
Development of a Novel CD26-Targeted Chimeric Antigen Receptor T-Cell Therapy for CD26-Expressing T-Cell Malignancies

 , , , , , ,
, , , , , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Patient Samples
2.3. Ethics Approval and Consent to Participate
2.4. Luciferase-Expression Vector
2.5. Flow Cytometry
2.6. Immunohistochemical Staining
2.7. Construction of CD26 CAR
2.8. Transduction and Expression of CD26 CAR
2.9. Activation of CD26 CAR-T-Cells
2.10. In Vitro Anti-Tumor Activity of CD26 CAR-T-Cells
2.11. In Vivo Anti-Tumor Activity of CD26 CAR-T-Cells
2.12. In Vitro Anti-Tumor Activity of CD26 CAR-T-Cells against Patient Samples
2.13. Statistical Analysis
3. Results
3.1. Transduction and Expression of CD26 CAR
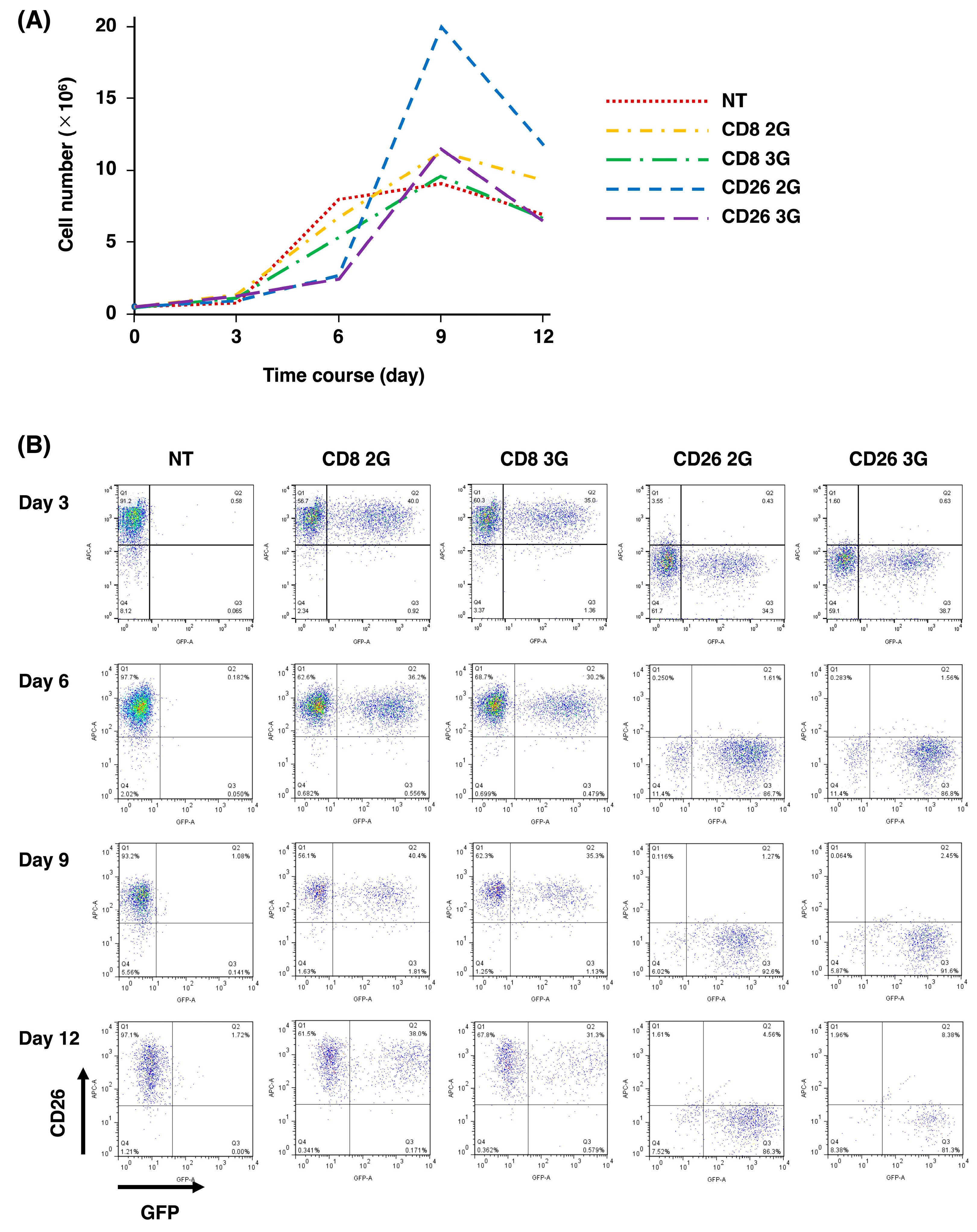
3.2. Expansion of CD26 CAR-T-Cells
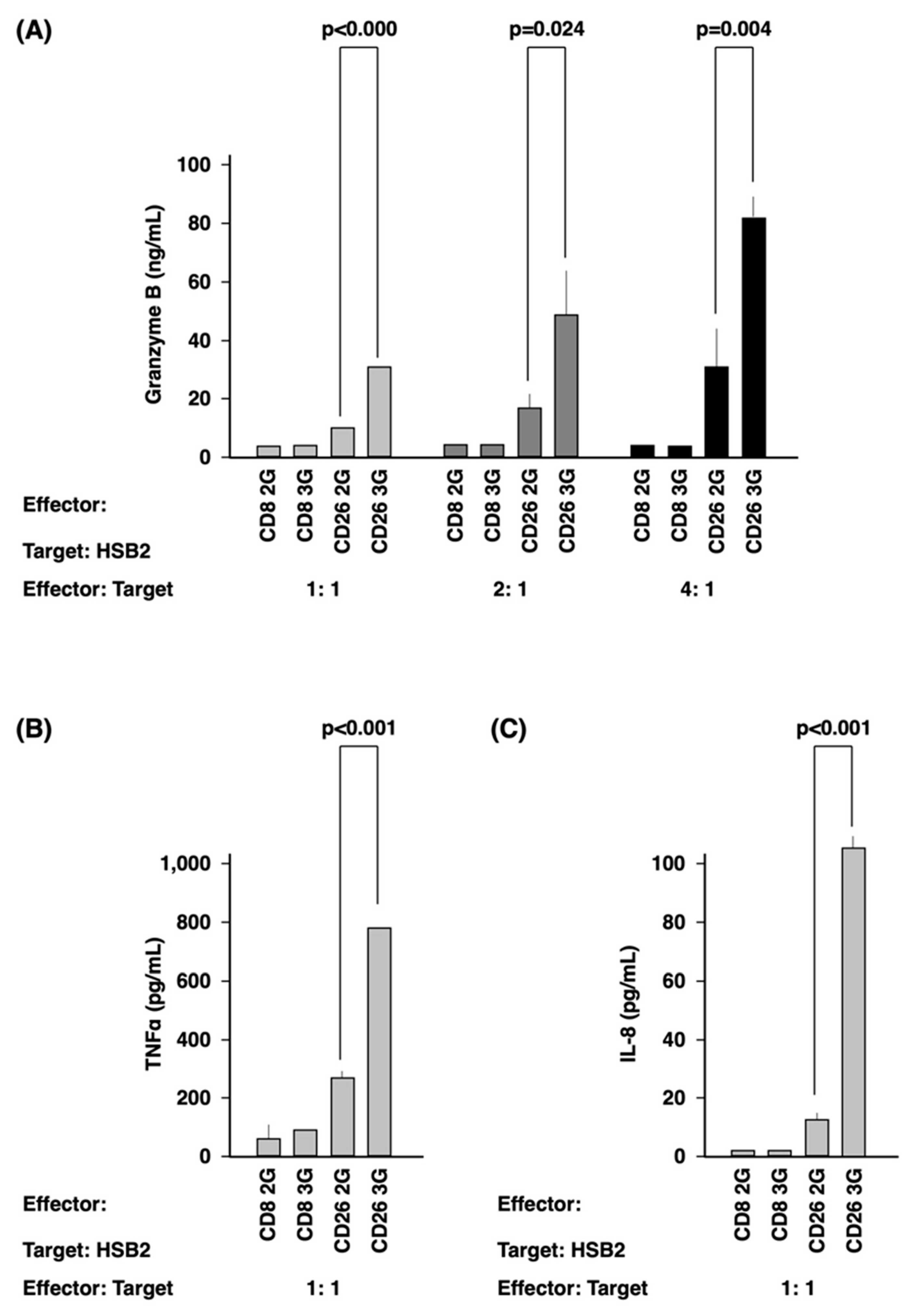
3.3. Activation of CD26 CAR-T-Cells
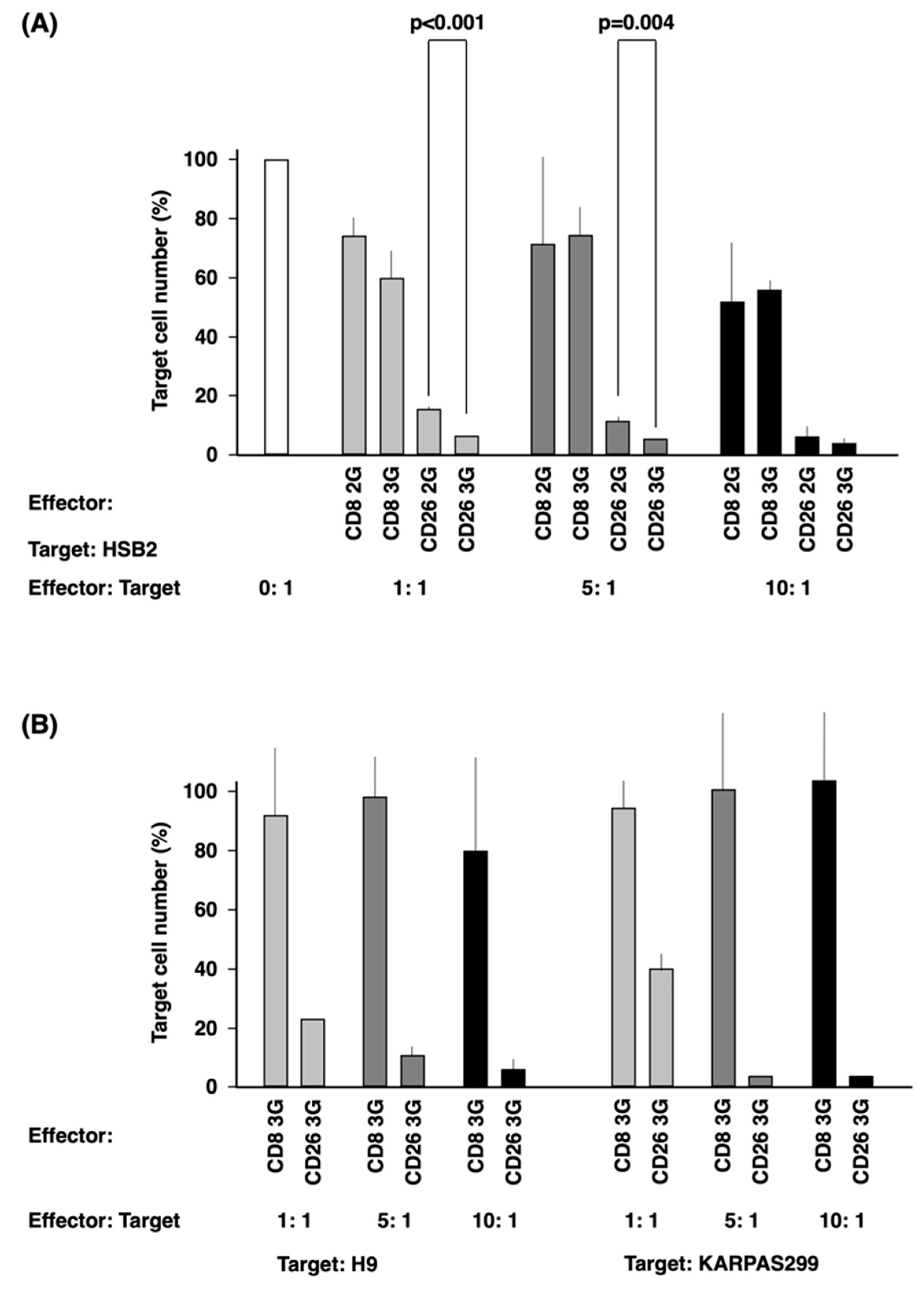
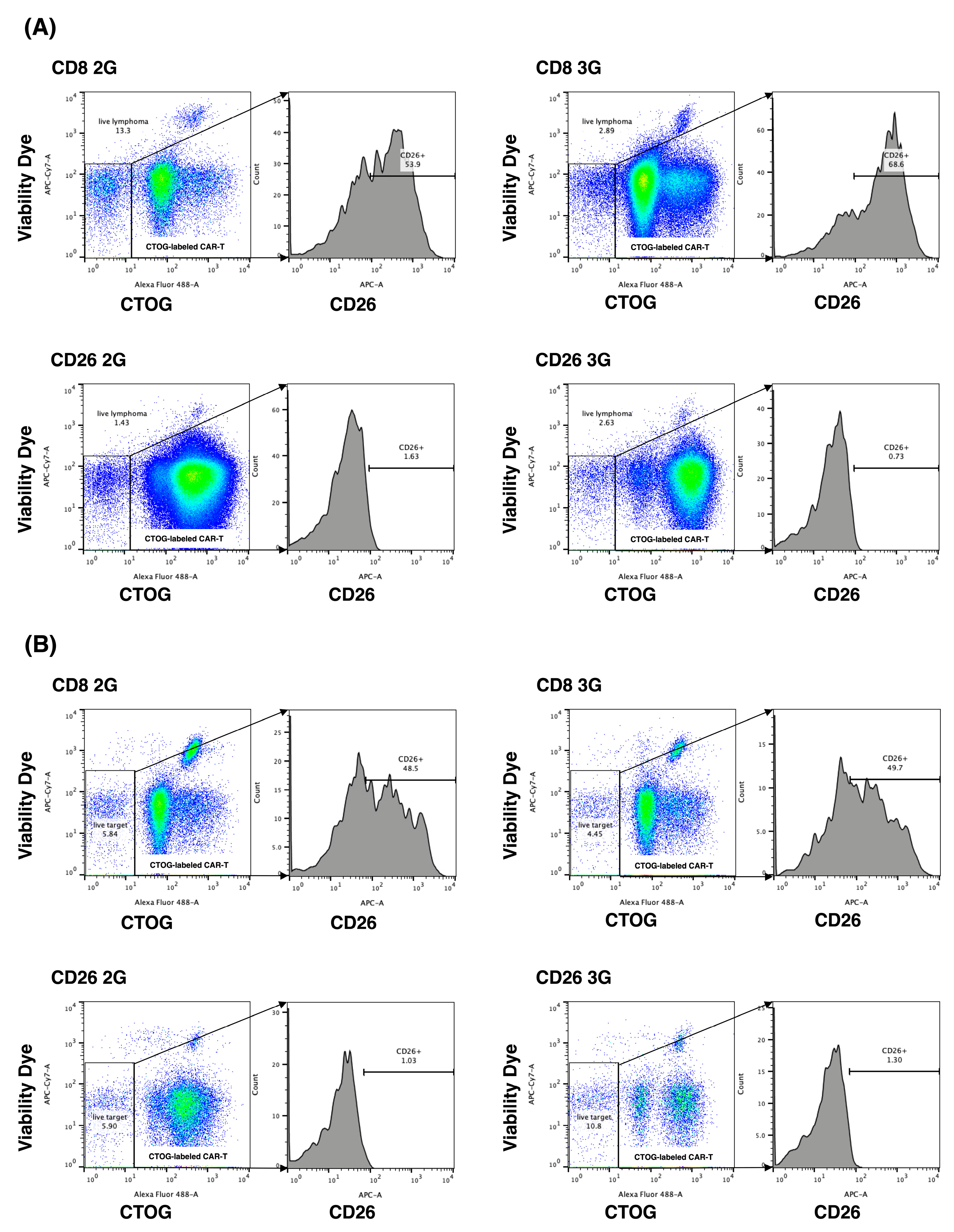
3.4. In Vitro Anti-Tumor Activity of CD26 CAR-T-Cells
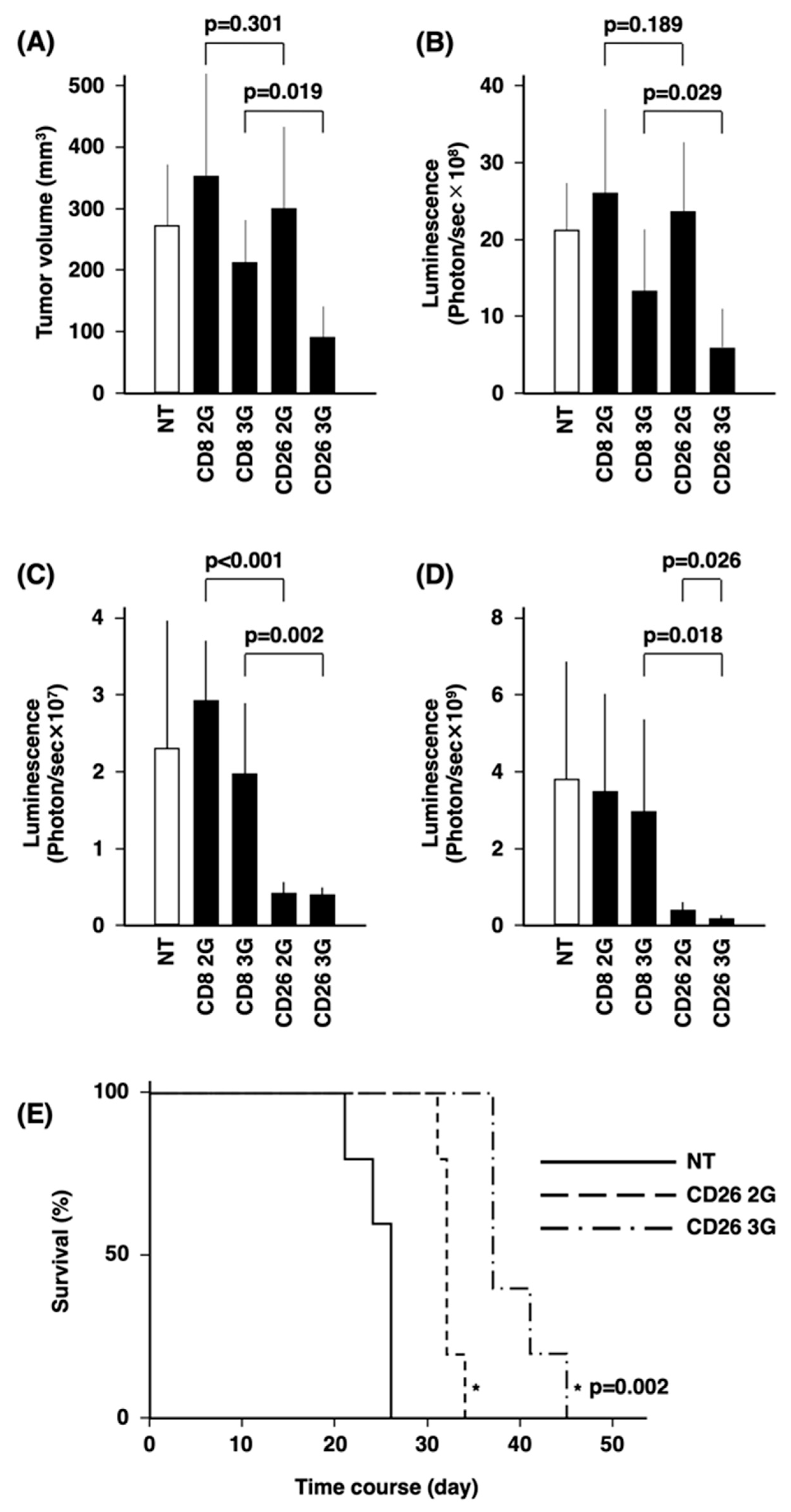
3.5. In Vivo Anti-Tumor Activity of CD26 CAR-T-Cells
3.6. Anti-Tumor Activity of CD26 CAR-T-Cells against Patient Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ohnuma, K.; Hosono, O.; Dang, N.H.; Morimoto, C. Dipeptidyl Peptidase in Autoimmune Pathophysiology. Adv. Clin. Chem. 2011, 53, 51–84. [Google Scholar]
- Thompson, M.; Ohnuma, K.; Abe, M.; Morimoto, C.; Dang, N. CD26/Dipeptidyl Peptidase IV as a Novel Therapeutic Target for Cancer and Immune Disorders. Mini-Rev. Med. Chem. 2007, 7, 253–273. [Google Scholar] [CrossRef] [PubMed]
- Cordero, O.J.; Rafael-Vidal, C.; Varela-Calviño, R.; Calviño-Sampedro, C.; Malvar-Fernández, B.; García, S.; Viñuela, J.E.; Pego-Reigosa, J.M. Distinctive CD26 Expression on CD4 T-Cell Subsets. Biomolecules 2021, 11, 1446. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, K.; Hatano, R.; Komiya, E.; Otsuka, H.; Itoh, T.; Iwao, N.; Kaneko, Y.; Yamada, T.; Dang, N.H.; Morimoto, C. A Novel Role for CD26/Dipeptidyl Peptidase IV as a Therapeutic Target. Front. Biosci. 2018, 23, 1754–1779. [Google Scholar] [CrossRef] [PubMed]
