

Mitochondrial Cholesterol Metabolites in a Bile Acid Synthetic Pathway Drive Nonalcoholic Fatty Liver Disease: A Revised “Two-Hit” Hypothesis


Abstract
1. Introduction
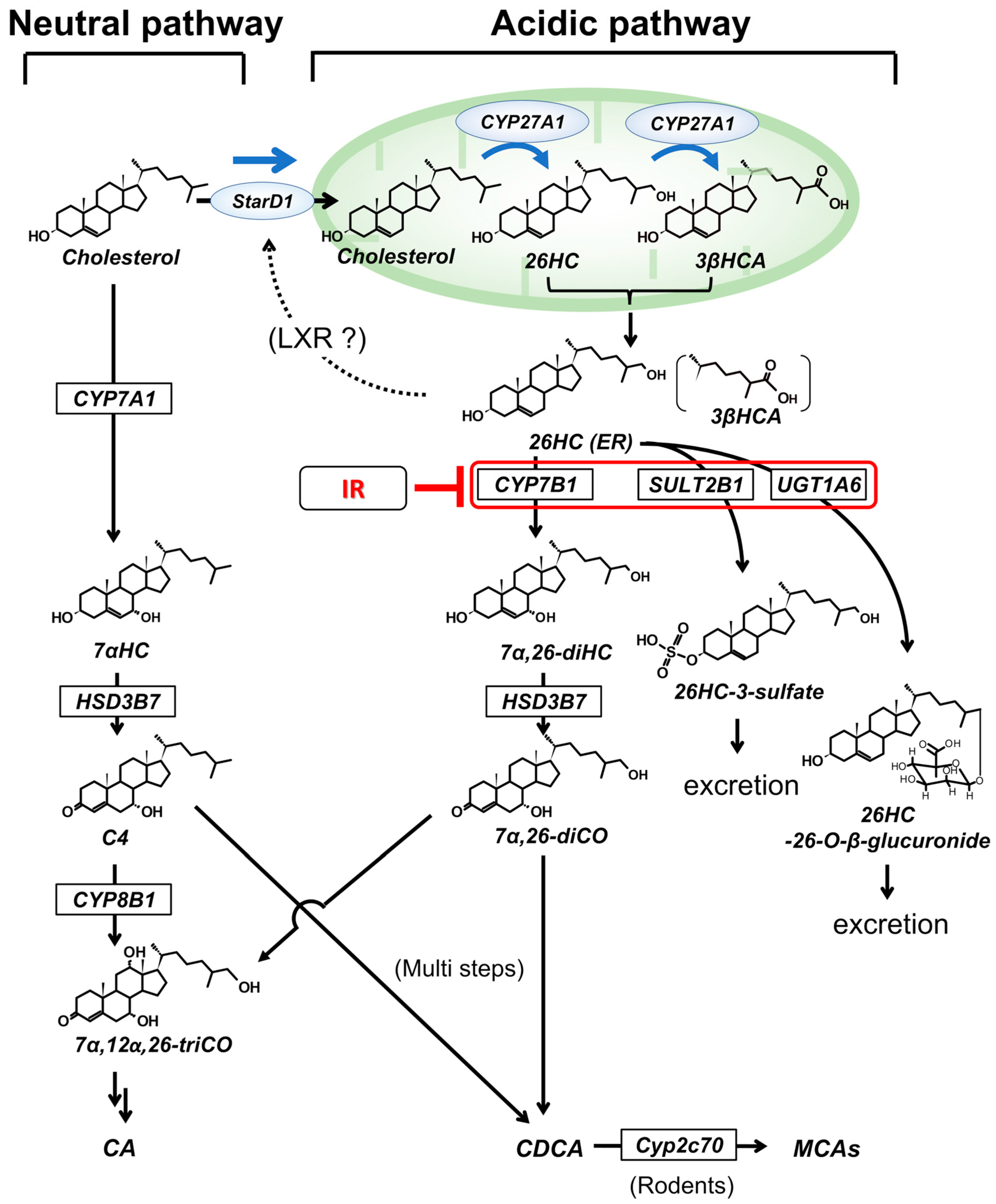
2. Hepatic Cholesterol Metabolism
3. Key Concept of the Two-Hit Hypothesis
4. Insulin Regulation of Mitochondrial Bile Acid Intermediates
5. Cholesterol Metabolites in Mitochondria Impairment
6. Cholesterol Metabolites and Endoplasmic Reticulum Function
7. Targeting Mitochondrial Cholesterol Metabolites for NASH Intervention
8. Conclusions and Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Younossi, Z.M.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, E.F.; King, A.J.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.-A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef]
- Younossi, Z.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.-S.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, A.M.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.E.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef]
- O’Neil, P.M.; Birkenfeld, A.L.; McGowan, B.; Mosenzon, O.; Pedersen, S.D.; Wharton, S.; Carson, C.G.; Jepsen, C.H.; Kabisch, M.; Wilding, J.P.H. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: A randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet 2018, 392, 637–649. [Google Scholar] [CrossRef]
- Kushner, R.F.; Calanna, S.; Davies, M.; Dicker, D.; Garvey, W.T.; Goldman, B.; Lingvay, I.; Thomsen, M.; Wadden, T.A.; Wharton, S.; et al. Semaglutide 2.4 mg for the Treatment of Obesity: Key Elements of the STEP Trials 1 to 5. Obesity 2020, 28, 1050–1061. [Google Scholar] [CrossRef]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.-S.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef] [PubMed]
- Ferrell, J.M.; Chiang, J.Y.L. Understanding Bile Acid Signaling in Diabetes: From Pathophysiology to Therapeutic Targets. Diabetes Metab. J. 2019, 43, 257–272. [Google Scholar] [CrossRef]
- Pandak, W.M.; Kakiyama, G. The acidic pathway of bile acid synthesis: Not just an alternative pathway. Liver Res. 2019, 3, 88–98. [Google Scholar] [CrossRef]
- Jia, W.; Wei, M.; Rajani, C.; Zheng, X. Targeting the alternative bile acid synthetic pathway for metabolic diseases. Protein Cell 2021, 12, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Horn, C.L.; Morales, A.L.; Savard, C.; Farrell, G.C.; Ioannou, G.N. Role of Cholesterol-Associated Steatohepatitis in the Development of NASH. Hepatol. Commun. 2021, 6, 12–35. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Marques, D.; Takei, H.; Nittono, H.; Erickson, S.; Fuchs, M.; Rodriguez-Agudo, D.; Gil, G.; Hylemon, P.B.; Zhou, H.; et al. Mitochondrial oxysterol biosynthetic pathway gives evidence for CYP7B1 as controller of regulatory oxysterols. J. Steroid Biochem. Mol. Biol. 2019, 189, 36–47. [Google Scholar] [CrossRef]
- Javitt, N.B. Cholesterol, Hydroxycholesterols, and Bile Acids. Biochem. Biophys. Res. Commun. 2002, 292, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Wang, Y. Cholesterol metabolism: From lipidomics to immunology. J. Lipid Res. 2022, 63, 100165. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Marques, D.; Martin, R.; Takei, H.; Rodriguez-Agudo, D.; LaSalle, S.A.; Hashiguchi, T.; Liu, X.; Green, R.; Erickson, S.; et al. Insulin resistance dysregulates CYP7B1 leading to oxysterol accumulation: A pathway for NAFL to NASH transition. J. Lipid Res. 2020, 61, 1629–1644. [Google Scholar] [CrossRef]
- Kakiyama, G.; Minowa, K.; Rodriguez-Agudo, D.; Martin, R.; Takei, H.; Mitamura, K.; Ikegawa, S.; Mitsuyoshi, S.; Nittono, H.; Fuchs, M.; et al. Coffee modulates insulin-HNF-4alpha-Cyp7b1 pathway and reduces oxysterol driven liver toxicity in a NAFLD mouse model. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 323, G488–G500. [Google Scholar] [CrossRef] [PubMed]
- Minowa, K.; Rodriguez-Agudo, D.; Suzuki, M.; Muto, Y.; Hirai, S.; Wang, Y.; Su, L.; Zhou, H.; Chen, Q.; Lesnefsky, E.J.; et al. Insulin dysregulation drives mitochondrial cholesterol metabolite accumulation: Initiating hepatic toxicity in NAFLD. J. Lipid Res. 2023, 64, 100363. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.S.; Bril, F.; Cusi, K.; Newsome, P.N. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 70, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M. The Liver as an Endocrine Organ—Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef]
- Christenson, L.K.; McAllister, J.M.; Martin, K.O.; Javitt, N.B.; Osborne, T.F.; Strauss, J.F. Oxysterol Regulation of Steroidogenic Acute Regulatory Protein Gene Expression. J. Biol. Chem. 1998, 273, 30729–30735. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Woollett, L.A.; Spady, D.K. The Interaction of Dietary Cholesterol and Specific Fatty Acids in the Regulation of LDL Receptor Activity and Plasma LDL-Cholesterol Concentrations. Ann. New York Acad. Sci. 1993, 676, 11–26. [Google Scholar] [CrossRef]
- Honda, A.; Miyazaki, T.; Iwamoto, J.; Hirayama, T.; Morishita, Y.; Monma, T.; Ueda, H.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J. Lipid Res. 2020, 61, 54–69. [Google Scholar] [CrossRef]
- Li, J.; Dawson, P.A. Animal models to study bile acid metabolism. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1865, 895–911. [Google Scholar] [CrossRef]
- Elustondo, P.; Martin, L.A.; Karten, B. Mitochondrial cholesterol import. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2017, 1862, 90–101. [Google Scholar] [CrossRef]
