

The Role of Swelling in the Regulation of OPA1-Mediated Mitochondrial Function in the Heart In Vitro

 and
and 
Abstract
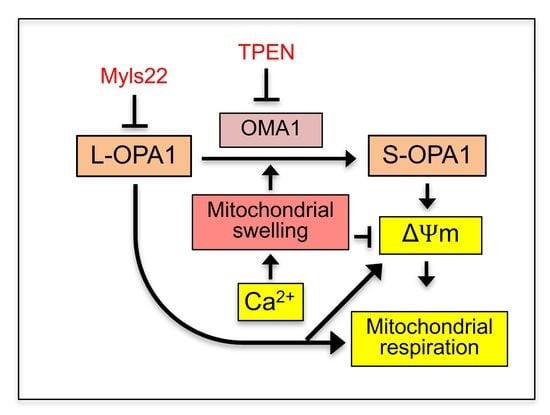
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cardiac Mitochondria Isolation
2.3. Analysis of mPTP Opening
2.4. Calcium Retention Capacity (CRC) Assay
2.5. Analysis of Mitochondrial Respiration and Membrane Potential
2.6. SDS-PAGE and Western Blotting
2.7. Statistical Analysis
3. Results
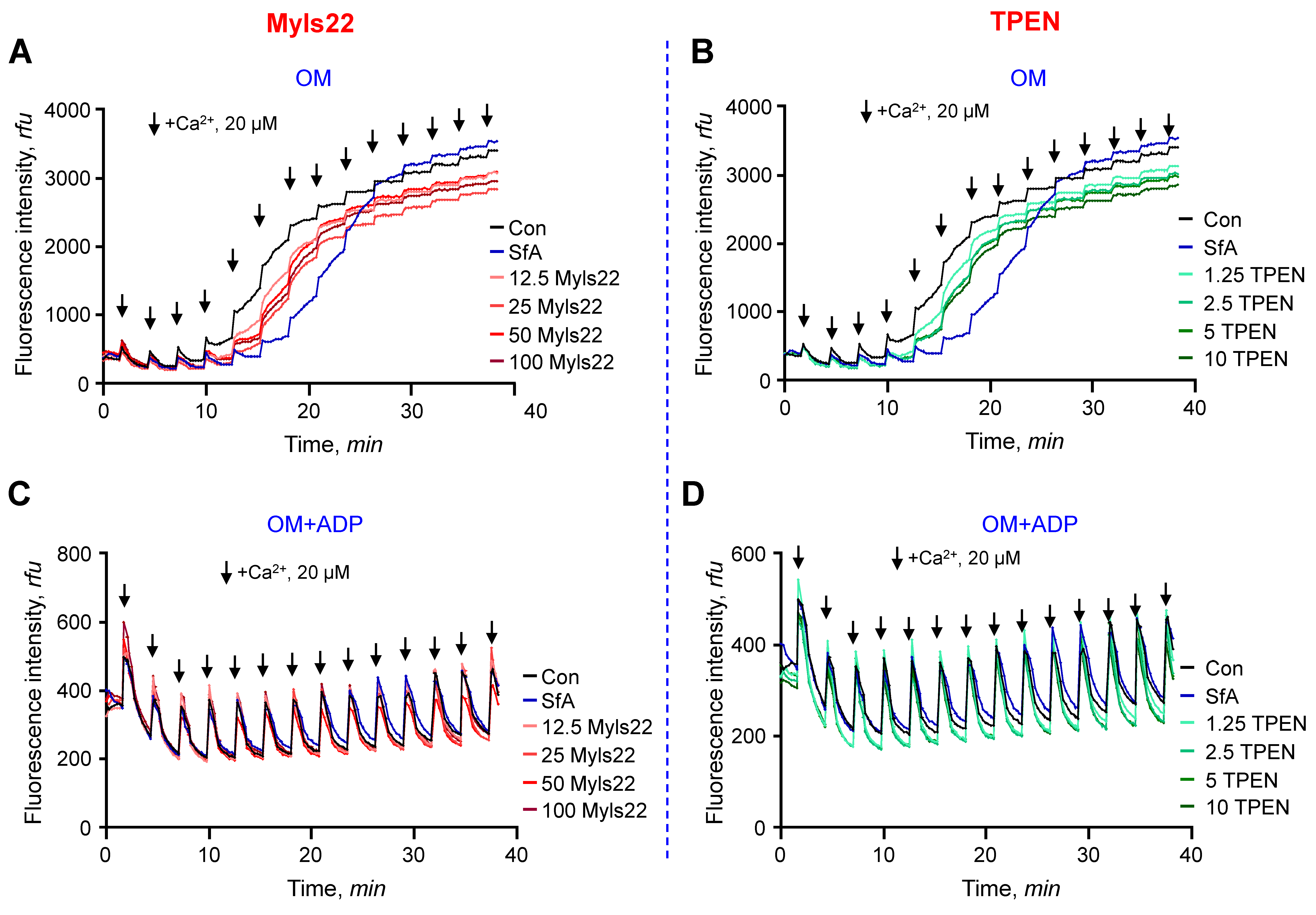
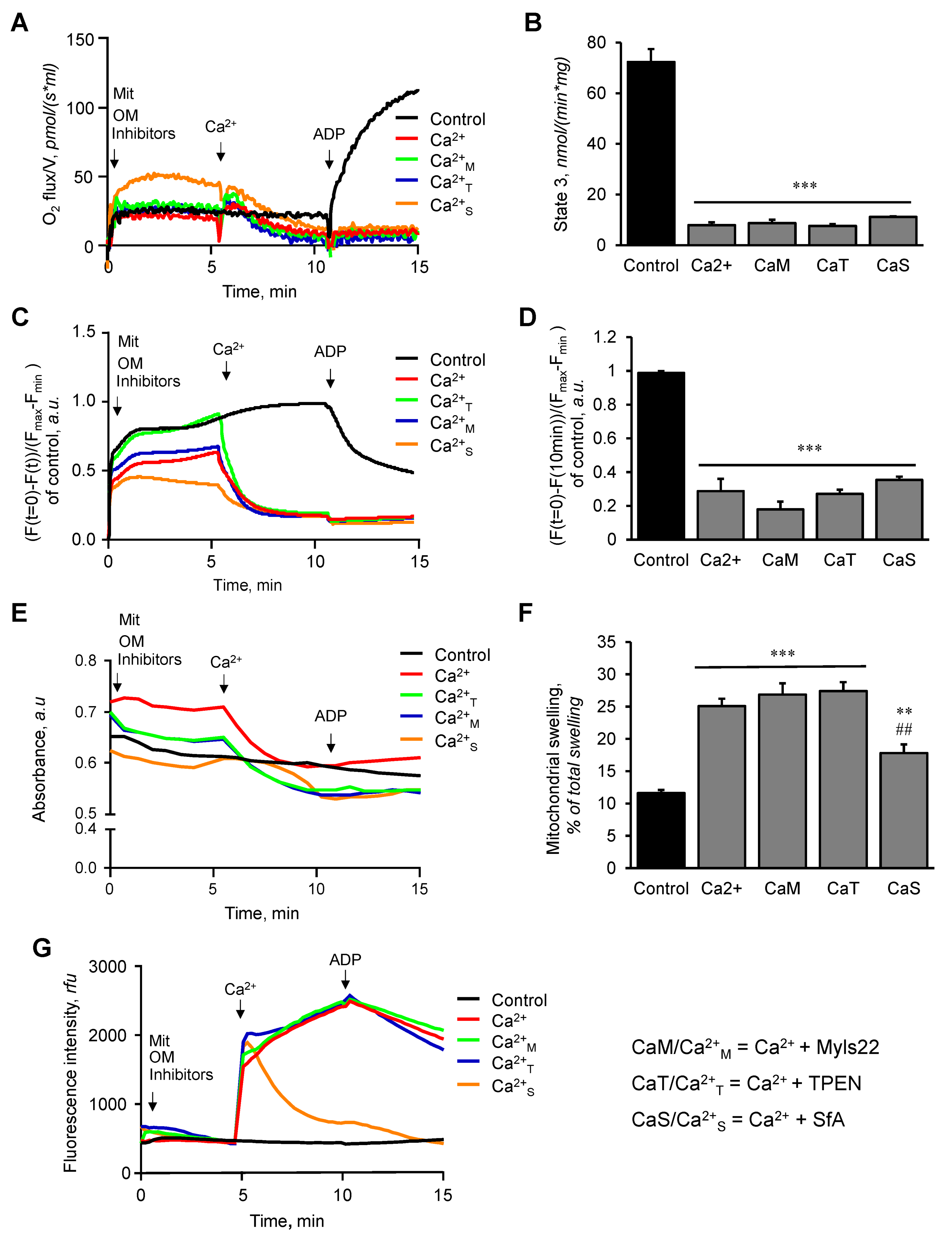
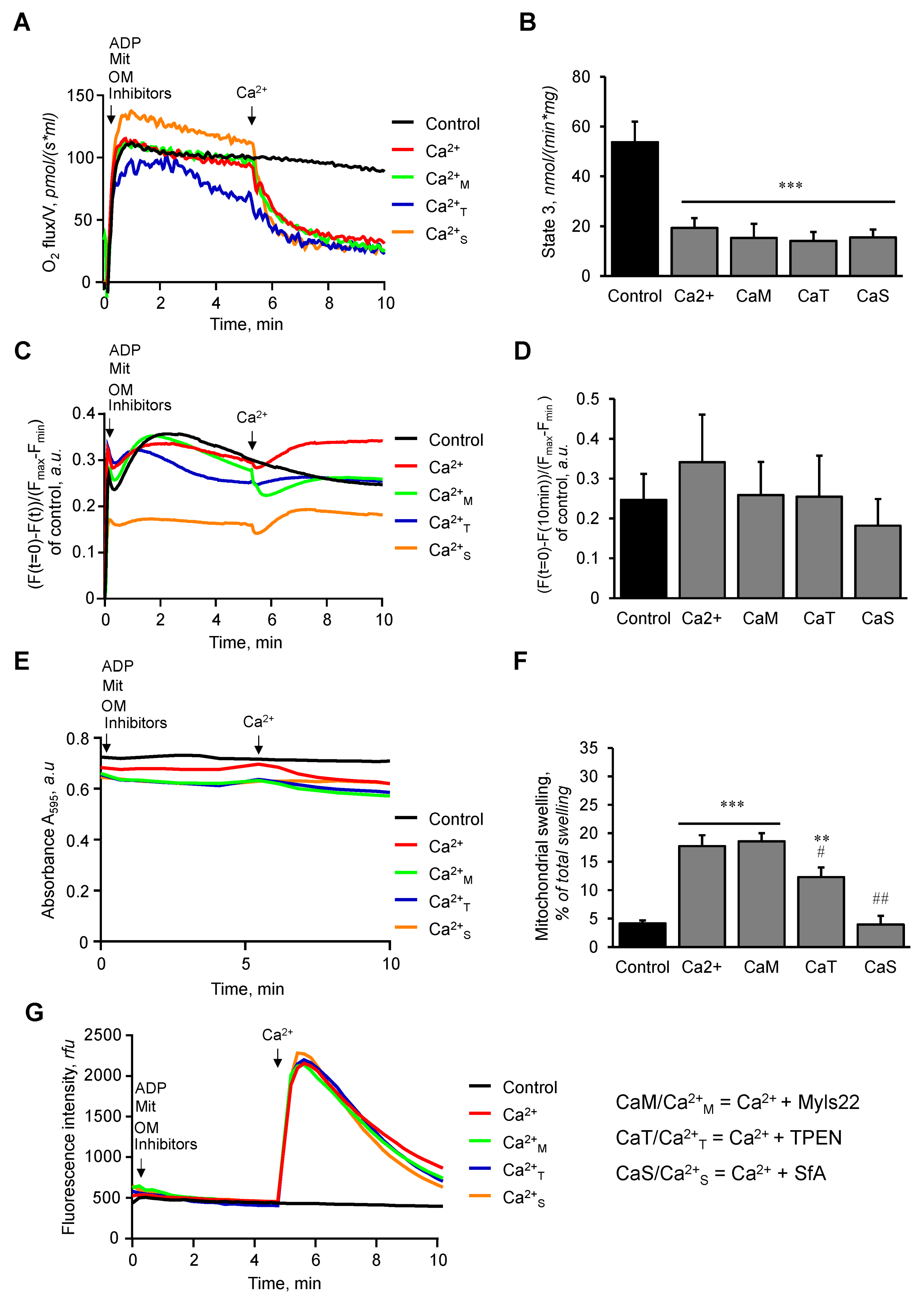
3.1. The Effects of Myls22 and TPEN on Mitochondrial CRC
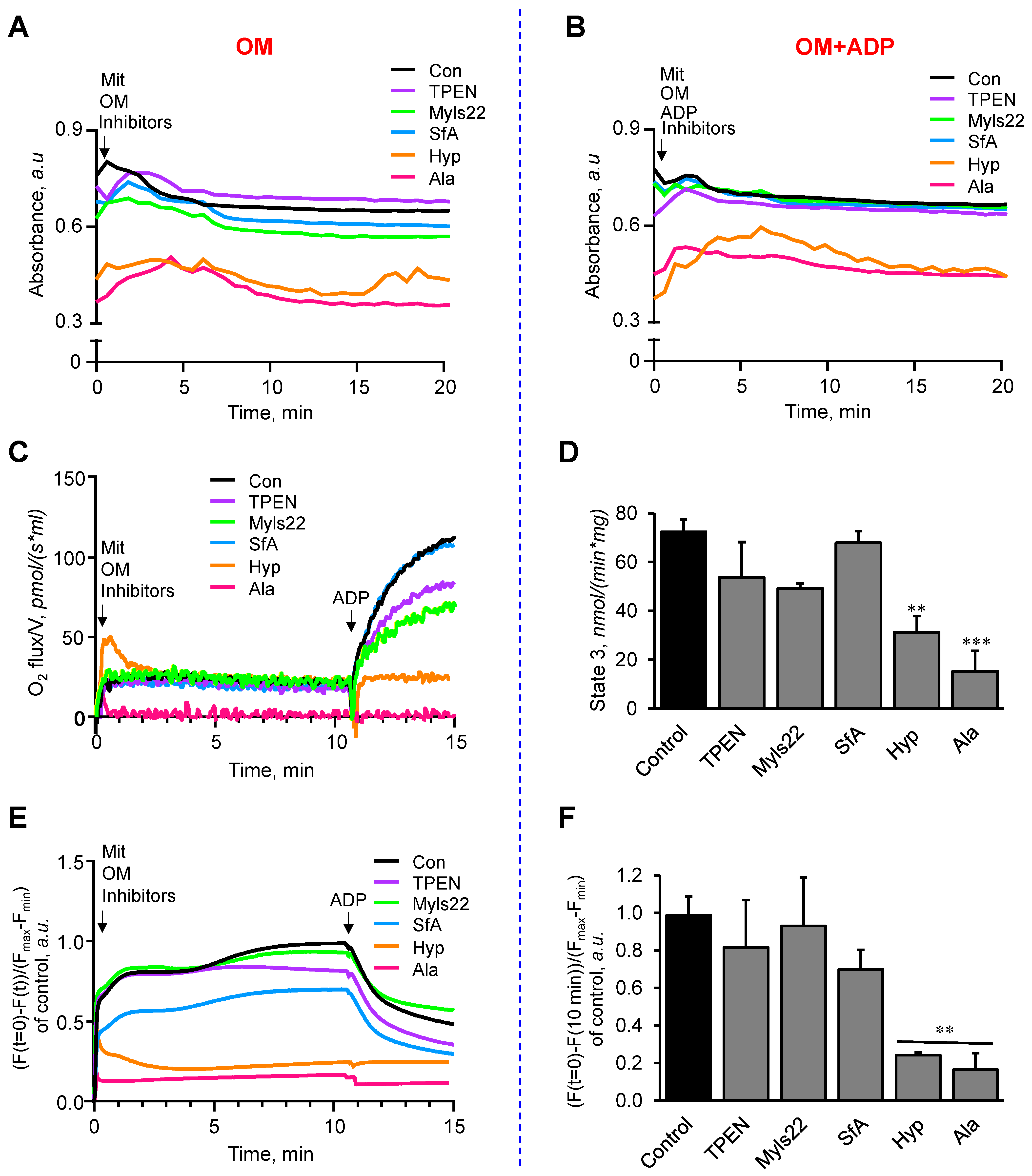
3.2. The Effects of Myls22 and TPEN on Mitochondrial Function
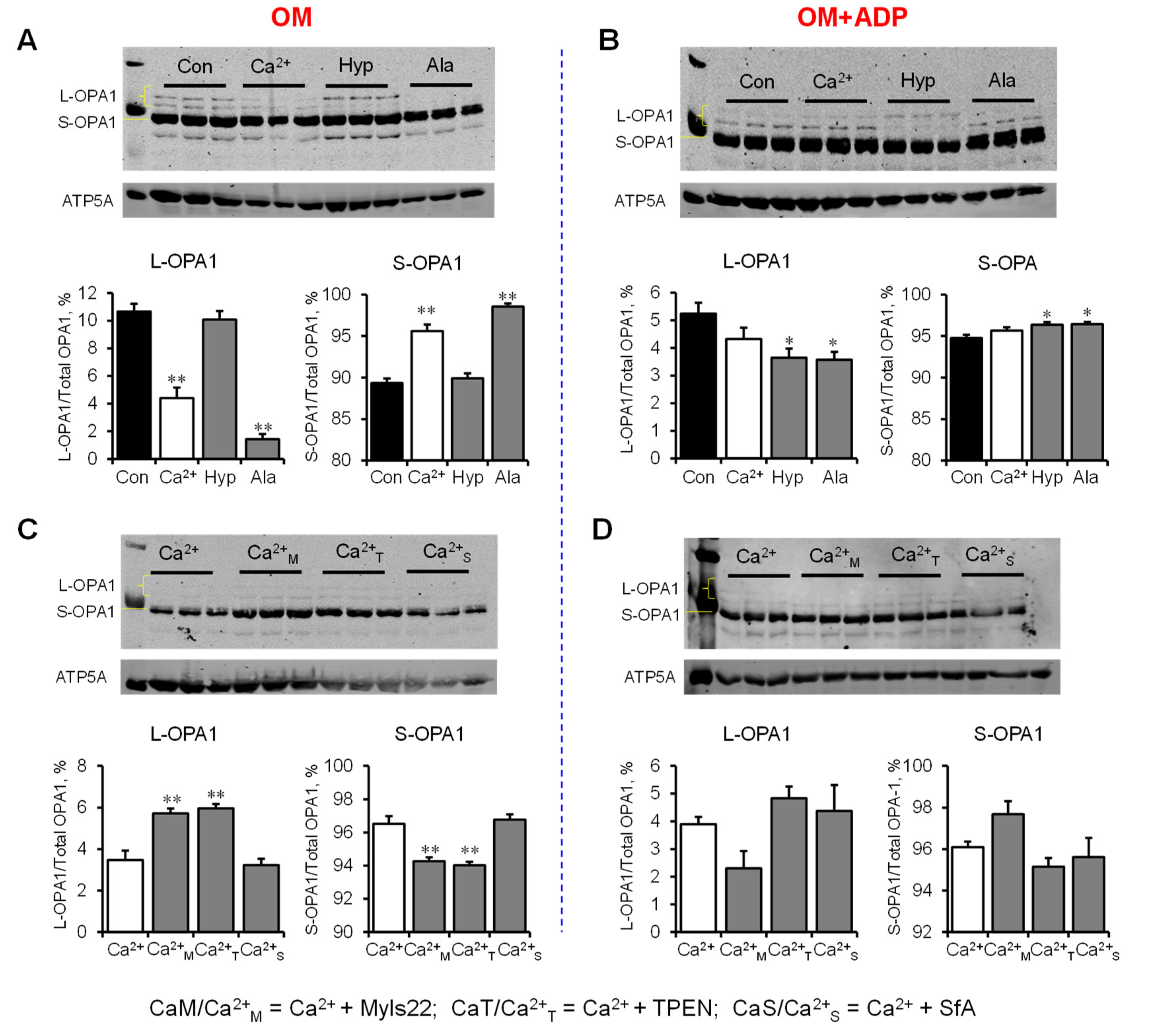
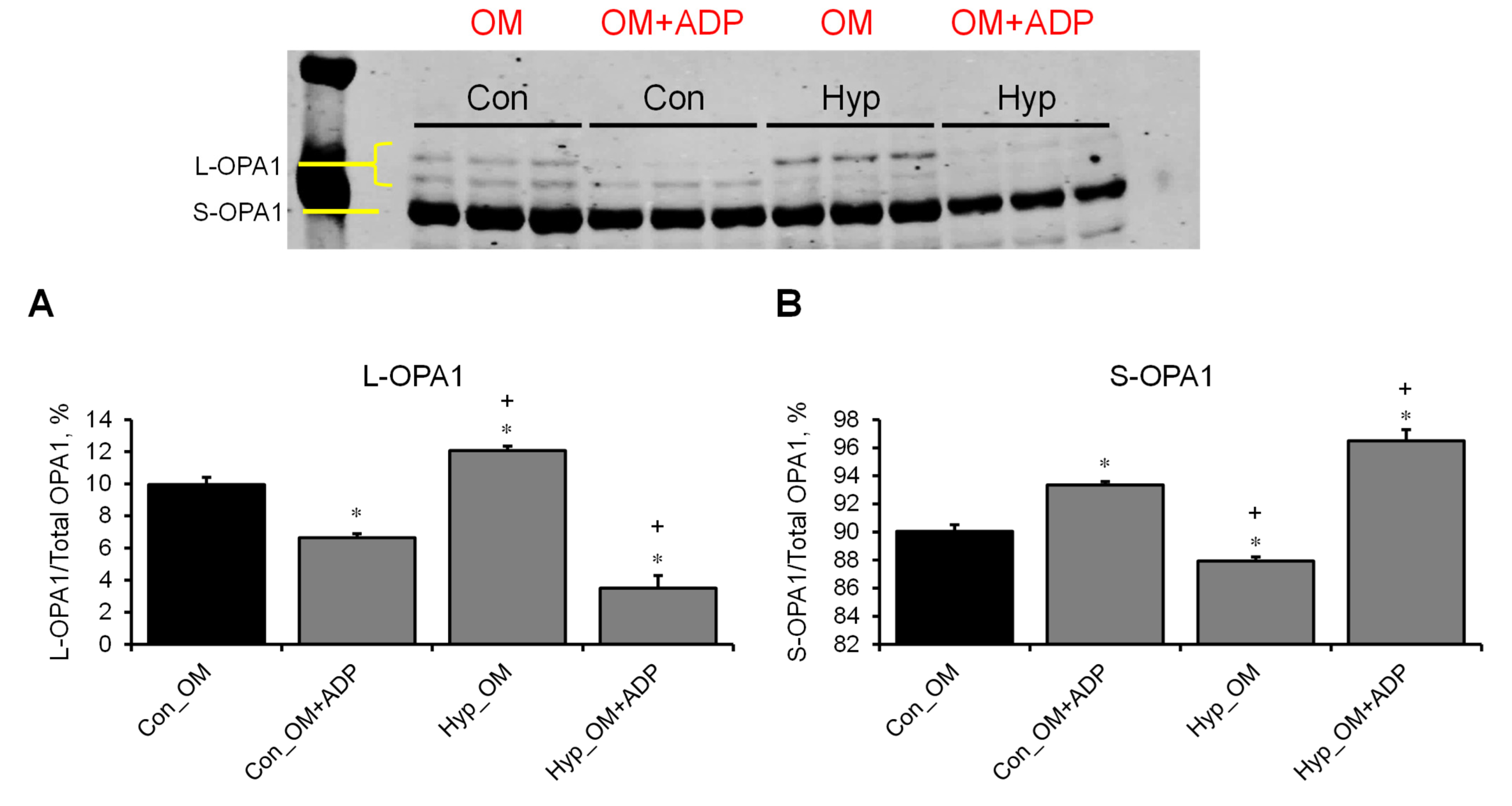
3.3. L-OPA1 Cleavage under Distinct Mitochondrial Energetic and Swelling Conditions
4. Discussion
5. Conclusions
Limitations
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kuhlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Del Dotto, V.; Mishra, P.; Vidoni, S.; Fogazza, M.; Maresca, A.; Caporali, L.; McCaffery, J.M.; Cappelletti, M.; Baruffini, E.; Lenaers, G.; et al. OPA1 Isoforms in the Hierarchical Organization of Mitochondrial Functions. Cell Rep. 2017, 19, 2557–2571. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Ge, Y.; Shi, X.; Boopathy, S.; McDonald, J.; Smith, A.W.; Chao, L.H. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. eLife 2020, 9, e50973. [Google Scholar] [CrossRef]
