

Myeloid-CITED2 Deficiency Exacerbates Diet-Induced Obesity and Pro-Inflammatory Macrophage Response

 and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Resources
2.2. Experimental Mouse Models
2.3. Cell Culture
2.4. RNAseq Analysis
2.5. RNA Extraction, Real-Time Quantitative PCR, and Western Blot
2.6. Transient Transfection and Luciferase Reporter Assay
2.7. Quantitative and Statistical Analysis
3. Results
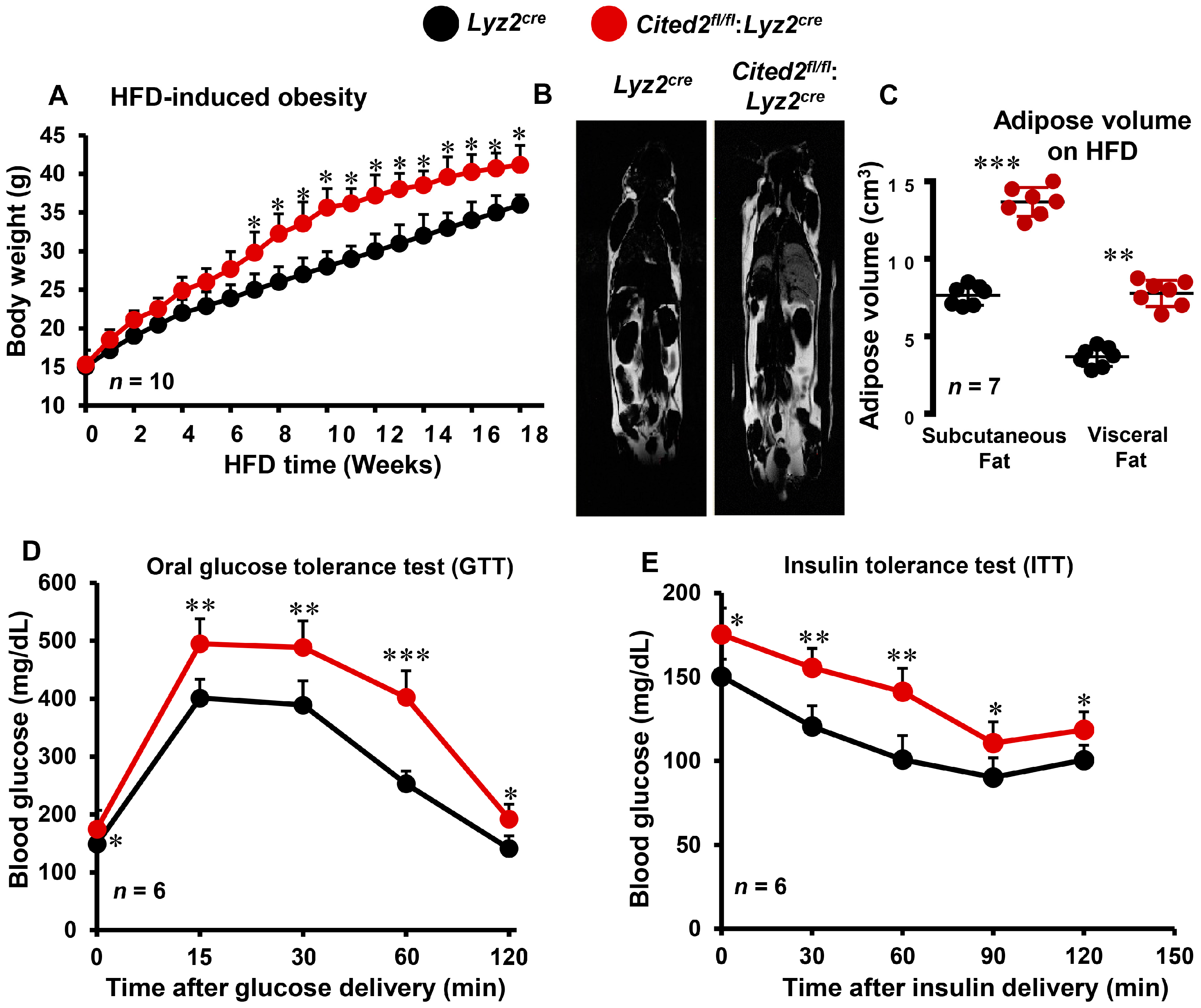
3.1. Myeloid-CITED2 Deficiency Exacerbates HFD-Induced Obesity and Insulin Resistance
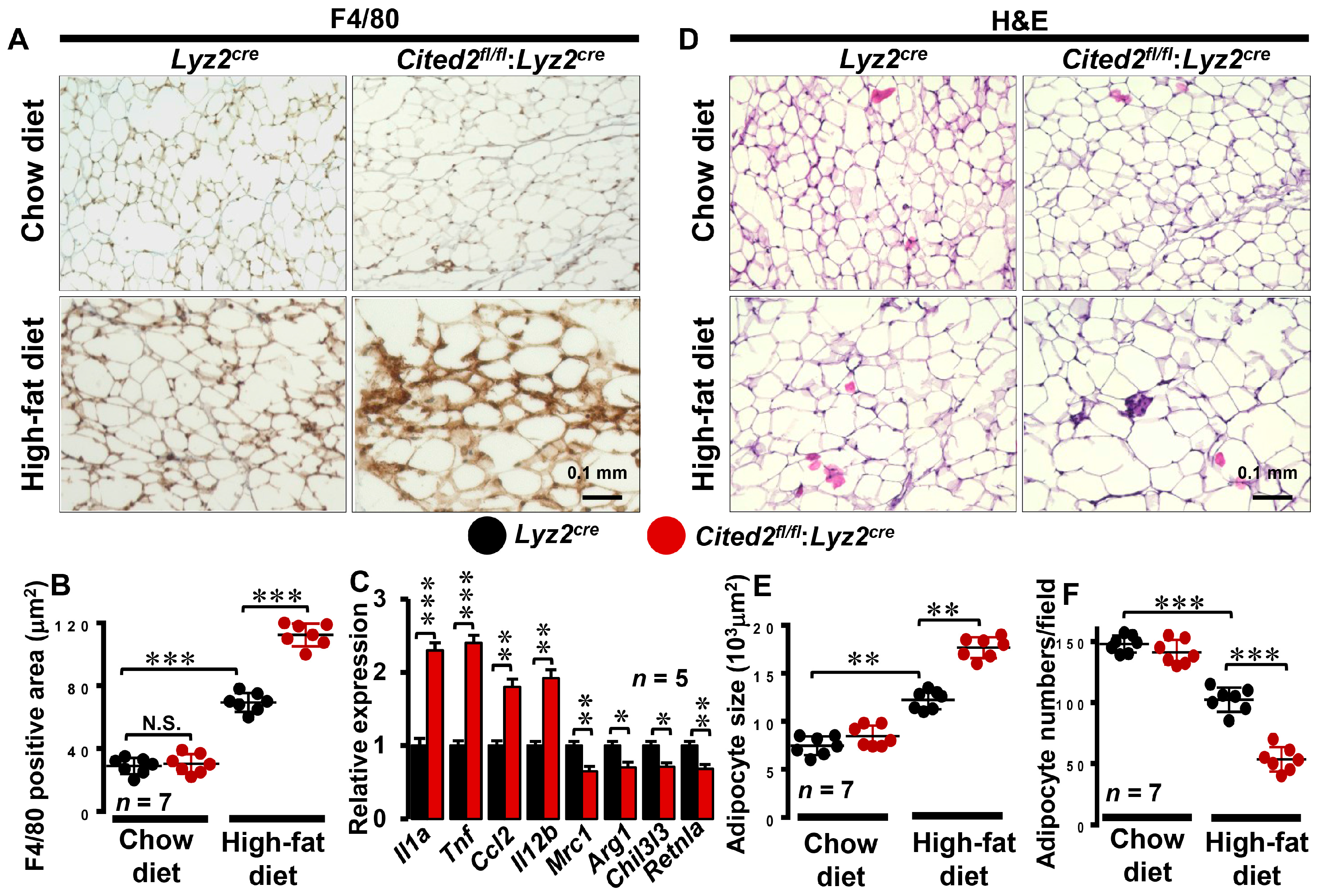
3.2. Myeloid-CITED2 Deficiency Enhances HFD-Induced Adipose Tissue Macrophage Abundance
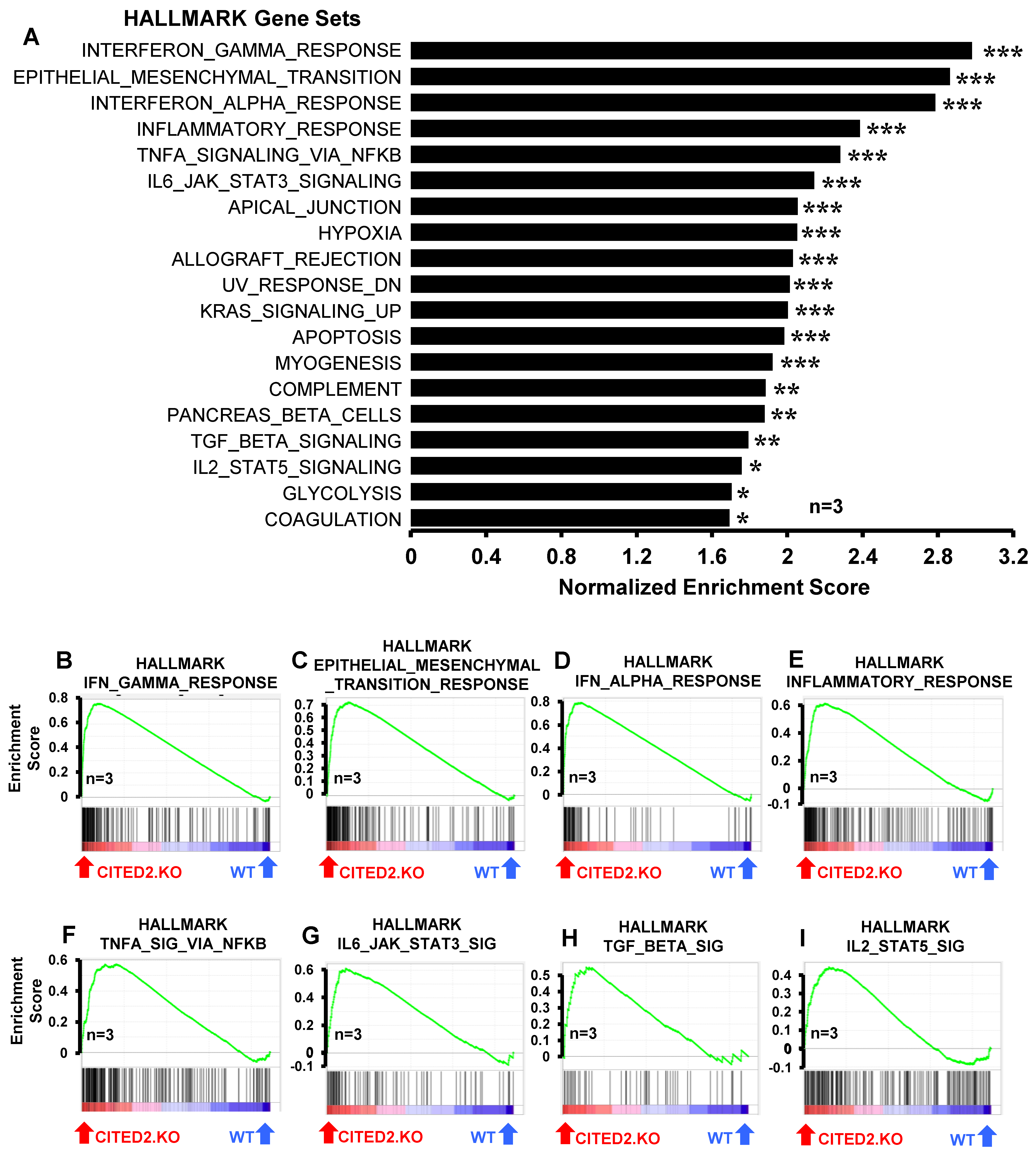
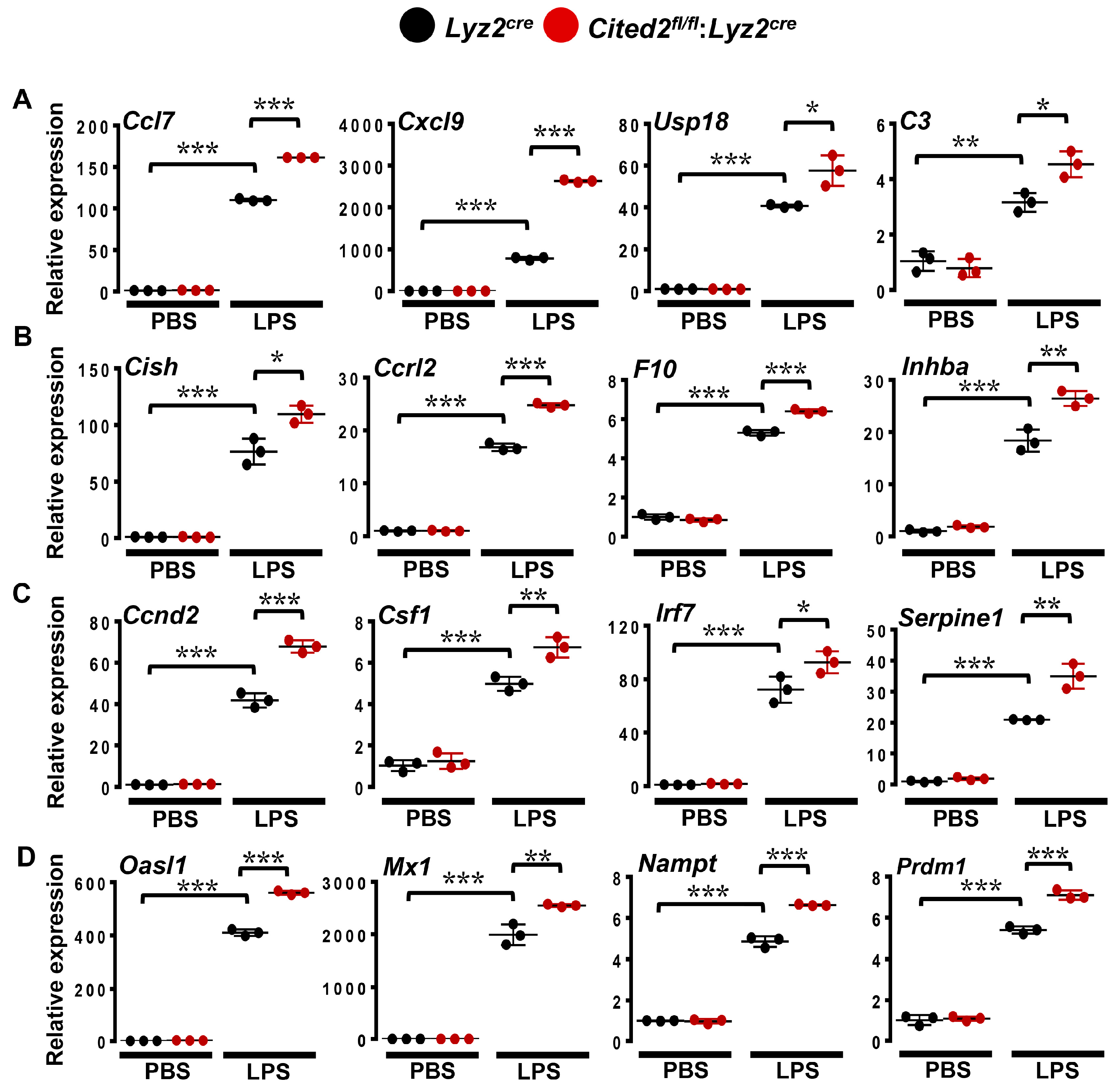
3.3. CITED2 Deficiency Augments Broad Pro-Inflammatory Gene Expression in Macrophages
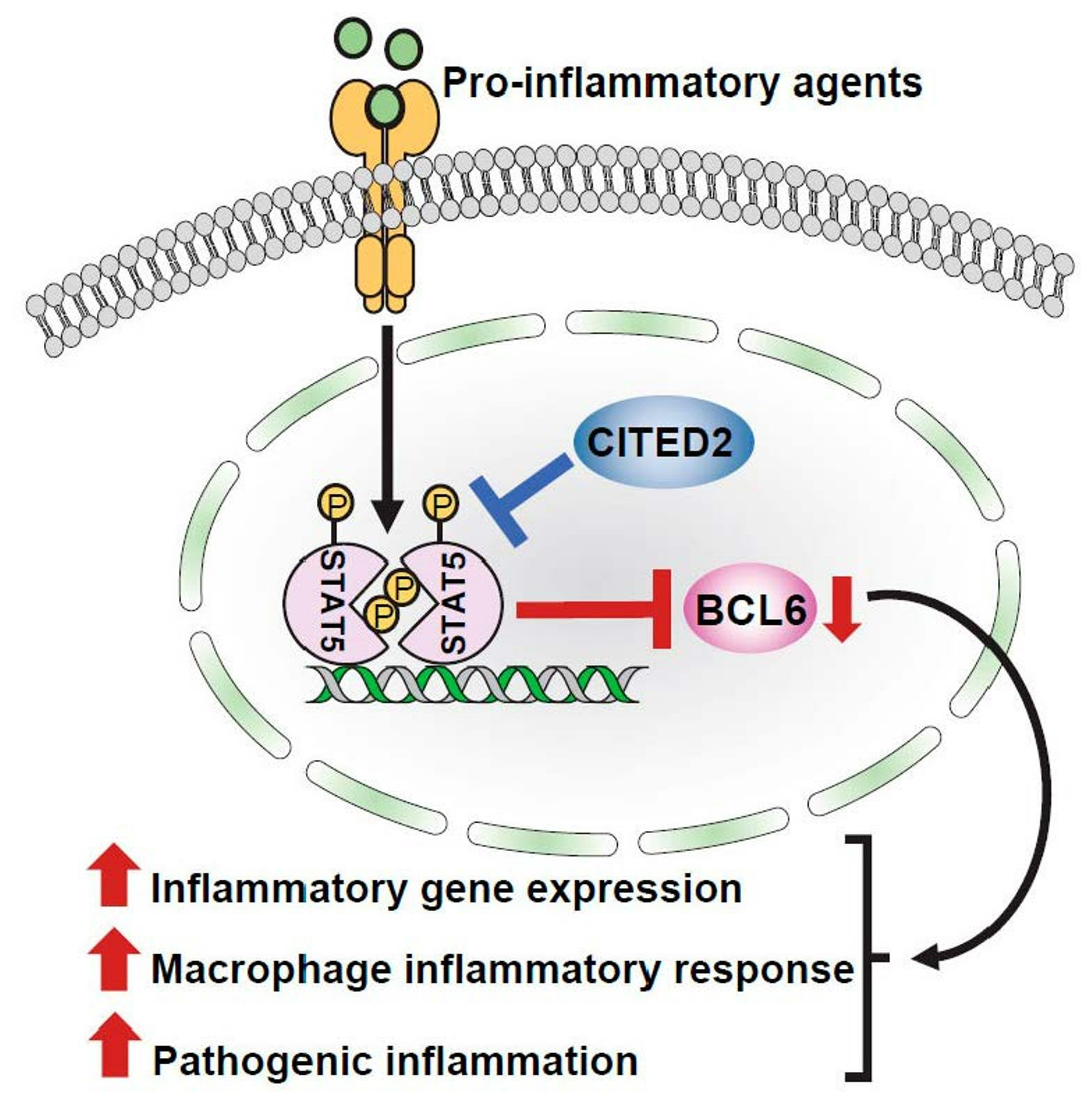
3.4. CITED2 Deficiency Derepresses Classical BCL6 Gene Targets
3.5. CITED2 Promotes BCL6 Expression in Macrophages
3.6. CITED2 Deficiency Boosts STAT5-Mediated BCL6 Repression in Macrophages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Taylor, P.R.; Martinez-Pomares, L.; Stacey, M.; Lin, H.H.; Brown, G.D.; Gordon, S. Macrophage receptors and immune recognition. Annu. Rev. Immunol. 2005, 23, 901–944. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Heilbronn, L.K.; Campbell, L.V. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr. Pharm. Des. 2008, 14, 1225–1230. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Rohm, T.V.; Meier, D.T.; Olefsky, J.M.; Donath, M.Y. Inflammation in obesity, diabetes, and related disorders. Immunity 2022, 55, 31–55. [Google Scholar] [CrossRef]
- Dent, A.L.; Shaffer, A.L.; Yu, X.; Allman, D.; Staudt, L.M. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science 1997, 276, 589–592. [Google Scholar] [CrossRef]
- Ye, B.H.; Rao, P.H.; Chaganti, R.S.; Dalla-Favera, R. Cloning of bcl-6, the locus involved in chromosome translocations affecting band 3q27 in B-cell lymphoma. Cancer Res. 1993, 53, 2732–2735. [Google Scholar]
- Barish, G.D.; Yu, R.T.; Karunasiri, M.; Ocampo, C.B.; Dixon, J.; Benner, C.; Dent, A.L.; Tangirala, R.K.; Evans, R.M. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 2010, 24, 2760–2765. [Google Scholar] [CrossRef]
- Basso, K.; Dalla-Favera, R. BCL6: Master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv. Immunol. 2010, 105, 193–210. [Google Scholar] [PubMed]
- Barish, G.D.; Yu, R.T.; Karunasiri, M.S.; Becerra, D.; Kim, J.; Tseng, T.W.; Tai, L.J.; Leblanc, M.; Diehl, C.; Cerchietti, L.; et al. The Bcl6-SMRT/NCoR cistrome represses inflammation to attenuate atherosclerosis. Cell Metab. 2012, 15, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Zhang, C.; Huang, K.; Zhao, J.; Le, S.; Jiang, L.; Liu, H.; Yang, P.; Xiao, X.; et al. B-cell lymphoma 6 alleviates nonalcoholic fatty liver disease in mice through suppression of fatty acid transporter CD36. Cell Death Dis. 2022, 13, 359. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.B.; Zhu, Y.X.; Yin, T.; Sledge, G.; Yang, Y.C. MRG1, the product of a melanocyte-specific gene related gene, is a cytokine-inducible transcription factor with transformation activity. Proc. Natl. Acad. Sci. USA 1998, 95, 13555–13560. [Google Scholar] [CrossRef]
- Dunwoodie, S.L.; Rodriguez, T.A.; Beddington, R.S. Msg1 and Mrg1, founding members of a gene family, show distinct patterns of gene expression during mouse embryogenesis. Mech. Dev. 1998, 72, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Lopes Floro, K.; Artap, S.T.; Preis, J.I.; Fatkin, D.; Chapman, G.; Furtado, M.B.; Harvey, R.P.; Hamada, H.; Sparrow, D.B.; Dunwoodie, S.L. Loss of Cited2 causes congenital heart disease by perturbing left-right patterning of the body axis. Hum. Mol. Genet. 2011, 20, 1097–1110. [Google Scholar] [CrossRef]