- Pandak, W.M.; Ren, S.; Marques, D.; Hall, E.; Redford, K.; Mallonee, D.; Bohdan, P.; Heuman, D.; Gil, G.; Hylemon, P. Transport of Cholesterol into Mitochondria Is Rate-limiting for Bile Acid Synthesis via the Alternative Pathway in Primary Rat Hepatocytes. J. Biol. Chem. 2002, 277, 48158–48164. [Google Scholar] [CrossRef]
- Ren, S.; Hylemon, P.B.; Marques, D.; Gurley, E.; Bodhan, P.; Hall, E.; Redford, K.; Gil, G.; Pandak, W.M. Overexpression of cholesterol transporter StAR increasesin vivo rates of bile acid synthesis in the rat and mouse. Hepatology 2004, 40, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Caron, K.M.; Soo, S.-C.; Wetsel, W.C.; Stocco, D.M.; Clark, B.J.; Parker, K.L. Targeted disruption of the mouse gene encoding steroidogenic acute regulatory protein provides insights into congenital lipoid adrenal hyperplasia. Proc. Natl. Acad. Sci. USA 1997, 94, 11540–11545. [Google Scholar] [CrossRef]
- Ren, S.; Hylemon, P.; Marques, D.; Hall, E.; Redford, K.; Gil, G.; Pandak, W.M. Effect of increasing the expression of cholesterol transporters (StAR, MLN64, and SCP-2) on bile acid synthesis. J. Lipid Res. 2004, 45, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Balboa, E.; Castro, J.; Pinochet, M.-J.; Cancino, G.I.; Matías, N.; Sáez, P.J.; Martínez, A.; Álvarez, A.R.; Garcia-Ruiz, C.; Fernández-Checa, J.C.; et al. MLN64 induces mitochondrial dysfunction associated with increased mitochondrial cholesterol content. Redox Biol. 2017, 12, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Fakheri, R.J.; Javitt, N.B. 27-Hydroxycholesterol, does it exist? On the nomenclature and stereochemistry of 26-hydroxylated sterols. Steroids 2012, 77, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Axelson, M.; Mörk, B.; Sjövall, J. Occurrence of 3 beta-hydroxy-5-cholestenoic acid, 3 beta,7 alpha-dihydroxy-5-cholestenoic acid, and 7 alpha-hydroxy-3-oxo-4-cholestenoic acid as normal constituents in human blood. J. Lipid Res. 1988, 29, 629–641. [Google Scholar] [CrossRef]
- Axelson, M.; Mörk, B.; Aly, A.; Wisén, O.; Sjövall, J. Concentrations of cholestenoic acids in plasma from patients with liver disease. J. Lipid Res. 1989, 30, 1877–1882. [Google Scholar] [CrossRef]
- Axelson, M.; Mörk, B.; Aly, A.; Walldius, G.; Sjövall, J. Concentrations of cholestenoic acids in plasma from patients with reduced intestinal reabsorption of bile acids. J. Lipid Res. 1989, 30, 1883–1887. [Google Scholar] [CrossRef]
- Taylor, J.M.; Borthwick, F.; Bartholomew, C.; Graham, A. Overexpression of steroidogenic acute regulatory protein increases macrophage cholesterol efflux to apolipoprotein AI. Cardiovasc. Res. 2010, 86, 526–534. [Google Scholar] [CrossRef]
- Song, C.; Liao, S. Cholestenoic Acid Is a Naturally Occurring Ligand for Liver X Receptor α**This work was supported by NIH grants. Endocrinology 2000, 141, 4180–4184. [Google Scholar] [CrossRef]
- Fu, X.; Menke, J.G.; Chen, Y.; Zhou, G.; MacNaul, K.L.; Wright, S.D.; Sparrow, C.P.; Lund, E.G. 27-Hydroxycholesterol Is an Endogenous Ligand for Liver X Receptor in Cholesterol-loaded Cells. J. Biol. Chem. 2001, 276, 38378–38387. [Google Scholar] [CrossRef]
- Saito, H.; Tachiura, W.; Nishimura, M.; Shimizu, M.; Sato, R.; Yamauchi, Y. Hydroxylation site–specific and production-dependent effects of endogenous oxysterols on cholesterol homeostasis: Implications for SREBP-2 and LXR. J. Biol. Chem. 2023, 299, 102733. [Google Scholar] [CrossRef] [PubMed]
- Heverin, M.; Ali, Z.; Olin, M.; Tillander, V.; Joibari, M.M.; Makoveichuk, E.; Leitersdorf, E.; Warner, M.; Olivercrona, G.; Gustafsson, J.; et al. On the regulatory importance of 27-hydroxycholesterol in mouse liver. J. Steroid Biochem. Mol. Biol. 2017, 169, 10–21. [Google Scholar] [CrossRef]
- Javitt, N.B.; Kok, E.; Cohen, B.; Burstein, S. Cerebrotendinous xanthomatosis: Reduced serum 26-hydroxycholesterol. J. Lipid Res. 1982, 23, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Björkhem, I.; Hansson, M. Cerebrotendinous xanthomatosis: An inborn error in bile acid synthesis with defined mutations but still a challenge. Biochem. Biophys. Res. Commun. 2010, 396, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.-H.; Wang, Z.-X. Liver transplantation due to cerebrotendinous xanthomatosis end-stage liver disease. World J. Pediatr. 2018, 14, 414–415. [Google Scholar] [CrossRef]
- Pietrobattista, A.; Spada, M.; Candusso, M.; Boenzi, S.; Dionisi-Vici, C.; Francalanci, P.; Morrone, A.; Ferri, L.; Indolfi, G.; Agolini, E.; et al. Liver transplantation in an infant with cerebrotendinous xanthomatosis, cholestasis, and rapid evolution of liver failure. Pediatr. Transplant. 2022, 26, e14318. [Google Scholar] [CrossRef]
- Lipiński, P.; Klaudel-Dreszler, M.; Ciara, E.; Jurkiewicz, D.; Płoski, R.; Cielecka-Kuszyk, J.; Socha, P.; Jankowska, I. Sterol 27-Hydroxylase Deficiency as a Cause of Neonatal Cholestasis: Report of 2 Cases and Review of the Literature. Front. Pediatr. 2020, 8, 616582. [Google Scholar] [CrossRef]
- Gong, J.-Y.; Setchell, K.D.; Zhao, J.; Zhang, W.; Wolfe, B.; Lu, Y.; Lackner, K.; Knisely, A.; Wang, N.-L.; Hao, C.-Z.; et al. Severe Neonatal Cholestasis in Cerebrotendinous Xanthomatosis: Genetics, Immunostaining, Mass Spectrometry. J. Craniofacial Surg. 2017, 65, 561–568. [Google Scholar] [CrossRef]
- Heubi, J.E.; Setchell, K.D.R.; Bove, K.E. Inborn Errors of Bile Acid Metabolism. Semin. Liver Dis. 2007, 27, 282–294. [Google Scholar] [CrossRef]