- Rainbolt, T.K.; Saunders, J.M.; Wiseman, R.L. YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress. EMBO Rep. 2015, 16, 97–106. [Google Scholar] [CrossRef]
- Wang, R.; Mishra, P.; Garbis, S.D.; Moradian, A.; Sweredoski, M.J.; Chan, D.C. Identification of new OPA1 cleavage site reveals that short isoforms regulate mitochondrial fusion. Mol. Biol. Cell 2021, 32, 157–168. [Google Scholar] [CrossRef]
- Merkwirth, C.; Dargazanli, S.; Tatsuta, T.; Geimer, S.; Lower, B.; Wunderlich, F.T.; von Kleist-Retzow, J.C.; Waisman, A.; Westermann, B.; Langer, T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes. Dev. 2008, 22, 476–488. [Google Scholar] [CrossRef]
- Lee, H.; Smith, S.B.; Yoon, Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J. Biol. Chem. 2017, 292, 7115–7130. [Google Scholar] [CrossRef]
- Wai, T.; Garcia-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Ruperez, F.J.; Barbas, C.; Ibanez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease—The long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef]
- Noone, J.; O’Gorman, D.J.; Kenny, H.C. OPA1 regulation of mitochondrial dynamics in skeletal and cardiac muscle. Trends Endocrinol. Metab. 2022, 33, 710–721. [Google Scholar] [CrossRef]
