

Pharmacological Cardioprotection against Ischemia Reperfusion Injury—The Search for a Clinical Effective Therapy

 , ,
, ,  , and
, and
Abstract

1. Introduction
2. Mechanisms of Cardiac IRI
2.1. Decreased ΔGATP, Acidosis, and Ion Disturbances as Initial Processes Driving IRI
2.2. Oxidative Stress Involvement in IRI
2.3. Involvement of Intermediate Metabolites in IRI
2.3.1. Succinate
2.3.2. Glycogen and Glucose-6-Phosphate
2.3.3. Acylcarnitines
2.4. Cofactors Involved in IRI
3. Novel Pharmacological Strategies Targeting IRI in Cardiomyocytes
3.1. NAD+ Precursors
3.2. Malonate
3.3. NLRP3 Inflammasome Inhibitors
3.4. Caspase and Calpain Inhibitors
4. Translating Preclinical Cardioprotection into the Clinical Arena: Role of Risk Factors, Comorbidities, Comedications, Peri-Operative Care, and Ischemia Duration
5. Volatile Anesthetics and Noble Gases for Cardioprotection against IRI
5.1. Volatile Anesthetics
5.2. Noble Gases
6. Translating Preclinical Cardioprotection by Volatile Anesthetics and Noble Gases into the Clinical Arena
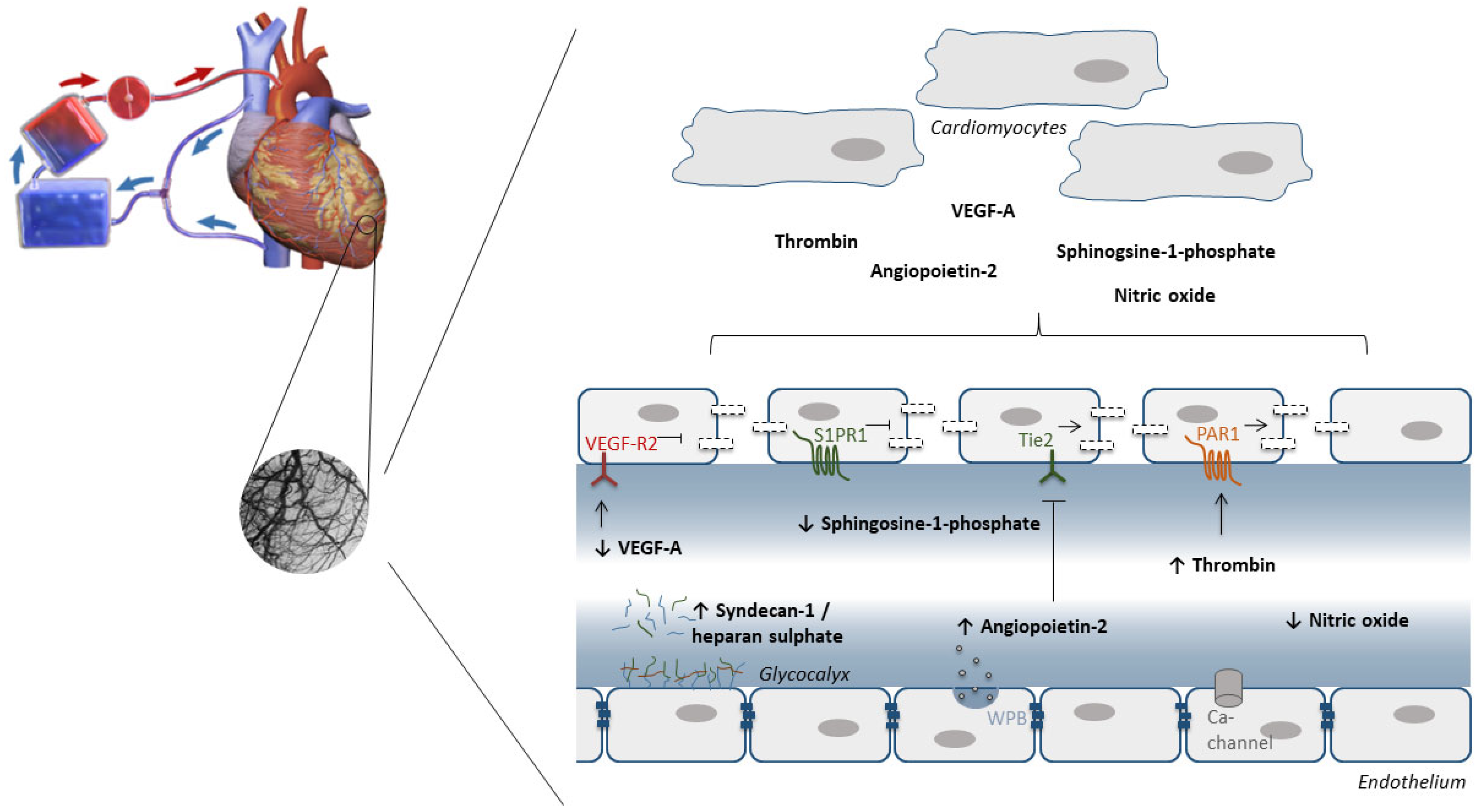
7. The Endothelium as Target to Protect against IRI during Cardiac Surgery with Cardiopulmonary Bypass
7.1. Oxidative Stress
7.2. Glycocalyx
7.3. Endothelial Barrier Function
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferdinandy, P.; Andreadou, I.; Baxter, G.F.; Bøtker, H.E.; Davidson, S.M.; Dobrev, D.; Gersh, B.J.; Heusch, G.; Lecour, S.; Ruiz-Meana, M.; et al. Interaction of Cardiovascular Nonmodifiable Risk Factors, Comorbidities and Comedications with Ischemia/Reperfusion Injury and Cardioprotection by Pharmacological Treatments and Ischemic Conditioning. Pharmacol. Rev. 2022, 75, 159–216. [Google Scholar] [CrossRef]
- Makkos, A.; Ágg, B.; Petrovich, B.; Varga, Z.V.; Görbe, A.; Ferdinandy, P. Systematic review and network analysis of microRNAs involved in cardioprotection against myocardial ischemia/reperfusion injury and infarction: Involvement of redox signalling. Free. Radic. Biol. Med. 2021, 172, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.V.; Zvara, Á.; Faragó, N.; Kocsis, G.F.; Pipicz, M.; Gaspar, R.; Bencsik, P.; Görbe, A.; Csonka, C.; Puskás, L.G.; et al. MicroRNAs associated with ischemia-reperfusion injury and cardioprotection by ischemic pre- and postconditioning: ProtectomiRs. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H216–H227. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Bell, R.M.; Bøtker, H.E.; Zuurbier, C.J. SGLT2 inhibitors reduce infarct size in reperfused ischemic heart and improve cardiac function during ischemic episodes in preclinical models. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165770. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Q.; Christodoulou, A.; Mylonas, N.; Bakker, D.; Nederlof, R.