- Kim, G.D.; Das, R.; Rao, X.; Zhong, J.; Deiuliis, J.A.; Ramirez-Bergeron, D.L.; Rajagopalan, S.; Mahabeleshwar, G.H. CITED2 Restrains Proinflammatory Macrophage Activation and Response. Mol. Cell. Biol. 2018, 38, e00452-17. [Google Scholar] [CrossRef]
- Zafar, A.; Pong Ng, H.; Diamond-Zaluski, R.; Kim, G.D.; Ricky Chan, E.; Dunwoodie, S.L.; Smith, J.D.; Mahabeleshwar, G.H. CITED2 inhibits STAT1-IRF1 signaling and atherogenesis. FASEB J. 2021, 35, e21833. [Google Scholar] [CrossRef]
- Pong Ng, H.; Kim, G.D.; Ricky Chan, E.; Dunwoodie, S.L.; Mahabeleshwar, G.H. CITED2 limits pathogenic inflammatory gene programs in myeloid cells. FASEB J. 2020, 34, 12100–12113. [Google Scholar] [CrossRef]
- Sakai, M.; Matsumoto, M.; Tujimura, T.; Yongheng, C.; Noguchi, T.; Inagaki, K.; Inoue, H.; Hosooka, T.; Takazawa, K.; Kido, Y.; et al. CITED2 links hormonal signaling to PGC-1α acetylation in the regulation of gluconeogenesis. Nat. Med. 2012, 18, 612–617. [Google Scholar] [CrossRef]
- Preis, J.I.; Wise, N.; Solloway, M.J.; Harvey, R.P.; Sparrow, D.B.; Dunwoodie, S.L. Generation of conditional Cited2 null alleles. Genesis 2006, 44, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chawla, A.; Nguyen, K.D.; Goh, Y.P. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 2011, 11, 738–749. [Google Scholar]