- Lee, H.; Smith, S.B.; Sheu, S.S.; Yoon, Y. The short variant of optic atrophy 1 (OPA1) improves cell survival under oxidative stress. J. Biol. Chem. 2020, 295, 6543–6560. [Google Scholar] [CrossRef]
- Jang, S.; Javadov, S. OPA1 regulates respiratory supercomplexes assembly: The role of mitochondrial swelling. Mitochondrion 2020, 51, 30–39. [Google Scholar] [CrossRef]
- Zamberlan, M.; Boeckx, A.; Muller, F.; Vinelli, F.; Ek, O.; Vianello, C.; Coart, E.; Shibata, K.; Christian, A.; Grespi, F.; et al. Inhibition of the mitochondrial protein Opa1 curtails breast cancer growth. J. Exp. Clin. Cancer Res. 2022, 41, 95. [Google Scholar] [CrossRef]
- Tang, J.; Liu, Z.; Han, J.; Xue, J.; Liu, L.; Lin, J.; Wu, C.; Zhang, Q.; Wu, S.; Liu, C.; et al. Increased Mobile Zinc Regulates Retinal Ganglion Cell Survival via Activating Mitochondrial OMA1 and Integrated Stress Response. Antioxidants 2022, 11, 2001. [Google Scholar] [CrossRef]
- Chapa-Dubocq, X.R.; Rodriguez-Graciani, K.M.; Guzman-Hernandez, R.A.; Jang, S.; Brookes, P.S.; Javadov, S. Cardiac Function is not Susceptible to Moderate Disassembly of Mitochondrial Respiratory Supercomplexes. Int. J. Mol. Sci. 2020, 21, 1555. [Google Scholar] [CrossRef]
- Chapa-Dubocq, X.; Makarov, V.; Javadov, S. Simple kinetic model of mitochondrial swelling in cardiac cells. J. Cell Physiol. 2018, 233, 5310–5321. [Google Scholar] [CrossRef]