- Duell, P.B.; Salen, G.; Eichler, F.S.; DeBarber, A.E.; Connor, S.L.; Casaday, L.; Jayadev, S.; Kisanuki, Y.; Lekprasert, P.; Malloy, M.J.; et al. Diagnosis, treatment, and clinical outcomes in 43 cases with cerebrotendinous xanthomatosis. J. Clin. Lipidol. 2018, 12, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Martin, K.; Javitt, N.; Chiang, J. Structure and functions of human oxysterol 7α-hydroxylase cDNAs and gene CYP7B1. J. Lipid Res. 1999, 40, 2195–2203. [Google Scholar] [CrossRef] [PubMed]
- Stiles, A.R.; McDonald, J.G.; Bauman, D.R.; Russell, D.W. CYP7B1: One Cytochrome P450, Two Human Genetic Diseases, and Multiple Physiological Functions. J. Biol. Chem. 2009, 284, 28485–28489. [Google Scholar] [CrossRef]
- Ayaki, Y.; Kok, E.; Javitt, N.B. Cholic Acid Synthesis from 26-Hydroxycholesterol and 3-Hydroxy-5-cholestenoic Acid in the Rabbit. J. Biol. Chem. 1989, 264, 3818–3821. [Google Scholar] [CrossRef] [PubMed]
- Setchell, K.D.R.; Schwarz, M.; O’Connell, N.C.; Lund, E.G.; Davis, D.L.; Lathe, R.; Thompson, H.R.; Tyson, R.W.; Sokol, R.J.; Russell, D.W. Identification of a new inborn error in bile acid synthesis: Mutation of the oxysterol 7alpha-hydroxylase gene causes severe neonatal liver disease. J. Clin. Investig. 1998, 102, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.; Mills, P.B.; Footitt, E.; Gissen, P.; McClean, P.; Stahlschmidt, J.; Coupry, I.; Lavie, J.; Mochel, F.; Goizet, C.; et al. Liver disease in infancy caused by oxysterol 7α-hydroxylase deficiency: Successful treatment with chenodeoxycholic acid. J. Inherit. Metab. Dis. 2014, 37, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Mizuochi, T.; Kimura, A.; Suzuki, M.; Ueki, I.; Takei, H.; Nittono, H.; Kakiuchi, T.; Shigeta, T.; Sakamoto, S.; Fukuda, A.; et al. Successful heterozygous living donor liver transplantation for an oxysterol 7α-hydroxylase deficiency in a Japanese patient. Liver Transplant. 2011, 17, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Koike, M.; Sakiyama, M.; Okuda, S.; Okuda, M.; Tanaka, T.; Unno, A.; Nittono, H.; Takei, H.; Murai, T.; et al. 3beta-Hydroxy-Delta5-C27-steroid dehydrogenase/isomerase deficiency in a 23-year-old woman. Pediatr. Int. 2000, 42, 685–688. [Google Scholar] [CrossRef]
- Kimura, A.; Mizuochi, T.; Takei, H.; Ohtake, A.; Mori, J.; Shinoda, K.; Hashimoto, T.; Kasahara, M.; Togawa, T.; Murai, T.; et al. Bile Acid Synthesis Disorders in Japan: Long-Term Outcome and Chenodeoxycholic Acid Treatment. Dig. Dis. Sci. 2021, 66, 3885–3892. [Google Scholar] [CrossRef]
- Li-Hawkins, J.; Lund, E.G.; Turley, S.D.; Russell, D.W. Disruption of the Oxysterol 7α-Hydroxylase Gene in Mice. J. Biol. Chem. 2000, 275, 16536–16542. [Google Scholar] [CrossRef]
- Minowa, K.; Rodriguez-Agudo, D.; Suzuki, M.; Hirai, S.; Muto, Y.; Su, L.; Mitamura, K.; Ikegawa, S.; Heuman, D.M.; Zhou, H. Modulation of Mitochondrial Cholesterol Metabolites in Cyp7b1 Deficiency: A New Insight into a Mechanism of Initiating Nash. In Hepatology; WILEY: Hoboken, NJ, USA, 2022; pp. 697–698. [Google Scholar]
- Houben, T.; Bitorina, A.V.; Oligschlaeger, Y.; Jeurissen, M.L.; Rensen, S.; Köhler, S.E.; Westerterp, M.; Lütjohann, D.; Theys, J.; Romano, A.; et al. Sex-opposed inflammatory effects of 27-hydroxycholesterol are mediated via differences in estrogen signaling. J. Pathol. 2020, 251, 429–439. [Google Scholar] [CrossRef]
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2018, 69, 2241–2257. [Google Scholar] [CrossRef] [PubMed]
- Raselli, T.; Hearn, T.; Wyss, A.; Atrott, K.; Peter, A.; Frey-Wagner, I.; Spalinger, M.R.; Maggio, E.M.; Sailer, A.W.; Schmitt, J.; et al. Elevated oxysterol levels in human and mouse livers reflect nonalcoholic steatohepatitis. J. Lipid Res. 2019, 60, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Na, J.; Choi, S.A.; Khan, A.; Huh, J.Y.; Piao, L.; Hwang, I.; Ha, H.; Park, Y.H. Integrative Omics Reveals Metabolic and Transcriptomic Alteration of Nonalcoholic Fatty Liver Disease in Catalase Knockout Mice. Biomol. Ther. 2019, 27, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Evangelakos, I.; Schwinge, D.; Worthmann, A.; John, C.; Roeder, N.; Pertzborn, P.; Behrens, J.; Schramm, C.; Scheja, L.; Heeren, J. Oxysterol 7-α Hydroxylase (CYP7B1) Attenuates Metabolic-Associated Fatty Liver Disease in Mice at Thermoneutrality. Cells 2021, 10, 2656. [Google Scholar] [CrossRef]
- Shoji, S.; Maekawa, M.; Ogura, J.; Sato, T.; Mano, N. Identification cholesterol metabolites altered before the onset of nonalcoholic steatohepatitis by targeted metabolomics. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2022, 1867, 159135. [Google Scholar] [CrossRef]
- Suga, T.; Yamaguchi, H.; Ogura, J.; Shoji, S.; Maekawa, M.; Mano, N. Altered bile acid composition and disposition in a mouse model of non-alcoholic steatohepatitis. Toxicol. Appl. Pharmacol. 2019, 379, 114664. [Google Scholar] [CrossRef]
- Wu, Z.; Chiang, J.Y. Transcriptional regulation of human oxysterol 7α-hydroxylase gene (CYP7B1) by Sp1. Gene 2001, 272, 191–197. [Google Scholar] [CrossRef]
- Inoue, Y.; Yu, A.-M.; Yim, S.H.; Ma, X.; Krausz, K.W.; Inoue, J.; Xiang, C.C.; Brownstein, M.J.; Eggertsen, G.; Björkhem, I.; et al. Regulation of bile acid biosynthesis by hepatocyte nuclear factor 4α. J. Lipid Res. 2006, 47, 215–227. [Google Scholar] [CrossRef]
- Gupta, R.K.; Kaestner, K.H. HNF-4α: From MODY to late-onset type 2 diabetes. Trends Mol. Med. 2004, 10, 521–524. [Google Scholar] [CrossRef]