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; Wakker, V.; et al. Sodium Glucose Cotransporter-2 Inhibitor Empagliflozin Reduces Infarct Size Independently of Sodium Glucose Cotransporter-2. Circulation 2023, 147, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Yellon, D.M.; Sivaraman, V.; Das, D.; Boi-Doku, C.; Arjun, S.; Zheng, Y.; Riquelme, J.A.; Kearney, J.; Sharma, V.; et al. Plasma Exosomes Protect the Myocardium from Ischemia-Reperfusion Injury. J. Am. Coll. Cardiol. 2015, 65, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; E Crespo-Avilan, G.; Hernandez-Resendiz, S.; Ong, S.-G.; Hausenloy, D.J. Extracellular vesicles-mediating and delivering cardioprotection in acute myocardial infarction and heart failure. Cond. Med. 2020, 3, 227–238. [Google Scholar]
- Bell, R.M.; Yellon, D.M. There is More to Life than Revascularization: Therapeutic Targeting of Myocardial Ischemia/Reperfusion Injury. Cardiovasc. Ther. 2010, 29, e67–e79. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dorado, D.; Théroux, P.; Duran, J.M.; Solares, J.; Alonso, J.; Sanz, E.; Munoz, R.; Elizaga, J.; Botas, J.; Fernandez-Avilés, F. Selective inhibition of the contractile apparatus. A new approach to modification of infarct size, infarct composition, and infarct geometry during coronary artery occlusion and reperfusion. Circulation 1992, 85, 1160–1174. [Google Scholar] [CrossRef]
- Fiolet, J.; Baartscheer, A.; Schumacher, C.; Coronel, R.; ter Welle, H. The change of the free energy of ATP hydrolysis during global ischemia and anoxia in the rat heart: Its possible role in the regulation of transsarcolemmal sodium and potassium gradients. J. Mol. Cell. Cardiol. 1984, 16, 1023–1036. [Google Scholar] [CrossRef]
- Steenbergen, C.; Murphy, E.; Watts, J.A.; London, R.E. Correlation between cytosolic free calcium, contracture, ATP, and irreversible ischemic injury in perfused rat heart. Circ. Res. 1990, 66, 135–146. [Google Scholar] [CrossRef]
- Van Borren, M.M.; Zegers, J.G.; Baartscheer, A.; Ravesloot, J.H. Contribution of NHE-1 to cell length shortening of normal and failing rabbit cardiac myocytes. J. Mol. Cell. Cardiol. 2006, 41, 706–715. [Google Scholar] [CrossRef]
- Karmazyn, M.; Gan, X.T.; Humphreys, R.A.; Yoshida, H.; Kusumoto, K. The myocardial Na(+)-H(+) exchange: Structure, regulation, and its role in heart disease. Circ. Res. 1999, 85, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Miyata, N.; Morris, L.L.; Chen, Q.; Thorne, C.; Singla, A.; Zhu, W.; Winter, M.; Melton, S.D.; Li, H.; Sifuentes-Dominguez, L.; et al. Microbial Sensing by Intestinal Myeloid Cells Controls Carcinogenesis and Epithelial Differentiation. Cell Rep. 2018, 24, 2342–2355. [Google Scholar] [CrossRef]