- Jang, S.; Chapa-Dubocq, X.R.; Fossati, S.; Javadov, S. Analysis of Mitochondrial Calcium Retention Capacity in Cultured Cells: Permeabilized Cells Versus Isolated Mitochondria. Front. Physiol. 2021, 12, 773839. [Google Scholar] [CrossRef]
- Scaduto, R.C., Jr.; Grotyohann, L.W. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 1999, 76, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Ban, T.; Heymann, J.A.; Song, Z.; Hinshaw, J.E.; Chan, D.C. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet. 2010, 19, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.L.; Lee, J.; Pendleton, K.E.; Berner, M.J.; Goff, E.B.; Tan, L.; Martinez, S.A.; Mahmud, I.; Wang, T.; Meyer, M.D.; et al. Mitochondrial structure and function adaptation in residual triple negative breast cancer cells surviving chemotherapy treatment. Oncogene 2023, 42, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. TPEN induces apoptosis independently of zinc chelator activity in a model of acute lymphoblastic leukemia and ex vivo acute leukemia cells through oxidative stress and mitochondria caspase-3- and AIF-dependent pathways. Oxid. Med. Cell Longev. 2012, 2012, 313275. [Google Scholar] [CrossRef]
- Garlid, K.D.; Paucek, P. The mitochondrial potassium cycle. IUBMB Life 2001, 52, 153–158. [Google Scholar] [CrossRef]