- Havre, P.A.; Abe, M.; Urasaki, Y.; Ohnuma, K.; Morimoto, C.; Dang, N.H. The Role of CD26/Dipeptidyl Peptidase IV in Cancer. Front. Biosci. 2008, 13, 1345–1351. [Google Scholar] [CrossRef]
- Inamoto, T.; Yamochi, T.; Ohnuma, K.; Iwata, S.; Kina, S.; Inamoto, S.; Tachibana, M.; Katsuoka, Y.; Dang, N.H.; Morimoto, C. Anti-CD26 Monoclonal Antibody-Mediated G1-S Arrest of Human Renal Clear Cell Carcinoma Caki-2 Is Associated with Retinoblastoma Substrate Dephosphorylation, Cyclin-Dependent Kinase 2 Reduction, P27kip1 Enhancement, and Disruption of Binding to the Extracellular Matrix. Clin. Cancer Res. 2006, 12, 3470–3477. [Google Scholar] [PubMed]
- Inamoto, T.; Yamada, T.; Ohnuma, K.; Kina, S.; Takahashi, N.; Yamochi, T.; Inamoto, S.; Katsuoka, Y.; Hosono, O.; Tanaka, H.; et al. Humanized Anti-CD26 Monoclonal Antibody as a Treatment for Malignant Mesothelioma Tumors. Clin. Cancer Res. 2007, 13, 4191–4200. [Google Scholar] [CrossRef]
- Angevin, E.; Isambert, N.; Trillet-Lenoir, V.; You, B.; Alexandre, J.; Zalcman, G.; Vielh, P.; Farace, F.; Valleix, F.; Podoll, T.; et al. First-in-Human Phase 1 of YS110, a Monoclonal Antibody Directed against CD26 in Advanced CD26-Expressing Cancers. Br. J. Cancer 2017, 116, 1126–1137. [Google Scholar] [CrossRef]
- Nakagawa, K.; Kijima, T.; Okada, M.; Morise, M.; Kato, M.; Hirano, K.; Fujimoto, N.; Takenoyama, M.; Yokouchi, H.; Ohe, Y.; et al. Phase 2 Study of YS110, a Recombinant Humanized Anti-CD26 Monoclonal Antibody, in Japanese Patients With Advanced Malignant Pleural Mesothelioma. JTO Clin. Res. Rep. 2021, 2, 100178. [Google Scholar] [CrossRef]
- Takeda, M.; Ohe, Y.; Horinouchi, H.; Hida, T.; Shimizu, J.; Seto, T.; Nosaki, K.; Kishimoto, T.; Miyashita, I.; Yamada, M.; et al. Phase I Study of YS110, a Recombinant Humanized Monoclonal Antibody to CD26, in Japanese Patients with Advanced Malignant Pleural Mesothelioma. Lung Cancer 2019, 137, 64–70. [Google Scholar] [CrossRef]