- Glass, C.K.; Olefsky, J.M. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012, 15, 635–645. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity 2017, 47, 406–420. [Google Scholar]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef]
- Walker, S.R.; Nelson, E.A.; Yeh, J.E.; Pinello, L.; Yuan, G.C.; Frank, D.A. STAT5 outcompetes STAT3 to regulate the expression of the oncogenic transcriptional modulator BCL6. Mol. Cell. Biol. 2013, 33, 2879–2890. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T.; Natoli, G. Transcriptional control of inflammatory responses. Cold Spring Harb. Perspect. Biol. 2014, 6, a016261. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Hatzi, K.; Melnick, A. Lineage-specific functions of Bcl-6 in immunity and inflammation are mediated by distinct biochemical mechanisms. Nat. Immunol. 2013, 14, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Kunz, H.E.; Hart, C.R.; Gries, K.J.; Parvizi, M.; Laurenti, M.; Dalla Man, C.; Moore, N.; Zhang, X.; Ryan, Z.; Polley, E.C.; et al. Adipose tissue macrophage populations and inflammation are associated with systemic inflammation and insulin resistance in obesity. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E105–E121. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef]
- Ricardo-Gonzalez, R.R.; Red Eagle, A.; Odegaard, J.I.; Jouihan, H.; Morel, C.R.; Heredia, J.E.; Mukundan, L.; Wu, D.; Locksley, R.M.; Chawla, A. IL-4/STAT6 immune axis regulates peripheral nutrient metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2010, 107, 22617–22622. [Google Scholar] [CrossRef]
- Li, A.C.; Binder, C.J.; Gutierrez, A.; Brown, K.K.; Plotkin, C.R.; Pattison, J.W.; Valledor, A.F.; Davis, R.A.; Willson, T.M.; Witztum, J.L.; et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J. Clin. Investig. 2004, 114, 1564–1576. [Google Scholar] [CrossRef]
- Bunting, K.L.; Melnick, A.M. New effector functions and regulatory mechanisms of BCL6 in normal and malignant lymphocytes. Curr. Opin. Immunol. 2013, 25, 339–346. [Google Scholar] [CrossRef]
- Pfitzner, E.; Jähne, R.; Wissler, M.; Stoecklin, E.; Groner, B. p300/CREB-binding protein enhances the prolactin-mediated transcriptional induction through direct interaction with the transactivation domain of Stat5, but does not participate in the Stat5-mediated suppression of the glucocorticoid response. Mol. Endocrinol. 1998, 12, 1582–1593. [Google Scholar]
- De Guzman, R.N.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Interaction of the TAZ1 domain of the CREB-binding protein with the activation domain of CITED2: Regulation by competition between intrinsically unstructured ligands for non-identical binding sites. J. Biol. Chem. 2004, 279, 3042–3049. [Google Scholar] [CrossRef] [PubMed]