- Rodriguez-Graciani, K.M.; Chapa-Dubocq, X.R.; MacMillan-Crow, L.A.; Javadov, S. Association between L-OPA1 Cleavage and Cardiac Dysfunction During Ischemia-Reperfusion Injury in Rats. Cell Physiol. Biochem. 2020, 54, 1101–1114. [Google Scholar] [CrossRef]
- Li, H.; Qin, S.; Liang, Q.; Xi, Y.; Bo, W.; Cai, M.; Tian, Z. Exercise Training Enhances Myocardial Mitophagy and Improves Cardiac Function via Irisin/FNDC5-PINK1/Parkin Pathway in MI Mice. Biomedicines 2021, 9, 701. [Google Scholar] [CrossRef]
- Boissan, M.; Montagnac, G.; Shen, Q.; Griparic, L.; Guitton, J.; Romao, M.; Sauvonnet, N.; Lagache, T.; Lascu, I.; Raposo, G.; et al. Membrane trafficking. Nucleoside diphosphate kinases fuel dynamin superfamily proteins with GTP for membrane remodeling. Science 2014, 344, 1510–1515. [Google Scholar] [CrossRef]
- Chapa-Dubocq, X.R.; Garcia-Baez, J.F.; Bazil, J.N.; Javadov, S. Crosstalk between adenine nucleotide transporter and mitochondrial swelling: Experimental and computational approaches. Cell Biol. Toxicol. 2022, 39, 435–450. [Google Scholar] [CrossRef]
- Rahn, J.J.; Stackley, K.D.; Chan, S.S. Opa1 is required for proper mitochondrial metabolism in early development. PLoS ONE 2013, 8, e59218. [Google Scholar] [CrossRef]
- Hu, C.; Shu, L.; Huang, X.; Yu, J.; Li, L.; Gong, L.; Yang, M.; Wu, Z.; Gao, Z.; Zhao, Y.; et al. OPA1 and MICOS Regulate mitochondrial crista dynamics and formation. Cell Death Dis. 2020, 11, 940. [Google Scholar] [CrossRef]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef]