- Pass, G.J.; Becker, W.; Kluge, R.; Linnartz, K.; Plum, L.; Giesen, K.; Joost, H.-G.; Anderson, J.J.; Rao, S.P.; Rowe, B.; et al. Effect of Hyperinsulinemia and Type 2 Diabetes-Like Hyperglycemia on Expression of Hepatic Cytochrome P450 and GlutathioneS-Transferase Isoforms in a New Zealand Obese-Derived Mouse Backcross Population. Experiment 2002, 302, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Nojima, K.; Sugimoto, K.; Ueda, H.; Babaya, N.; Ikegami, H.; Rakugi, H. Analysis of hepatic gene expression profile in a spontaneous mouse model of type 2 diabetes under a high sucrose diet. Endocr. J. 2013, 60, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, S.B.; Haas, J.T.; Yu, B.B.; Bezy, O.; Jing, E.; Zhang, W.; Unterman, T.G.; Carey, M.C.; Kahn, C.R. Hepatic insulin resistance directly promotes formation of cholesterol gallstones. Nat. Med. 2008, 14, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Pettersson, H.; Norlin, M. Involvement of the PI3K/Akt pathway in estrogen-mediated regulation of human CYP7B1: Identification of CYP7B1 as a novel target for PI3K/Akt and MAPK signalling. J. Steroid Biochem. Mol. Biol. 2008, 112, 63–73. [Google Scholar] [CrossRef]
- Bi, Y.; Shi, X.; Zhu, J.; Guan, X.; Garbacz, W.G.; Huang, Y.; Gao, L.; Yan, J.; Xu, M.; Ren, S.; et al. Regulation of Cholesterol Sulfotransferase SULT2B1b by Hepatocyte Nuclear Factor 4α Constitutes a Negative Feedback Control of Hepatic Gluconeogenesis. Mol. Cell Biol. 2018, 38, e00654-17. [Google Scholar] [CrossRef]
- Coll, O.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology 2003, 38, 692–702. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Caballero, F.; Fernández, A.; De Lacy, A.M.; Fernández-Checa, J.C.; Caballería, J.; García-Ruiz, C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J. Hepatol. 2009, 50, 789–796. [Google Scholar] [CrossRef]
- Chen, W.; Chiang, J.Y. Regulation of human sterol 27-hydroxylase gene (CYP27A1) by bile acids and hepatocyte nuclear factor 4α (HNF4α). Gene 2003, 313, 71–82. [Google Scholar] [CrossRef]
- Hall, E.; Ren, S.; Hylemon, P.; Rodriguez-Agudo, D.; Redford, K.; Marques, D.; Kang, D.; Gil, G.; Pandak, W. Detection of the steroidogenic acute regulatory protein, StAR, in human liver cells. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2005, 1733, 111–119. [Google Scholar] [CrossRef]
- de la Rosa, L.C.; Garcia-Ruiz, C.; Vallejo, C.; Baulies, A.; Nuñez, S.; Monte, M.J.; Marin, J.J.; Baila-Rueda, L.; Cenarro, A.; Civeira, F.; et al. STARD1 promotes NASH-driven HCC by sustaining the generation of bile acids through the alternative mitochondrial pathway. J. Hepatol. 2021, 74, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef]
- Tichauer, M.J.E.; Morales, L.A.A.G.; Amigo, L.; Galdames, L.; Klein, V.S.; Quiñones, C.F.N.; Ferrada, C.; Alvarez, A.; Rio, M.-C.; Miquel, J.F.; et al. Overexpression of the cholesterol-binding protein MLN64 induces liver damage in the mouse. World J. Gastroenterol. 2007, 13, 3071–3079. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; García-Ruiz, C.; Lluis, J.M.; Coll, O.; Mari, M.; Fernández-Checa, J.C. Cholesterol Impairs the Adenine Nucleotide Translocator-mediated Mitochondrial Permeability Transition through Altered Membrane Fluidity. J. Biol. Chem. 2003, 278, 33928–33935. [Google Scholar] [CrossRef]
- Wu, M.K.; Cohen, D.E. Altered hepatic cholesterol metabolism compensates for disruption of phosphatidylcholine transfer protein in mice. Am. J. Physiol. Liver Physiol. 2005, 289, G456–G461. [Google Scholar] [CrossRef]
- Echegoyen, S.; Oliva, E.B.; Sepulveda, J.; Díaz-Zagoya, J.C.; Espinosa-García, M.T.; Pardo, J.P.; Martínez, F. Cholesterol increase in mitochondria: Its effect on inner-membrane functions, submitochondrial localization and ultrastructural morphology. Biochem. J. 1993, 289, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, C.; Mari, M.; Colell, A.; Morales, A.; Caballero, F.; Montero, J.; Terrones, O.; Basañez, G.; Fernández-Checa, J.C. Mitochondrial cholesterol in health and disease. Histol Histopathol. 2009, 24, 117–132. [Google Scholar] [CrossRef]
- Ha, S.-D.; Park, S.; Han, C.Y.; Nguyen, M.L.; Kim, S.O. Cellular Adaptation to Anthrax Lethal Toxin-Induced Mitochondrial Cholesterol Enrichment, Hyperpolarization, and Reactive Oxygen Species Generation through Downregulating MLN64 in Macrophages. Mol. Cell Biol. 2012, 32, 4846–4860. [Google Scholar] [CrossRef]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial Glutathione, a Key Survival Antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Solsona-Vilarrasa, E.; Fucho, R.; Torres, S.; Nuñez, S.; Nuño-Lámbarri, N.; Enrich, C.; García-Ruiz, C.; Fernández-Checa, J.C. Cholesterol enrichment in liver mitochondria impairs oxidative phosphorylation and disrupts the assembly of respiratory supercomplexes. Redox Biol. 2019, 24, 101214. [Google Scholar] [CrossRef]
- Zurkinden, L.; Mansour, Y.T.; Rohrbach, B.; Vogt, B.; Mistry, H.D.; Escher, G. Hepatic caveolin-1 is enhanced in Cyp27a1/ApoE double knockout mice. FEBS Open Bio 2016, 6, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Zurkinden, L.; Sviridov, D.; Vogt, B.; Escher, G. Sterol 27-hydroxylase gene dosage and the antiatherosclerotic effect of Rifampicin in mice. Biosci. Rep. 2018, 38, BSR20171162. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Xing, Y.; Liu, L.; Fan, X.; Liu, L.; Geng, T.; Gong, D. GC-TOF-MS-Based Metabolomics Analyses of Liver and Intestinal Contents in the Overfed vs. Normally-Fed Geese. Animals 2020, 10, 2375. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef]