- Yang, X.-M.; Cohen, M.V.; Sayner, S.; Audia, J.P.; Downey, J.M. Lethal Caspase-1/4-Dependent Injury Occurs in the First Minutes of Coronary Reperfusion and Requires Calpain Activity. Int. J. Mol. Sci. 2023, 24, 3801. [Google Scholar] [CrossRef]
- Tani, M.; Hasegawa, H.; Suganuma, Y.; Shinmura, K.; Kayashi, Y.; Nakamura, Y. Protection of ischemic myocardium by inhibition of contracture in isolated rat heart. Am. J. Physiol. Content 1996, 271, H2515–H2519. [Google Scholar] [CrossRef] [PubMed]
- Zweier, J.L.; Flaherty, J.T.; Weisfeldt, M.L. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Natl. Acad. Sci. USA 1987, 84, 1404–1407. [Google Scholar] [CrossRef]
- Maupoil, V.; Rochette, L.; Tabard, A.; Clauser, P.; Harpey, C. Evolution of free radical formation during low-flow ischemia and reperfusion in isolated rat heart. Cardiovasc. Drugs Ther. 1990, 4, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Timoshin, A.A.; Tskitishvili, O.V.; Serebryakova, L.I.; Kuzmin, A.I.; Medvedev, O.; Ruuge, E.K. Microdialysis study of ischemia-induced hydroxyl radicals in the canine heart. Experientia 1994, 50, 677–679. [Google Scholar] [CrossRef]
- Vanden Hoek, T.L.; Li, C.; Shao, Z.; Schumacker, P.T.; Becker, L.B. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J. Mol. Cell. Cardiol. 1997, 29, 2571–2583. [Google Scholar] [CrossRef]
- Eaton, P.; Li, J.-M.; Hearse, D.J.; Shattock, M.J. Formation of 4-hydroxy-2-nonenal-modified proteins in ischemic rat heart. Am. J. Physiol. Content 1999, 276, H935–H943. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yim, K.; Zhang, H.; Bakker, D.; Nederlof, R.; Smeitink, J.A.M.; Renkema, H.; Hollmann, M.W.; Weber, N.C.; Zuurbier, C.J. The Redox Modulating Sonlicromanol Active Metabolite KH176m and the Antioxidant MPG Protect Against Short-Duration Cardiac Ischemia-Reperfusion Injury. Cardiovasc. Drugs Ther. 2021, 35, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Eaton, P.; Hearse, D.J.; Shattock, M.J. Lipid hydroperoxide modification of proteins during myocardial ischaemia. Cardiovasc. Res. 2001, 51, 294–303. [Google Scholar] [CrossRef]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 133, 230–255. [Google Scholar] [CrossRef] [PubMed]
- Garlick, P.B.; Davies, M.J.; Hearse, D.J.; Slater, T.F. Direct detection of free radicals in the reperfused rat heart using electron spin resonance spectroscopy. Circ. Res. 1987, 61, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Nederlof, R.; Gürel-Gurevin, E.; Eerbeek, O.; Xie, C.; Deijs, G.S.; Konkel, M.; Hu, J.; Weber, N.C.; Schumacher, C.A.; Baartscheer, A.; et al. Reducing mitochondrial bound hexokinase II mediates transition from non-injurious into injurious ischemia/reperfusion of the intact heart. J. Physiol. Biochem. 2016, 73, 323–333. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Matsushima, S.; Sadoshima, J. Yin and Yang of NADPH Oxidases in Myocardial Ischemia-Reperfusion. Antioxidants 2022, 11, 1069. [Google Scholar] [CrossRef]