- Griparic, L.; Kanazawa, T.; van der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef]
- Sprenger, H.G.; Wani, G.; Hesseling, A.; Konig, T.; Patron, M.; MacVicar, T.; Ahola, S.; Wai, T.; Barth, E.; Rugarli, E.I.; et al. Loss of the mitochondrial i-AAA protease YME1L leads to ocular dysfunction and spinal axonopathy. EMBO Mol. Med. 2019, 11, e9288. [Google Scholar] [CrossRef]
- Quiros, P.M.; Ramsay, A.J.; Sala, D.; Fernandez-Vizarra, E.; Rodriguez, F.; Peinado, J.R.; Fernandez-Garcia, M.S.; Vega, J.A.; Enriquez, J.A.; Zorzano, A.; et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012, 31, 2117–2133. [Google Scholar] [CrossRef]
- Larrue, C.; Mouche, S.; Lin, S.; Simonetta, F.; Scheidegger, N.K.; Poulain, L.; Birsen, R.; Sarry, J.E.; Stegmaier, K.; Tamburini, J. Mitochondrial fusion is a therapeutic vulnerability of acute myeloid leukemia. Leukemia 2023, 37, 765–775. [Google Scholar] [CrossRef]
- Tobacyk, J.; Parajuli, N.; Shrum, S.; Crow, J.P.; MacMillan-Crow, L.A. The first direct activity assay for the mitochondrial protease OMA1. Mitochondrion 2019, 46, 1–5. [Google Scholar] [CrossRef]