- Serviddio, G.; Blonda, M.; Bellanti, F.; Villani, R.; Iuliano, L.; Vendemiale, G. Oxysterols and redox signaling in the pathogenesis of non-alcoholic fatty liver disease. Free. Radic. Res. 2013, 47, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Villani, R.; Tamborra, R.; Blonda, M.; Iannelli, G.; di Bello, G.; Facciorusso, A.; Poli, G.; Iuliano, L.; Avolio, C.; et al. Synergistic interaction of fatty acids and oxysterols impairs mitochondrial function and limits liver adaptation during nafld progression. Redox Biol. 2017, 15, 86–96. [Google Scholar] [CrossRef]
- Bellanti, F.; Mitarotonda, D.; Tamborra, R.; Blonda, M.; Iannelli, G.; Petrella, A.; Sanginario, V.; Iuliano, L.; Vendemiale, G.; Serviddio, G. Oxysterols induce mitochondrial impairment and hepatocellular toxicity in non-alcoholic fatty liver disease. Free. Radic. Biol. Med. 2014, 75, S16–S17. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, X.; Hu, Q.; Wu, J.; Wang, G.; Hong, Z.; Ren, J. Mitochondrial DNA in liver inflammation and oxidative stress. Life Sci. 2019, 236, 116464. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2019, 129, 546–555. [Google Scholar] [CrossRef]
- Umetani, M.; Ghosh, P.; Ishikawa, T.; Umetani, J.; Ahmed, M.; Mineo, C.; Shaul, P.W. The Cholesterol Metabolite 27-Hydroxycholesterol Promotes Atherosclerosis via Proinflammatory Processes Mediated by Estrogen Receptor Alpha. Cell Metab. 2014, 20, 172–182. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Kaplowitz, N.; Lebeaupin, C.; Kroemer, G.; Kaufman, R.J.; Malhi, H.; Ren, J. Endoplasmic reticulum stress in liver diseases. Hepatology 2023, 77, 619–639. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Gentile, C.L.; Frye, M.; Pagliassotti, M.J. Endoplasmic Reticulum Stress and the Unfolded Protein Response in Nonalcoholic Fatty Liver Disease. Antioxid. Redox Signal. 2011, 15, 505–521. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef]
- Park, S.W.; Zhou, Y.; Lee, J.; Lee, J.; Ozcan, U. Sarco(endo)plasmic reticulum Ca2+ -ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 19320–19325. [Google Scholar] [CrossRef]
- Wang, X.; Cai, B.; Yang, X.; Sonubi, O.O.; Zheng, Z.; Ramakrishnan, R.; Shi, H.; Valenti, L.; Pajvani, U.B.; Sandhu, J.; et al. Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 969–986.e7. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, Z.; Caviglia, J.M.; Corey, K.E.; Herfel, T.M.; Cai, B.; Masia, R.; Chung, R.T.; Lefkowitch, J.H.; Schwabe, R.F.; et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2016, 24, 848–862. [Google Scholar] [CrossRef]
- Rodriguez-Agudo, D.; Ren, S.; Hylemon, P.B.; Redford, K.; Natarajan, R.; Del Castillo, A.; Gil, G.; Pandak, W.M. Human StarD5, a cytosolic StAR-related lipid binding protein. J. Lipid Res. 2005, 46, 1615–1623. [Google Scholar] [CrossRef]
- Rodriguez-Agudo, D.; Ren, S.; Hylemon, P.B.; Montañez, R.; Redford, K.; Natarajan, R.; Medina, M.A.; Gil, G.; Pandak, W.M. Localization of StarD5 cholesterol binding protein. J. Lipid Res. 2006, 47, 1168–1175. [Google Scholar] [CrossRef]
- Rodriguez-Agudo, D.; Malacrida, L.; Kakiyama, G.; Sparrer, T.; Fortes, C.; Maceyka, M.; Subler, M.A.; Windle, J.J.; Gratton, E.; Pandak, W.M.; et al. StarD5: An ER stress protein regulates plasma membrane and intracellular cholesterol homeostasis. J. Lipid Res. 2019, 60, 1087–1098. [Google Scholar] [CrossRef]
- Rodriguez-Agudo, D.; Kakiyama, G.; Pandak Jr, W.M. StarD5, Maintains Cholesterol and Lipid Homeostasis through its ability to Transport Cholesterol to the Pm and Increase Vldl Secretion: Protecting against Transition from Nafl to Nash. In Hepatology; Willey: Hoboken, NJ, USA, 2022; pp. S843–S844. [Google Scholar]
- Fromenty, B.; Roden, M. Mitochondrial alterations in fatty liver diseases. J. Hepatol. 2022, 78, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Zhang, X.; Xu, L.; Kakiyama, G.; Heuman, D.; Sanyal, A.; Pandak, W.M.; Yin, L.; Xie, W.; Ren, S. Oxysterol sulfation by cytosolic sulfotransferase suppresses liver X receptor/sterol regulatory element binding protein–1c signaling pathway and reduces serum and hepatic lipids in mouse models of nonalcoholic fatty liver disease. Metabolism 2012, 61, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, Y.; Bai, Q.; Li, X.; Han, J.; Hou, Y.; Ji, Y.; Zhang, Z. Inhibition of LXR signaling by SULT2B1b promotes liver regeneration after partial hepatectomy in mouse models of nonalcoholic fatty liver disease. Am. J. Physiol. Liver Physiol. 2020, 319, G87–G96. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Kim, J.K.; Bai, Q.; Zhang, X.; Kakiyama, G.; Min, H.-K.; Sanyal, A.J.; Pandak, W.M.; Ren, S. 5-Cholesten-3β,25-Diol 3-Sulfate Decreases Lipid Accumulation in Diet-Induced Nonalcoholic Fatty Liver Disease Mouse Model. Mol. Pharmacol. 2012, 83, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tai, Y.-L.; Zhao, D.; Zhang, Y.; Yan, J.; Kakiyama, G.; Wang, X.; Gurley, E.C.; Liu, J.; Liu, J.; et al. Berberine Prevents Disease Progression of Nonalcoholic Steatohepatitis through Modulating Multiple Pathways. Cells 2021, 10, 210. [Google Scholar] [CrossRef]
- Deng, Z.; Meng, C.; Huang, H.; Song, S.; Fu, L.; Fu, Z. The different effects of psyllium husk and orlistat on weight control, the amelioration of hypercholesterolemia and non-alcohol fatty liver disease in obese mice induced by a high-fat diet. Food Funct. 2022, 13, 8829–8849. [Google Scholar] [CrossRef]
- Zhao, W.-W.; Xiao, M.; Wu, X.; Li, X.-W.; Li, X.-X.; Zhao, T.; Yu, L.; Chen, X.-Q. Ilexsaponin A1 Ameliorates Diet-Induced Nonalcoholic Fatty Liver Disease by Regulating Bile Acid Metabolism in Mice. Front. Pharmacol. 2021, 12, 771976. [Google Scholar] [CrossRef]