- Da-Silva, W.S.; Gómez-Puyou, A.; de Gómez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855. [Google Scholar] [CrossRef]
- Ong, S.-G.; Lee, W.H.; Theodorou, L.; Kodo, K.; Lim, S.Y.; Shukla, D.H.; Briston, T.; Kiriakidis, S.; Ashcroft, M.; Davidson, S.M.; et al. HIF-1 reduces ischaemia–reperfusion injury in the heart by targeting the mitochondrial permeability transition pore. Cardiovasc. Res. 2014, 104, 24–36. [Google Scholar] [CrossRef]
- Yang, M.; Sun, J.; Stowe, D.F.; Tajkhorshid, E.; Kwok, W.-M.; Camara, A.K.S. Knockout of VDAC1 in H9c2 Cells Promotes Oxidative Stress-Induced Cell Apoptosis through Decreased Mitochondrial Hexokinase II Binding and Enhanced Glycolytic Stress. Cell. Physiol. Biochem. 2020, 54, 853–874. [Google Scholar] [CrossRef] [PubMed]
- Liepinsh, E.; Makrecka-Kuka, M.; Volska, K.; Kuka, J.; Makarova, E.; Antone, U.; Sevostjanovs, E.; Vilskersts, R.; Strods, A.; Tars, K.; et al. Long-chain acylcarnitines determine ischaemia/reperfusion-induced damage in heart mitochondria. Biochem. J. 2016, 473, 1191–1202. [Google Scholar] [CrossRef]
- Dambrova, M.; Zuurbier, C.J.; Borutaite, V.; Liepinsh, E.; Makrecka-Kuka, M. Energy substrate metabolism and mitochondrial oxidative stress in cardiac ischemia/reperfusion injury. Free. Radic. Biol. Med. 2021, 165, 24–37. [Google Scholar] [CrossRef]
- Dumitrescu, C.; Biondi, R.; Xia, Y.; Cardounel, A.J.; Druhan, L.J.; Ambrosio, G.; Zweier, J.L. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc. Natl. Acad. Sci. USA 2007, 104, 15081–15086. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Richardson, A.P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 129–141. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Bertrand, L.; Beauloye, C.R.; Andreadou, I.; Ruiz-Meana, M.; Jespersen, N.R.; Kula-Alwar, D.; Prag, H.A.; Eric Botker, H.; Dambrova, M.; et al. Cardiac metabolism as a driver and therapeutic target of myocardial infarction. J. Cell. Mol. Med. 2020, 24, 5937–5954. [Google Scholar] [CrossRef]
- Taegtmeyer, H. Metabolic responses to cardiac hypoxia. Increased production of succinate by rabbit papillary muscles. Circ. Res. 1978, 43, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.T.; Miller, J.H.; Day, M.M.; Munger, J.C.; Brookes, P.S. Accumulation of Succinate in Cardiac Ischemia Primarily Occurs via Canonical Krebs Cycle Activity. Cell Rep. 2018, 23, 2617–2628. [Google Scholar] [CrossRef]
- Vogt, A.M.; Poolman, M.; Ackermann, C.; Yildiz, M.; Schoels, W.; Fell, D.A.; Kübler, W. Regulation of Glycolytic Flux in Ischemic Preconditioning. J. Biol. Chem. 2002, 277, 24411–24419. [Google Scholar] [CrossRef]