- Garcia-Perez, C.; Roy, S.S.; Naghdi, S.; Lin, X.; Davies, E.; Hajnoczky, G. Bid-induced mitochondrial membrane permeabilization waves propagated by local reactive oxygen species (ROS) signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 4497–4502. [Google Scholar] [CrossRef]
- Mishra, P.; Varuzhanyan, G.; Pham, A.H.; Chan, D.C. Mitochondrial Dynamics is a Distinguishing Feature of Skeletal Muscle Fiber Types and Regulates Organellar Compartmentalization. Cell Metab. 2015, 22, 1033–1044. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Labbe, K.; Murley, A.; Nunnari, J. Determinants and functions of mitochondrial behavior. Annu. Rev. Cell Dev. Biol. 2014, 30, 357–391. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. Structural and functional remodeling of mitochondria as an adaptive response to energy deprivation. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148393. [Google Scholar] [CrossRef]
- Loor, G.; Schumacker, P.T. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ. 2008, 15, 686–690. [Google Scholar] [CrossRef]
- Javadov, S.; Rajapurohitam, V.; Kilic, A.; Hunter, J.C.; Zeidan, A.; Said Faruq, N.; Escobales, N.; Karmazyn, M. Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: The effect of NHE-1 inhibition. Basic. Res. Cardiol. 2011, 106, 99–109. [Google Scholar] [CrossRef]
- Ban, T.; Kohno, H.; Ishihara, T.; Ishihara, N. Relationship between OPA1 and cardiolipin in mitochondrial inner-membrane fusion. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 951–957. [Google Scholar] [CrossRef]