- Torres, S.; Baulies, A.; Insausti-Urkia, N.; Alarcón-Vila, C.; Fucho, R.; Solsona-Vilarrasa, E.; Núñez, S.T.; Robles, D.; Ribas, V.; Wakefield, L.; et al. Endoplasmic Reticulum Stress-Induced Upregulation of STARD1 Promotes Acetaminophen-Induced Acute Liver Failure. Gastroenterology 2019, 157, 552–568. [Google Scholar] [CrossRef]
- Torres, S.; Solsona-Vilarrasa, E.; Nuñez, S.; Matías, N.; Insausti-Urkia, N.; Castro, F.; Casasempere, M.; Fabriás, G.; Casas, J.; Enrich, C.; et al. Acid ceramidase improves mitochondrial function and oxidative stress in Niemann-Pick type C disease by repressing STARD1 expression and mitochondrial cholesterol accumulation. Redox Biol. 2021, 45, 102052. [Google Scholar] [CrossRef]
- Fernandez, A.; Matias, N.; Fucho, R.; Ribas, V.; Von Montfort, C.; Nuño, N.; Baulies, A.; Martinez, L.; Tarrats, N.; Mari, M.; et al. ASMase is required for chronic alcohol induced hepatic endoplasmic reticulum stress and mitochondrial cholesterol loading. J. Hepatol. 2013, 59, 805–813. [Google Scholar] [CrossRef]
- Akula, N.; Midzak, A.; Lecanu, L.; Papadopoulos, V. Identification of small-molecule inhibitors of the steroidogenic acute regulatory protein (STARD1) by structure-based design. Bioorganic Med. Chem. Lett. 2012, 22, 4139–4143. [Google Scholar] [CrossRef] [PubMed]
- Bhangoo, A.; Anhalt, H.; Ten, S.; King, S.R. Phenotypic variations in lipoid congenital adrenal hyperplasia. Pediatr. Endocrinol. Rev. 2006, 3, 258–271. [Google Scholar] [PubMed]
- Baker, B.Y.; Lin, L.; Kim, C.J.; Raza, J.; Smith, C.P.; Miller, W.L.; Achermann, J.C. Nonclassic Congenital Lipoid Adrenal Hyperplasia: A New Disorder of the Steroidogenic Acute Regulatory Protein with Very Late Presentation and Normal Male Genitalia. J. Clin. Endocrinol. Metab. 2006, 91, 4781–4785. [Google Scholar] [CrossRef] [PubMed]
- Metherell, L.A.; Naville, D.; Halaby, G.; Begeot, M.; Huebner, A.; Nürnberg, G.; Nürnberg, P.; Green, J.; Tomlinson, J.; Krone, N.; et al. Nonclassic Lipoid Congenital Adrenal Hyperplasia Masquerading as Familial Glucocorticoid Deficiency. J. Clin. Endocrinol. Metab. 2009, 94, 3865–3871. [Google Scholar] [CrossRef]
- Khoury, K.; Barbar, E.; AinMelk, Y.; Ouellet, A.; Lavigne, P.; Lehoux, J.-G. Thirty-Eight-Year Follow-Up of Two Sibling Lipoid Congenital Adrenal Hyperplasia Patients Due to Homozygous Steroidogenic Acute Regulatory (STARD1) Protein Mutation. Molecular Structure and Modeling of the STARD1 L275P Mutation. Front. Neurosci. 2016, 10, 527. [Google Scholar] [CrossRef] [PubMed]
- Going, C.C.; Alexandrova, L.; Lau, K.; Yeh, C.Y.; Feldman, D.; Pitteri, S.J. Vitamin D supplementation decreases serum 27-hydroxycholesterol in a pilot breast cancer trial. Breast Cancer Res. Treat. 2017, 167, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Mast, N.; Lin, J.B.; Pikuleva, I.A. Marketed Drugs Can Inhibit Cytochrome P450 27A1, a Potential New Target for Breast Cancer Adjuvant Therapy. Mol. Pharmacol. 2015, 88, 428–436. [Google Scholar] [CrossRef]
- Mast, N.; Reem, R.; Bederman, I.; Huang, S.; DiPatre, P.L.; Björkhem, I.; Pikuleva, I. Cholestenoic Acid Is an Important Elimination Product of Cholesterol in the Retina: Comparison of Retinal Cholesterol Metabolism with That in the Brain. Investig. Opthalmol. Vis. Sci. 2011, 52, 594–603. [Google Scholar] [CrossRef]
- Tang, Y.-P.; Gong, J.-Y.; Setchell, K.D.R.; Zhang, W.; Zhao, J.; Wang, J.-S. Successful treatment of infantile oxysterol 7α-hydroxylase deficiency with oral chenodeoxycholic acid. BMC Gastroenterol. 2021, 21, 163. [Google Scholar] [CrossRef]
- Björkhem, I. Cerebrotendinous xanthomatosis. Curr. Opin. Infect. Dis. 2013, 24, 283–287. [Google Scholar] [CrossRef]
- Lam, M.; Mast, N.; Pikuleva, I.A. Drugs and Scaffold That Inhibit Cytochrome P450 27A1 In Vitro and In Vivo. Mol. Pharmacol. 2018, 93, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Xu, L.; Kakiyama, G.; Runge-Morris, M.A.; Hylemon, P.B.; Yin, L.; Pandak, W.M.; Ren, S. Sulfation of 25-hydroxycholesterol by SULT2B1b decreases cellular lipids via the LXR/SREBP-1c signaling pathway in human aortic endothelial cells. Atherosclerosis 2011, 214, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, X.; Ren, S. Cholesterol Metabolites 25-Hydroxycholesterol and 25-Hydroxycholesterol 3-Sulfate Are Potent Paired Regulators: From Discovery to Clinical Usage. Metabolites 2020, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, W.; Brown, J.E.; Chen, L.; Pandak, W.M.; Hylemon, P.B.; Ren, S. 25-Hydroxycholesterol 3-sulfate is an endogenous ligand of DNA methyltransferases in hepatocytes. J. Lipid Res. 2021, 62, 100063. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pandak, W.M.; Lesnefsky, E.J.; Hylemon, P.B.; Ren, S. 25-Hydroxycholesterol 3-Sulfate Recovers Acetaminophen Induced Acute Liver Injury via Stabilizing Mitochondria in Mouse Models. Cells 2021, 10, 3027. [Google Scholar] [CrossRef]
- Fu, S.; Fan, J.; Blanco, J.; Giménez-Cassina, A.; Danial, N.N.; Watkins, S.M.; Hotamisligil, G.S. Polysome Profiling in Liver Identifies Dynamic Regulation of Endoplasmic Reticulum Translatome by Obesity and Fasting. PLoS Genet. 2012, 8, e1002902. [Google Scholar] [CrossRef]
- Ma, H.; Sales, V.M.; Wolf, A.R.; Subramanian, S.; Matthews, T.J.; Chen, M.; Sharma, A.; Gall, W.; Kulik, W.; Cohen, D.E.; et al. Attenuated Effects of Bile Acids on Glucose Metabolism and Insulin Sensitivity in a Male Mouse Model of Prenatal Undernutrition. Endocrinology 2017, 158, 2441–2452. [Google Scholar] [CrossRef]