- Sato, T.; Yamochi, T.; Yamochi, T.; Aytac, U.; Ohnuma, K.; McKee, K.S.; Morimoto, C.; Dang, N.H. CD26 Regulates P38 Mitogen-Activated Protein Kinase-Dependent Phosphorylation of Integrin Β1, Adhesion to Extracellular Matrix, and Tumorigenicity of T-Anaplastic Large Cell Lymphoma Karpas 299. Cancer Res. 2005, 65, 6950–6956. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Aytac, U.; Stephens, L.C.; Ohnuma, K.; Mills, G.B.; McKee, K.S.; Neumann, C.; LaPushin, R.; Cabanillas, F.; Abbruzzese, J.L.; et al. In Vitro and in Vivo Antitumor Effect of the Anti-CD26 Monoclonal Antibody 1F7 on Human CD30+ Anaplastic Large Cell T-Cell Lymphoma Karpas 299. Clin. Cancer Res. 2001, 7, 2031–2040. [Google Scholar] [PubMed]
- Escalón, M.P.; Liu, N.S.; Yang, Y.; Hess, M.; Walker, P.L.; Smith, T.L.; Dang, N.H. Prognostic Factors and Treatment of Patients with T-Cell Non-Hodgkin Lymphoma: The, M.D. Anderson Cancer Center Experience. Cancer 2005, 103, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Tam, C.S.; Borchmann, P.; Worel, N.; McGuirk, J.P.; Holte, H.; Waller, E.K.; Jaglowski, S.; Bishop, M.R.; Damon, L.E.; et al. Long-Term Clinical Outcomes of Tisagenlecleucel in Patients with Relapsed or Refractory Aggressive B-Cell Lymphomas (JULIET): A Multicentre, Open-Label, Single-Arm, Phase 2 Study. Lancet Oncol. 2021, 22, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Flugel, C.L.; Majzner, R.G.; Krenciute, G.; Dotti, G.; Riddell, S.R.; Wagner, D.L.; Abou-el-Enein, M. Overcoming On-Target, off-Tumour Toxicity of CAR T Cell Therapy for Solid Tumours. Nat. Rev. Clin. Oncol. 2023, 20, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Safarzadeh Kozani, P.; Safarzadeh Kozani, P.; Rahbarizadeh, F. CAR-T Cell Therapy in T-Cell Malignancies: Is Success a Low-Hanging Fruit? Stem Cell Res. Ther. 2021, 12, 527. [Google Scholar] [CrossRef]