- Jennings, R.B.; Murry, C.E.; Reimer, K.A. Energy metabolism in preconditioned and control myocardium: Effect of total ischemia. J. Mol. Cell. Cardiol. 1991, 23, 1449–1458. [Google Scholar] [CrossRef]
- Pasdois, P.; Parker, J.E.; Halestrap, A.P. Extent of mitochondrial hexokinase II dissociation during ischemia correlates with mitochondrial cytochrome c release, reactive oxygen species production, and infarct size on reperfusion. J. Am. Heart Assoc. 2012, 2, e005645. [Google Scholar] [CrossRef] [PubMed]
- Smeele, K.M.A.; Southworth, R.; Wu, R.; Xie, C.; Nederlof, R.; Warley, A.; Nelson, J.K.; Van Horssen, P.; Wijngaard, J.P.V.D.; Heikkinen, S.; et al. Disruption of Hexokinase II–Mitochondrial Binding Blocks Ischemic Preconditioning and Causes Rapid Cardiac Necrosis. Circ. Res. 2011, 108, 1165–1169. [Google Scholar] [CrossRef]
- Nederlof, R.; Eerbeek, O.; Hollmann, M.W.; Southworth, R.; Zuurbier, C.J. Targeting hexokinase II to mitochondria to modulate energy metabolism and reduce ischaemia-reperfusion injury in heart. Br. J. Pharmacol. 2014, 171, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Andrienko, T.N.; Pasdois, P.; Pereira, G.C.; Ovens, M.J.; Halestrap, A.P. The role of succinate and ROS in reperfusion injury—A critical appraisal. J. Mol. Cell. Cardiol. 2017, 110, 1–14. [Google Scholar] [CrossRef]
- Gürel, E.; Smeele, K.M.; Eerbeek, O.; Koeman, A.; Demirci, C.; Hollmann, M.W.; Zuurbier, C.J. Ischemic preconditioning affects hexokinase activity and HKII in different subcellular compartments throughout cardiac ischemia-reperfusion. J. Appl. Physiol. 2009, 106, 1909–1916. [Google Scholar] [CrossRef] [PubMed]
- Nederlof, R.; Denis, S.; Lauzier, B.; Rosiers, C.D.; Laakso, M.; Hagen, J.; Argmann, C.; Wanders, R.; Houtkooper, R.H.; Hollmann, M.W.; et al. Acute detachment of hexokinase II from mitochondria modestly increases oxygen consumption of the intact mouse heart. Metabolism 2017, 72, 66–74. [Google Scholar] [CrossRef]
- Chiara, F.; Castellaro, D.; Marin, O.; Petronilli, V.; Brusilow, W.S.; Juhaszova, M.; Sollott, S.J.; Forte, M.; Bernardi, P.; Rasola, A. Hexokinase II Detachment from Mitochondria Triggers Apoptosis through the Permeability Transition Pore Independent of Voltage-Dependent Anion Channels. PLoS ONE 2008, 3, e1852. [Google Scholar] [CrossRef]
- Perevoshchikova, I.V.; Zorov, S.D.; Kotova, E.A.; Zorov, D.B.; Antonenko, Y.N. Hexokinase inhibits flux of fluorescently labeled ATP through mitochondrial outer membrane porin. FEBS Lett. 2010, 584, 2397–2402. [Google Scholar] [CrossRef]