- Zhuang, Q.; Ye, X.; Shen, S.; Cheng, J.; Shi, Y.; Wu, S.; Xia, J.; Ning, M.; Dong, Z.; Wan, X. Astragalus Polysaccharides Ameliorate Diet-Induced Gallstone Formation by Modulating Synthesis of Bile Acids and the Gut Microbiota. Front. Pharmacol. 2021, 12, 701003. [Google Scholar] [CrossRef]
- Sun, L.; Pang, Y.; Wang, X.; Wu, Q.; Liu, H.; Liu, B.; Liu, G.; Ye, M.; Kong, W.; Jiang, C. Ablation of gut microbiota alleviates obesity-induced hepatic steatosis and glucose intolerance by modulating bile acid metabolism in hamsters. Acta Pharm. Sin. B 2019, 9, 702–710. [Google Scholar] [CrossRef]
- Meng, Y.; Meng, K.; Zhao, X.; Li, N.; Gao, Q.; Wu, S.; Cui, Y. Protective Effects of Yinchenhao Decoction on Cholesterol Gallstone in Mice Fed a Lithogenic Diet by Regulating LXR, CYP7A1, CYP7B1, and HMGCR Pathways. Evid. Based Complement Altern. Med. 2018, 2018, 8134918. [Google Scholar] [CrossRef]
- Duan, J.; Pan, J.; Sun, M.; Fang, Y. Comparative multiomics study of the effects of Ellagic acid on the gut environment in young and adult mice. Food Res. Int. 2022, 161, 111819. [Google Scholar] [CrossRef] [PubMed]
- Steinman, J.B.; Salomao, M.A.; Pajvani, U.B. Zonation in NASH—A key paradigm for understanding pathophysiology and clinical outcomes. Liver Int. 2021, 41, 2534–2546. [Google Scholar] [CrossRef]
- Rizzolo, D.; Kong, B.; Taylor, R.E.; Brinker, A.; Goedken, M.; Buckley, B.; Guo, G.L. Bile acid homeostasis in female mice deficient in Cyp7a1 and Cyp27a1. Acta Pharm. Sin. B 2021, 11, 3847–3856. [Google Scholar] [CrossRef]
- Takeshita, Y.; Takamura, T.; Honda, M.; Kita, Y.; Zen, Y.; Kato, K.-I.; Misu, H.; Ota, T.; Nakamura, M.; Yamada, K.; et al. The effects of ezetimibe on non-alcoholic fatty liver disease and glucose metabolism: A randomised controlled trial. Diabetologia 2014, 57, 878–890. [Google Scholar] [CrossRef]
- Cho, Y.; Rhee, H.; Kim, Y.-E.; Lee, M.; Lee, B.-W.; Kang, E.S.; Cha, B.-S.; Choi, J.-Y.; Lee, Y.-H. Ezetimibe combination therapy with statin for non-alcoholic fatty liver disease: An open-label randomized controlled trial (ESSENTIAL study). BMC Med. 2022, 20, 93. [Google Scholar] [CrossRef] [PubMed]
- Crisby, M.; Nilsson, J.; Kostulas, V.; Björkhem, I.; Diczfalusy, U. Localization of sterol 27-hydroxylase immuno-reactivity in human atherosclerotic plaques. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1997, 1344, 278–285. [Google Scholar] [CrossRef]
- Allen, A.M.; Taylor, J.M.W.; Graham, A. Mitochondrial (dys)function and regulation of macrophage cholesterol efflux. Clin. Sci. 2013, 124, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Hagey, L.R. Key discoveries in bile acid chemistry and biology and their clinical applications: History of the last eight decades. J. Lipid Res. 2014, 55, 1553–1595. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Model | Main Findings in Terms of Role Oxysterols (Cholestenoic Acids) in NAFLD Pathogenesis | Pitfalls and Unresolved Question | |
|---|---|---|---|
| Raselli et al. [64] | 1. Biopsy-proven NASH | 1. Elevated 24(R/S)HC and 7αHC but no changes in 25HC and 26HC levels in NASH livers compared with control livers without inflammation. | 1. The data compared advanced NASH (NAS score 3–5; NASH fibrosis) and normal livers. No comparison is provided between steatosis and a normal liver. |
| 2. C57Bl/6; Ch25h−/−; Ebi2−/−; Cyp7b1−/− mice fed with high-fat, high-cholesterol diet and high-fructose corn-syrup in their drinking water for 10–20 weeks. | 2. Elevated 24(R/S)HC and 7αHC but no changes in 25HC and 26HC levels in NASH livers (C57Bl/6). No genotype-related differences were found in the development of NASH; no essential role of these gene expressions and/or 26HC in NASH pathogenesis. | 2. The study focused on established NASH in which hepatic IR is no longer the key factor for oxysterol regulation. | |
| Kakiyama et al. [20] | 1. ♂ B6/129 mice fed with a Western diet for 2–6 weeks | 1. Elevated liver 26HC/3βHCA and suppressed Cyp7b1 mRNA in early fatty liver without histologic inflammation. Correlated levels of 26HC/3βHCA to HOMA-IR sores and liver enzymes (ALT). | 1. It remains unclear whether the elevated levels of 26HC/3βHCA themselves are direct causes of hepatotoxicity. Mechanistic study is missing. |
| 2. Streptozotocin-injected ♂ C57Bl/6 mice fed with LFD. | 2. Plasma insulin level is directly correlated to hepatic Cyp7b1 mRNA expression. | 2. Mechanism as to how insulin signal pathway regulates hepatic Cyp7b1 gene expression was not studied. | |
| Na et al. [65] | ♂ Catalase knockout mouse; C57Bl/6J fed with HFD for 11 weeks | Significantly reduced 3β,7α-diHCA secondary to Cyp7b1 mRNA downregulation in the livers of HFD-fed mice with higher serum ALT. The effect was more profound in Catalase knockout mice. | Histologic evaluation is missing and NAFLD stage is unclear; the presence of IR is unknown; the tissue 26HC/3βHCA level is unknown. The causative factor of liver injury cannot be proven solely by this study. |
| Evangelakos et al. [66] | ♂ Cyp7b1−/−; C57Bl/6 littermates fed with a choline-deficient HFD and housed in a thermoneutral temperature for 8 months. | The thermoneutral housing of Cyp7b1−/− mice promoted MAFLD more profoundly compared to the wild-type mice littermates. However, oxysterols did not correlate with the aggravation of MAFLD. | The model focused on established NASH, and oxysterol correlation with early disease progression is unknown. It is unclear whether choline-deficient HFD metabolically follows human metabolic disease. |