- Fleischer, L.C.; Spencer, H.T.; Raikar, S.S. Targeting T Cell Malignancies Using CAR-Based Immunotherapy: Challenges and Potential Solutions. J. Hematol. Oncol. 2019, 12, 141. [Google Scholar] [CrossRef]
- Safarzadeh Kozani, P.; Safarzadeh Kozani, P.; Rahbarizadeh, F. Optimizing the Clinical Impact of CAR-T Cell Therapy in B-Cell Acute Lymphoblastic Leukemia: Looking Back While Moving Forward. Front. Immunol. 2021, 12, 765097. [Google Scholar] [CrossRef]
- Mohanty, R.; Chowdhury, C.R.; Arega, S.; Sen, P.; Ganguly, P.; Ganguly, N. CAR T Cell Therapy: A New Era for Cancer Treatment (Review). Oncol. Rep. 2019, 42, 2183–2195. [Google Scholar] [CrossRef]
- Rinaldi, I.; Muthalib, A.; Edina, B.C.; Wiyono, L.; Winston, K. Role of Anti-B-Cell Maturation Antigen (BCMA) in the Management of Multiple Myeloma. Cancers 2022, 14, 3507. [Google Scholar] [CrossRef]
- Barros, L.R.C.; Couto, S.C.F.; da Silva Santurio, D.; Paixão, E.A.; Cardoso, F.; da Silva, V.J.; Klinger, P.; Ribeiro, P.d.A.C.; Rós, F.A.; Oliveira, T.G.M.; et al. Systematic Review of Available CAR-T Cell Trials around the World. Cancers 2022, 14, 2667. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef] [PubMed]
- Aytac, U.; Claret, F.X.; Ho, L.; Sato, K.; Ohnuma, K.; Mills, G.B.; Cabanillas, F.; Morimoto, C.; Dang, N.H. Expression of CD26 and Its Associated Dipeptidyl Peptidase IV Enzyme Activity Enhances Sensitivity to Doxorubicin-Induced Cell Cycle Arrest at the G(2)/M Checkpoint. Cancer Res. 2001, 61, 7204–7210. [Google Scholar]
- Aytac, U.; Sato, K.; Yamochi, T.; Yamochi, T.; Ohnuma, K.; Mills, G.B.; Morimoto, C.; Dang, N.H. Effect of CD26/Dipeptidyl Peptidase IV on Jurkat Sensitivity to G2/M Arrest Induced by Topoisomerase II Inhibitors. Br. J. Cancer 2003, 88, 455–462. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hatano, R.; Yamada, T.; Madokoro, H.; Otsuka, H.; Komiya, E.; Itoh, T.; Narita, Y.; Iwata, S.; Yamazaki, H.; Matsuoka, S.; et al. Development of Novel Monoclonal Antibodies with Specific Binding Affinity for Denatured Human CD26 in Formalin-Fixed Paraffinembedded and Decalcified Specimens. PLoS ONE 2019, 14, e0218330. [Google Scholar] [CrossRef] [PubMed]