- Uthman, L.; Nederlof, R.; Eerbeek, O.; Baartscheer, A.; Schumacher, C.; Buchholtz, N.; Hollmann, M.W.; Coronel, R.; Weber, N.C.; Zuurbier, C.J. Delayed ischaemic contracture onset by empagliflozin associates with NHE1 inhibition and is dependent on insulin in isolated mouse hearts. Cardiovasc. Res. 2019, 115, 1533–1545. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Eerbeek, O.; Meijer, A.J. Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H496–H499. [Google Scholar] [CrossRef]
- Cross, H.R.; Opie, L.H.; Radda, G.K.; Clarke, K. Is a high glycogen content beneficial or detrimental to the ischemic rat heart? A controversy resolved. Circ. Res. 1996, 78, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, J.T.; Idell-Wenger, J.A.; Rovetto, M.J.; Neely, J.R. Control of fatty acid metabolism in ischemic and hypoxic hearts. J. Biol. Chem. 1978, 253, 4305–4309. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.A.; Han, X.; Horner, C.C.; Gross, R.W. Accumulation of Unsaturated Acylcarnitine Molecular Species During Acute Myocardial Ischemia: Metabolic Compartmentalization of Products of Fatty Acyl Chain Elongation in the Acylcarnitine Pool. Biochemistry 1996, 35, 7903–7909. [Google Scholar] [CrossRef] [PubMed]
- van Weeghel, M.; Abdurrachim, D.; Nederlof, R.; Argmann, C.A.; Houtkooper, R.H.; Hagen, J.; Nabben, M.; Denis, S.; Ciapaite, J.; Kolwicz, S.C., Jr.; et al. Increased cardiac fatty acid oxidation in a mouse model with decreased malonyl-CoA sensitivity of CPT1B. Cardiovasc. Res. 2018, 114, 1324–1334. [Google Scholar] [CrossRef]
- Makrecka, M.; Kuka, J.; Volska, K.; Antone, U.; Sevostjanovs, E.; Cirule, H.; Grinberga, S.; Pugovics, O.; Dambrova, M.; Liepinsh, E. Long-chain acylcarnitine content determines the pattern of energy metabolism in cardiac mitochondria. Mol. Cell. Biochem. 2014, 395, 1–10. [Google Scholar] [CrossRef]
- Tominaga, H.; Katoh, H.; Odagiri, K.; Takeuchi, Y.; Kawashima, H.; Saotome, M.; Urushida, T.; Satoh, H.; Hayashi, H. Different effects of palmitoyl-L-carnitine and palmitoyl-CoA on mitochondrial function in rat ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H105–H112. [Google Scholar] [CrossRef]
- Moens, A.L.; Champion, H.C.; Claeys, M.J.; Tavazzi, B.; Kaminski, P.M.; Wolin, M.S.; Borgonjon, D.J.; Van Nassauw, L.; Haile, A.; Zviman, M.; et al. High-Dose Folic Acid Pretreatment Blunts Cardiac Dysfunction During Ischemia Coupled to Maintenance of High-Energy Phosphates and Reduces Postreperfusion Injury. Circulation 2008, 117, 1810–1819. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Heinen, A.; Koeman, A.; Stuifbergen, R.; Hakvoort, T.B.M.; Weber, N.C.; Hollmann, M.W. Cardioprotective efficacy depends critically on pharmacological dose, duration of ischaemia, health status of animals and choice of anaesthetic regimen: A case study with folic acid. J. Transl. Med. 2014, 12, 325. [Google Scholar] [CrossRef]
- Verdin, E. NAD⁺ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Chu, X.; Raju, R.P. Regulation of NAD(+) metabolism in aging and disease. Metabolism 2022, 126, 154923. [Google Scholar] [CrossRef]