- Fahrenkamp, D.; de Leur, H.S.; Küster, A.; Chatain, N.; Müller-Newen, G. Src family kinases interfere with dimerization of STAT5A through a phosphotyrosine-SH2 domain interaction. Cell Commun. Signal. CCS 2015, 13, 10. [Google Scholar] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zafar, A.; Ng, H.P.; Chan, E.R.; Dunwoodie, S.L.; Mahabeleshwar, G.H. Myeloid-CITED2 Deficiency Exacerbates Diet-Induced Obesity and Pro-Inflammatory Macrophage Response. Cells 2023, 12, 2136. https://doi.org/10.3390/cells12172136
Zafar A, Ng HP, Chan ER, Dunwoodie SL, Mahabeleshwar GH. Myeloid-CITED2 Deficiency Exacerbates Diet-Induced Obesity and Pro-Inflammatory Macrophage Response. Cells. 2023; 12(17):2136. https://doi.org/10.3390/cells12172136
Chicago/Turabian StyleZafar, Atif, Hang Pong Ng, E. Ricky Chan, Sally L. Dunwoodie, and Ganapati H. Mahabeleshwar. 2023. "Myeloid-CITED2 Deficiency Exacerbates Diet-Induced Obesity and Pro-Inflammatory Macrophage Response" Cells 12, no. 17: 2136. https://doi.org/10.3390/cells12172136
APA StyleZafar, A., Ng, H. P., Chan, E. R., Dunwoodie, S. L., & Mahabeleshwar, G. H. (2023). Myeloid-CITED2 Deficiency Exacerbates Diet-Induced Obesity and Pro-Inflammatory Macrophage Response. Cells, 12(17), 2136. https://doi.org/10.3390/cells12172136