| Shoji et al. [67] Suga et al. [68] | ♂ C57Bl/6J fed with choline-deficient, methionine-reduced high-fat diet for 3–21 days | When transitioning from NAFL to NASH, the hepatic desmosterol, 4βHC, secondary bile acid, etc., levels were significantly reduced. However, 26HC/3βHCA levels were unchanged. | Unclear whether choline-deficient HFD metabolically follows human metabolic disease. |
| Minowa et al. [22] | 1. Biopsy-proven NASH patients | 1. Elevated 26HC/3βHCA levels in NASH livers. | 1. NASH patient cohort and the 26HC/3βHCA levels in early fatty liver remain unknown. |
| 2. ♂ Cyp7b1−/− mice fed with WD and HCD for 4 weeks. | 2. Elevated 24(S),25EC, 26HC, and 3βHCA in the WD-fed Cyp7b1−/− mice with IR. Oxysterol sulfation and glucuronidation can also be impaired in early fatty liver with IR; contributing to the accumulation of 26HC/3βHCA. | 2 and 3. There were no significant changes in the enzyme activities of oxidative phosphorylation with early NAFL, although these mRNAs were significantly downregulated. It remains unclear if oxysterol-associated mitochondrial dysfunction is the initial cause of hepatocyte injury. | |
| 3. ♂ B6/129 mice fed with Western diet for 2–8 weeks | 3. Accumulated 26HC/3βHCA in the liver mitochondria of NAFL mice with elevation of ALT. RNA-seq data showed that genes in mitochondria oxidative phosphorylation and thermogenesis are impaired. |
| Agent or Models | Animal Model and Treatment | Results and Oxysterol-Related Mechanism |
|---|---|---|
| Liver-specific StarD1 knockout mouse; (StarD1Δhep) mice [82] | Diethylnitorisamine (DEN; 25 mg/kg)-injected StarD1Δhep (liver-specific StarD1 knockout mice) fed with a high-fat, high-cholesterol diet for 26 weeks. | StarD1Δhep mice were less sensitive to DEN + HFHC-diet-induced HCC. |
| Ad-Sult2b1 [114] | LDL−/− mice (♂/♀) and mice fed with a HFD for 10 weeks. Ad-Sult2b1 virus was injected (i.v., 1 × 108) 6 days before sacrifice. | Improved NAFLD condition (i.e., ALT/AST↓ *; cholesterol/triglyceride↓; Steatosis (Histology)↓; hepatic inflammation and stress responses (mRNA)). |
| Ad-Sult2b1 [115] | Ad-Sult2b1 virus was injected (i.v., 1 × 108) into C57Bl/6 mice, and the mice were fed a HFD for 8 weeks. | The Ad-Sult2b1-injected mice promoted liver regeneration after 70% partial hepatectomy. |
| 25-Hydroxycholesterol 3-Sulfate [116] | C57BL/6J mice (♀) fed with a HFD for 16 weeks. As of 10 weeks after the HFD feeding was initiated, 25HC3S (25 mg/kg, i.p.) was injected twice a day for 6 weeks. | Improved NAFLD condition (i.e., ALT/AST↓; cholesterol/triglyceride↓; steatosis (histology)↓; improved glucose tolerance test). |
| Coffee [21] | B6/129 mice (♂) fed with 1% or 4% (wt/wt) regular coffee or decaffeinated coffee blended WD for two weeks. | The coffee-fed mice had significantly lower serum ALT and hepatic inflammatory mRNAs and improved HOMA-IR scores compared to the WD-only fed mice. These mice had reduced 26HC/3βHCA secondary to upregulated hepatic Sult2b1 and Cyp7b1 mRNAs. |
| Berberine [117] | B6/129 mice (♂/♀) were fed with a Western diet–high-fructose diet to up to 21 weeks. As of 12 weeks of age, Berberine (50 mg/kg/day) was administered via gavage. | Significantly improved histology, NAS scores, and NASH-associated inflammation and stress responses in BBR-treated mice. Higher hepatic Cyp7b1 mRNA in BBR-treated mice compared to the mice without treatment. |
| Psyllium husk (lipase inhibitor) [118] | C57Bl/6 (♂) mice fed a high-fat diet for 16 weeks. Psyllium husk (140 mg/kg) was administered via oral gavage three times/day. | Improved NAFLD condition (i.e., body weight↓; ALT/AST↓; cholesterol/triglyceride↓; NAS score↓; steatosis (histology)↓). Higher hepatic Cyp7b1 mRNA in the Psyllium-husk-treated mice compared to the mice without treatment. |
| Ortlistat (lipase inhibitor) [118] | C57Bl/6 (♂) mice fed a high-fat diet for 16 weeks. Ortlistat (20 mg/kg × 1 and 10 mg/kg × 2) was administered via oral gavage. | Improved NAFLD condition (i.e., body weight↓; ALT/AST↓; cholesterol/triglyceride↓; NAS score↓; steatosis (histology)↓). Higher hepatic Cyp7b1 mRNA in the Ortlistat-treated mice compared to the mice without treatment. |
| Ilexaponin A1 [119] | C57BL/6 (♂) mice fed a high-fat diet for 8 weeks. Ilexaponin A1 (120 mg/kg/day) was administered via oral gavage. | Improved NAFLD condition (i.e., ALT/AST↓; NAS score↓; steatosis (histology)↓; hepatic inflammatory genes↓) of the Ilexaponin-A1-administered mice. Higher hepatic Cyp7b1 mRNA in the Ilexaponin-A1-treated mice compared to the mice without treatment. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kakiyama, G.; Rodriguez-Agudo, D.; Pandak, W.M. Mitochondrial Cholesterol Metabolites in a Bile Acid Synthetic Pathway Drive Nonalcoholic Fatty Liver Disease: A Revised “Two-Hit” Hypothesis. Cells 2023, 12, 1434. https://doi.org/10.3390/cells12101434
Kakiyama G, Rodriguez-Agudo D, Pandak WM. Mitochondrial Cholesterol Metabolites in a Bile Acid Synthetic Pathway Drive Nonalcoholic Fatty Liver Disease: A Revised “Two-Hit” Hypothesis. Cells. 2023; 12(10):1434. https://doi.org/10.3390/cells12101434
Chicago/Turabian StyleKakiyama, Genta, Daniel Rodriguez-Agudo, and William M. Pandak. 2023. "Mitochondrial Cholesterol Metabolites in a Bile Acid Synthetic Pathway Drive Nonalcoholic Fatty Liver Disease: A Revised “Two-Hit” Hypothesis" Cells 12, no. 10: 1434. https://doi.org/10.3390/cells12101434
APA StyleKakiyama, G., Rodriguez-Agudo, D., & Pandak, W. M. (2023). Mitochondrial Cholesterol Metabolites in a Bile Acid Synthetic Pathway Drive Nonalcoholic Fatty Liver Disease: A Revised “Two-Hit” Hypothesis. Cells, 12(10), 1434. https://doi.org/10.3390/cells12101434