- Abdellatif, M.; Sedej, S.; Kroemer, G. NAD(+) Metabolism in Cardiac Health, Aging, and Disease. Circulation 2021, 144, 1795–1817. [Google Scholar] [CrossRef]
- Di Lisa, F.; Menabò, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the Mitochondrial Permeability Transition Pore Causes Depletion of Mitochondrial and Cytosolic NAD+and Is a Causative Event in the Death of Myocytes in Postischemic Reperfusion of the Heart. J. Biol. Chem. 2001, 276, 2571–2575. [Google Scholar] [CrossRef] [PubMed]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Tran, D.H.; Wang, Z.V. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.C.; Sato, S.; Tsai, J.-Y.; Yan, S.; Bakr, S.; Zhang, H.; Oates, P.J.; Ramasamy, R. Aldose reductase activation is a key component of myocardial response to ischemia. FASEB J. 2001, 16, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.C.; Bakr, S.; Ellery, C.A.; Oates, P.J.; Ramasamy, R. Sorbitol dehydrogenase: A novel target for adjunctive protection of ischemic myocardium. FASEB J. 2003, 17, 2331–2333. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Byun, J.; Zhai, P.; Ikeda, Y.; Oka, S.; Sadoshima, J. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS ONE 2014, 9, e98972. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Wang, Y.T.; Nehrke, K.; Munger, J.; Brookes, P.S. Cardioprotection by nicotinamide mononucleotide (NMN): Involvement of glycolysis and acidic pH. J. Mol. Cell. Cardiol. 2018, 121, 155–162. [Google Scholar] [CrossRef]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Évora, P.; Castro-E-Silva, O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, B.; Fu, X.; Guan, S.; Han, W.; Zhang, J.; Gan, Q.; Fang, W.; Ying, W.; Qu, X. Exogenous NAD(+) administration significantly protects against myocardial ischemia/reperfusion injury in rat model. Am. J. Transl. Res. 2016, 8, 3342–3350. [Google Scholar]
- Zhang, Y.-J.; Zhang, M.; Zhao, X.; Shi, K.; Ye, M.; Tian, J.; Guan, S.; Ying, W.; Qu, X. NAD+ administration decreases microvascular damage following cardiac ischemia/reperfusion by restoring autophagic flux. Basic Res. Cardiol. 2020, 115, 57. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.J.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Phelp, P.; Wang, Q.; Bakker, D.; Nederlof, R.; Hollmann, M.W.; Zuurbier, C.J. Cardioprotecive Properties of Known Agents in Rat Ischemia-Reperfusion Model Under Clinically Relevant Conditions: Only the NAD Precursor Nicotinamide Riboside Reduces Infarct Size in Presence of Fentanyl, Midazolam and Cangrelor, but Not Propofol. Front. Cardiovasc. Med. 2021, 8, 712478. [Google Scholar] [CrossRef]