- Fedchenko, N.; Reifenrath, J. Different Approaches for Interpretation and Reporting of Immunohistochemistry Analysis Results in the Bone Tissue—A Review. Diagn. Pathol. 2014, 9, 221. [Google Scholar] [CrossRef]
- Kobayashi, E.; Kishi, H.; Ozawa, T.; Hamana, H.; Nakagawa, H.; Jin, A.; Lin, Z.; Muraguchi, A. A Chimeric Antigen Receptor for TRAIL-Receptor 1 Induces Apoptosis in Various Types of Tumor Cells. Biochem. Biophys. Res. Commun. 2014, 453, 798–803. [Google Scholar] [CrossRef]
- Ono, K.; Sato, T.; Iyama, S.; Tatekoshi, A.; Hashimoto, A.; Kamihara, Y.; Horiguchi, H.; Kikuchi, S.; Kawano, Y.; Takada, K.; et al. A Novel Strategy Inducing Autophagic Cell Death in Burkitt’s Lymphoma Cells with Anti-CD19-Targeted Liposomal Rapamycin. Blood Cancer J. 2014, 4, e180. [Google Scholar] [CrossRef]
- Sato, T.; Tatekoshi, A.; Takada, K.; Iyama, S.; Kamihara, Y.; Jawaid, P.; Rehman, M.U.; Noguchi, K.; Kondo, T.; Kajikawa, S.; et al. DPP8 Is a Novel Therapeutic Target for Multiple Myeloma. Sci. Rep. 2019, 9, 18094. [Google Scholar] [CrossRef]
- Mamonkin, M.; Mukherjee, M.; Srinivasan, M.; Sharma, S.; Gomes-Silva, D.; Mo, F.; Krenciute, G.; Orange, J.S.; Brenner, M.K. Reversible Transgene Expression Reduces Fratricide and Permits 4-1BB Costimulation of CAR T Cells Directed to T-Cell Malignancies. Cancer Immunol. Res. 2018, 6, 47–58. [Google Scholar] [CrossRef]
- Zhou, S.; Zhu, X.; Shen, N.; Li, Q.; Wang, N.; You, Y.; Zhong, Z.; Cheng, F.; Zou, P.; Zhu, X. T Cells Expressing CD26-Specific Chimeric Antigen Receptors Exhibit Extensive Self-Antigen-Driven Fratricide. Immunopharmacol. Immunotoxicol. 2019, 41, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Mamonkin, M.; Rouce, R.H.; Tashiro, H.; Brenner, M.K. A T-Cell-Directed Chimeric Antigen Receptor for the Selective Treatment of T-Cell Malignancies. Blood 2015, 126, 983–992. [Google Scholar] [CrossRef]