- Roy, M.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division and fusion in metabolism. Curr. Opin. Cell Biol. 2015, 33, 111–118. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chapa-Dubocq, X.R.; Rodríguez-Graciani, K.M.; García-Báez, J.; Vadovsky, A.; Bazil, J.N.; Javadov, S. The Role of Swelling in the Regulation of OPA1-Mediated Mitochondrial Function in the Heart In Vitro. Cells 2023, 12, 2017. https://doi.org/10.3390/cells12162017
Chapa-Dubocq XR, Rodríguez-Graciani KM, García-Báez J, Vadovsky A, Bazil JN, Javadov S. The Role of Swelling in the Regulation of OPA1-Mediated Mitochondrial Function in the Heart In Vitro. Cells. 2023; 12(16):2017. https://doi.org/10.3390/cells12162017
Chicago/Turabian StyleChapa-Dubocq, Xavier R., Keishla M. Rodríguez-Graciani, Jorge García-Báez, Alyssa Vadovsky, Jason N. Bazil, and Sabzali Javadov. 2023. "The Role of Swelling in the Regulation of OPA1-Mediated Mitochondrial Function in the Heart In Vitro" Cells 12, no. 16: 2017. https://doi.org/10.3390/cells12162017
APA StyleChapa-Dubocq, X. R., Rodríguez-Graciani, K. M., García-Báez, J., Vadovsky, A., Bazil, J. N., & Javadov, S. (2023). The Role of Swelling in the Regulation of OPA1-Mediated Mitochondrial Function in the Heart In Vitro. Cells, 12(16), 2017. https://doi.org/10.3390/cells12162017