef]
- Van de Linde, S.; Löschberger, A.; Klein, T.; Heidbreder, M.; Wolter, S.; Heilemann, M.; Sauer, M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 2011, 6, 991–1009. [Google Scholar] [CrossRef]
- Dempsey, G.T.; Vaughan, J.C.; Chen, K.H.; Bates, M.; Zhuang, X. evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Anal. Nat. methods 2011, 8, 1027–1036. [Google Scholar] [CrossRef]
- Ovesný, M.; Křížek, P.; Borkovec, J.; Svindrych, Z.; Hagen, G.M. ThunderSTORM: A comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 2014, 30, 2389–2390. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef]
- Ester, M.; Kriegel, H.-P.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In Proceedings of the KDD-96: The Second International Conference on Knowledge Discovery and Data Mining, Portland, Oregon, 2–4 August 1996; pp. 226–231. [Google Scholar]
- Nieves, D.J.; Pike, J.A.; Levet, F.; Williamson, D.J.; Baragilly, M.; Oloketuyi, S.; de Marco, A.; Griffié, J.; Sage, D.; Cohen, E.A.K.; et al. A framework for evaluating the performance of SMLM cluster analysis algorithms. Nat. Methods 2023, 20, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Götte, M. Syndecans in inflammation. FASEB J. 2003, 17, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Kind, S.; Merenkow, C.; Büscheck, F.; Möller, K.; Dum, D.; Chirico, V.; Luebke, A.M.; Höflmayer, D.; Hinsch, A.; Jacobsen, F.; et al. Prevalence of Syndecan-1 (CD138) Expression in Different Kinds of Human Tumors and Normal Tissues. Dis. Markers. 2019, 2019, 4928315. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mahendravada, A.; Ballard, B.; Kale, B.; Ramos, C.; West, J.; Maguire, T.; McKay, K.; Lichtman, E.; Tuchman, S.; et al. Safety and efficacy of targeting CD138 with a chimeric antigen receptor for the treatment of multiple myeloma. Oncotarget 2019, 10, 2369–2383. [Google Scholar] [CrossRef]
- Yakovian, O.; Schwarzer, R.; Sajman, J.; Neve-Oz, Y.; Razvag, Y.; Herrmann, A.; Sherman, E. Gp41 dynamically interacts with the TCR in the immune synapse and promotes early T cell activation. Sci. Rep. 2018, 8, 9747. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Riven, I.; Feigelson, S.W.; Kartvelishvily, E.; Tohya, K.; Miyasaka, M.; Alon, R.; Haran, G. Three-dimensional localization of T-cell receptors in relation to microvilli using a combination of superresolution microscopies. Proc. Natl. Acad. Sci. USA 2016, 113, E5916–E5924. [Google Scholar] [CrossRef]
- Jung, Y.; Wen, L.; Altman, A.; Ley, K. CD45 pre-exclusion from the tips of T cell microvilli prior to antigen recognition. Nat. Commun. 2021, 12, 3872. [Google Scholar] [CrossRef]
- Sherman, E. Resolving protein interactions and organization downstream the T cell antigen receptor using single-molecule localization microscopy: A review. Methods Appl. Fluoresc. 2016, 4, 022002. [Google Scholar] [CrossRef]
- Bridgeman, J.S.; Hawkins, R.E.; Bagley, S.; Blaylock, M.; Holland, M.; Gilham, D.E. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 2010, 184, 6938–6949. [Google Scholar] [CrossRef]
- Neve-Oz, Y.; Razvag, Y.; Sajman, J.; Sherman, E. Mechanisms of localized activation of the T cell antigen receptor inside clusters. Biochim. Biophys. Acta-Mol. Cell Res. 2015, 1853, 810–821. [Google Scholar] [CrossRef]
- Pageon, S.V.; Tabarin, T.; Yamamoto, Y.; Ma, Y.; Nicovich, P.R.; Bridgeman, J.S.; Cohnen, A.; Benzing, C.; Gao, Y.; Crowther, M.D.; et al. Functional role of T-cell receptor nanoclusters in signal initiation and antigen discrimination. Proc. Natl. Acad. Sci. USA 2016, 113, E5454–E5463. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhang, X.; Tu, L.; Cao, J.; Hinrichs, C.S.; Su, X. Size-dependent activation of CAR-T cells. Sci. Immunol. 2022, 7, eabl3995. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Segregation (20 nm) * | 0.996 | ||||
| Segregation (200 nm) * | 0.942 | 0.926 | |||
| Self-clustering | 0.898 | 0.911 | 0.944 | ||
| Ca++ influx | 0.999 | 0.991 | 0.935 | 0.876 | |
| Excess killing 24 H (MTT) | 0.862 | 0.832 | 0.724 | 0.557 | 0.887 |
| Ligand | Segregation (20 nm) | Segregation (200 nm) | Self-clustering | Ca++ influx |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sajman, J.; Yakovian, O.; Unger Deshet, N.; Almog, S.; Horn, G.; Waks, T.; Globerson Levin, A.; Sherman, E. Nanoscale CAR Organization at the Immune Synapse Correlates with CAR-T Effector Functions. Cells 2023, 12, 2261. https://doi.org/10.3390/cells12182261
Sajman J, Yakovian O, Unger Deshet N, Almog S, Horn G, Waks T, Globerson Levin A, Sherman E. Nanoscale CAR Organization at the Immune Synapse Correlates with CAR-T Effector Functions. Cells. 2023; 12(18):2261. https://doi.org/10.3390/cells12182261
Chicago/Turabian StyleSajman, Julia, Oren Yakovian, Naamit Unger Deshet, Shaked Almog, Galit Horn, Tova Waks, Anat Globerson Levin, and Eilon Sherman. 2023. "Nanoscale CAR Organization at the Immune Synapse Correlates with CAR-T Effector Functions" Cells 12, no. 18: 2261. https://doi.org/10.3390/cells12182261
APA StyleSajman, J., Yakovian, O., Unger Deshet, N., Almog, S., Horn, G., Waks, T., Globerson Levin, A., & Sherman, E. (2023). Nanoscale CAR Organization at the Immune Synapse Correlates with CAR-T Effector Functions. Cells, 12(18), 2261. https://doi.org/10.3390/cells12182261