- Flavell, D.J.; Boehm, D.A.; Okayama, K.; Kohler, J.A.; Flavell, S.N.U. Therapy of Human T-cell Acute Lymphoblastic Leukaemia in Severe Combined Immunodeficient Mice with Two Different Anti-CD7-saporin Immunotoxins Containing Hindered or Non-hindered Disulphide Cross-linkers. Int. J. Cancer 1994, 58, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Enblad, G.; Karlsson, H.; Gammelgård, G.; Wenthe, J.; Lövgren, T.; Amini, R.M.; Wikstrom, K.I.; Essand, M.; Savoldo, B.; Hallböök, H.; et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin. Cancer Res. 2018, 24, 6185–6194. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jensen, M.; Lin, Y.; Sui, X.; Chen, E.; Lindgren, C.G.; Till, B.; Raubitschek, A.; Forman, S.J.; Qian, X.; et al. Optimizing Adoptive Polyclonal T Cell Immunotherapy of Lymphomas, Using a Chimeric T Cell Receptor Possessing CD28 and CD137 Costimulatory Domains. Hum. Gene Ther. 2007, 18, 712–725. [Google Scholar] [CrossRef]
- Zhou, S.; Li, W.; Xiao, Y.; Zhu, X.; Zhong, Z.; Li, Q.; Cheng, F.; Zou, P.; You, Y.; Zhu, X. A Novel Chimeric Antigen Receptor Redirecting T-Cell Specificity towards CD26+ Cancer Cells. Leukemia 2021, 35, 119–129. [Google Scholar] [CrossRef]
- Yamada, K.; Hayashi, M.; Madokoro, H.; Nishida, H.; Du, W.; Ohnuma, K.; Sakamoto, M.; Morimoto, C.; Yamada, T. Nuclear Localization of CD26 Induced by a Humanized Monoclonal Antibody Inhibits Tumor Cell Growth by Modulating of POLR2A Transcription. PLoS ONE 2013, 8, e62304. [Google Scholar] [CrossRef]
- Dang, N.H.; Torimoto, Y.; Sugita, K.; Daley, J.F.; Schow, P.; Prado, C.; Schlossman, S.F.; Morimoto, C. Cell Surface Modulation of CD26 by Anti-1F7 Monoclonal Antibody. Analysis of Surface Expression and Human T Cell Activation. J. Immunol. 1990, 145, 3963–3971. [Google Scholar] [CrossRef]
- Hegen, M.; Kameoka, J.; Dong, R.P.; Schlossman, S.F.; Morimoto, C. Cross-Linking of CD26 Antibody Induces Tyrosine Phosphorylation and Activation of Mitogen-Activated Protein Kinase. Immunology 1997, 90, 257–264. [Google Scholar] [CrossRef]
- Morimoto, C.; Torimoto, Y.; Levinson, G.; Rudd, C.E.; Schrieber, M.; Dang, N.H.; Letvin, N.L.; Schlossman, S.F. 1F7, a Novel Cell Surface Molecule, Involved in Helper Function of CD4 Cells. J. Immunol. 1989, 143, 3430–3439. [Google Scholar] [CrossRef]
- Ohnuma, K.; Takahashi, N.; Yamochi, T.; Hosono, O.; Dang, N.H.; Morimoto, C. Role of CD26/Dipeptidyl Peptidase IV in Human T Cell Activation and Function. Front. Biosci. 2008, 13, 2299–2310. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Camerini, D.; Seed, B.; Torimoto, Y.; Dang, N.H.; Kameoka, J.; Dahlberg, H.N.; Schlossman, S.F.; Morimoto, C. Cloning and Functional Expression of the T Cell Activation Antigen CD26. J. Immunol. 1992, 149, 2090. [Google Scholar] [CrossRef]
- Philipson, B.I.; O’Connor, R.S.; May, M.J.; June, C.H.; Albelda, S.M.; Milone, M.C. 4-1BB Costimulation Promotes CAR T Cell Survival through Noncanonical NF-ΚB Signaling. Sci. Signal. 2020, 13, eaay8248. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, J.; Zhang, X.; Lu, X.A.; Xiong, M.; Zhang, J.; Zhou, X.; Qi, F.; He, T.; Ding, Y.; et al. Efficacy and Safety of CD28- or 4-1BB-Based CD19 CAR-T Cells in B Cell Acute Lymphoblastic Leukemia. Mol. Ther.-Oncolytics 2020, 18, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Tan, Y.; Wang, G.; Deng, B.; Ling, Z.; Song, W.; Seery, S.; Zhang, Y.; Peng, S.; Xu, J.; et al. Donor-Derived CD7 Chimeric Antigen Receptor T Cells for T-Cell Acute Lymphoblastic Leukemia: First-in-Human, Phase I Trial. J. Clin. Oncol. 2021, 39, 3340–3351. [Google Scholar] [CrossRef]
- Polgárová, K.; Otáhal, P.; Šálek, C.; Pytlík, R. Chimeric Antigen Receptor Based Cellular Therapy for Treatment Of T-Cell Malignancies. Front. Oncol. 2022, 12, 876758. [Google Scholar] [CrossRef]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A Gene Atlas of the Mouse and Human Protein-Encoding Transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [PubMed]
- Florentin, M.; Kostapanos, M.S.; Papazafiropoulou, A.K. Role of Dipeptidyl Peptidase 4 Inhibitors in the New Era of Antidiabetic Treatment. World J. Diabetes 2022, 13, 85–96. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Negative | Positive | |||
|---|---|---|---|---|
| Mild | Moderate | Strong | ||
| ALCL (n = 2) | 0 | 0 | 2 | 0 |
| AITL (n = 4) | 0 | 0 | 4 | 0 |
| NK/T (n = 5) | 0 | 3 | 0 | 2 |
| PTCL (n = 10) | 0 | 2 | 3 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, E.; Kamihara, Y.; Arai, M.; Wada, A.; Kikuchi, S.; Hatano, R.; Iwao, N.; Susukida, T.; Ozawa, T.; Adachi, Y.; et al. Development of a Novel CD26-Targeted Chimeric Antigen Receptor T-Cell Therapy for CD26-Expressing T-Cell Malignancies. Cells 2023, 12, 2059. https://doi.org/10.3390/cells12162059
Kobayashi E, Kamihara Y, Arai M, Wada A, Kikuchi S, Hatano R, Iwao N, Susukida T, Ozawa T, Adachi Y, et al. Development of a Novel CD26-Targeted Chimeric Antigen Receptor T-Cell Therapy for CD26-Expressing T-Cell Malignancies. Cells. 2023; 12(16):2059. https://doi.org/10.3390/cells12162059
Chicago/Turabian StyleKobayashi, Eiji, Yusuke Kamihara, Miho Arai, Akinori Wada, Shohei Kikuchi, Ryo Hatano, Noriaki Iwao, Takeshi Susukida, Tatsuhiko Ozawa, Yuichi Adachi, and et al. 2023. "Development of a Novel CD26-Targeted Chimeric Antigen Receptor T-Cell Therapy for CD26-Expressing T-Cell Malignancies" Cells 12, no. 16: 2059. https://doi.org/10.3390/cells12162059
APA StyleKobayashi, E., Kamihara, Y., Arai, M., Wada, A., Kikuchi, S., Hatano, R., Iwao, N., Susukida, T., Ozawa, T., Adachi, Y., Kishi, H., Dang, N. H., Yamada, T., Hayakawa, Y., Morimoto, C., & Sato, T. (2023). Development of a Novel CD26-Targeted Chimeric Antigen Receptor T-Cell Therapy for CD26-Expressing T-Cell Malignancies. Cells, 12(16), 2059. https://doi.org/10.3390/cells12162059