- Xiao, Y.; Phelp, P.; Bakker, D.; Nederlof, R.; Hollmann, M.W.; Weber, N.C.; Zuurbier, C.J. Only the NAD precursor nicotinamide riboside maintains cardioprotection under clinically relevant conditions, possibly through activation of glycolysis. Eur. Heart J. 2021, 42, ehab724.3248. [Google Scholar] [CrossRef]
- Kharechkina, E.; Nikiforova, A.; Kruglov, A. NAD(H) Regulates the Permeability Transition Pore in Mitochondria through an External Site. Int. J. Mol. Sci. 2021, 22, 8560. [Google Scholar] [CrossRef]
- Valls-Lacalle, L.; Barba, I.; Miró-Casas, E.; Ruiz-Meana, M.; Rodríguez-Sinovas, A.; García-Dorado, D. Selective Inhibition of Succinate Dehydrogenase in Reperfused Myocardium with Intracoronary Malonate Reduces Infarct Size. Sci. Rep. 2018, 8, 2442. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, N.R.; Hjortbak, M.V.; Lassen, T.R.; Støttrup, N.B.; Johnsen, J.; Tonnesen, P.T.; Larsen, S.; Kimose, H.-H.; Bøtker, H.E. Cardioprotective effect of succinate dehydrogenase inhibition in rat hearts and human myocardium with and without diabetes mellitus. Sci. Rep. 2020, 10, 10344. [Google Scholar] [CrossRef]
- Tonnesen, P.T.; Hjortbak, M.V.; Lassen, T.R.; Seefeldt, J.M.; Bøtker, H.E.; Jespersen, N.R. Myocardial salvage by succinate dehydrogenase inhibition in ischemia–reperfusion injury depends on diabetes stage in rats. Mol. Cell. Biochem. 2021, 476, 2675–2684. [Google Scholar] [CrossRef]
- Prag, H.A.; Aksentijevic, D.; Dannhorn, A.; Giles, A.V.; Mulvey, J.F.; Sauchanka, O.; Du, L.; Bates, G.; Reinhold, J.; Kula-Alwar, D.; et al. Ischemia-Selective Cardioprotection by Malonate for Ischemia/Reperfusion Injury. Circ. Res. 2022, 131, 528–541. [Google Scholar] [CrossRef]
- Arslan, F.; Smeets, M.B.; O’Neill, L.A.; Keogh, B.; McGuirk, P.; Timmers, L.; Tersteeg, C.; Hoefer, I.E.; Doevendans, P.A.; Pasterkamp, G.; et al. Myocardial Ischemia/Reperfusion Injury Is Mediated by Leukocytic Toll-Like Receptor-2 and Reduced by Systemic Administration of a Novel Anti–Toll-Like Receptor-2 Antibody. Circulation 2010, 121, 80–90. [Google Scholar] [CrossRef]
- Shimamoto, A.; Chong, A.J.; Yada, M.; Shomura, S.; Takayama, H.; Fleisig, A.J.; Agnew, M.L.; Hampton, C.R.; Rothnie, C.L.; Spring, D.J.; et al. Inhibition of Toll-like Receptor 4 With Eritoran Attenuates Myocardial Ischemia-Reperfusion Injury. Circulation 2006, 114, I270–I274. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mezzaroma, E.; Buckley, L.F.; Potere, N.; Di Nisio, M.; Biondi-Zoccai, G.; Van Tassell, B.W.; Abbate, A. Targeting the NLRP3 inflammasome in cardiovascular diseases. Pharmacol. Ther. 2021, 236, 108053. [Google Scholar] [CrossRef] [PubMed]
- Zuurbier, C.J.; Abbate, A.; Cabrera-Fuentes, H.A.; Cohen, M.V.; Collino, M.; De Kleijn, D.P.V.; Downey, J.M.; Pagliaro, P.; Preissner, K.T.; Takahashi, M.; et al. Innate immunity as a target for acute cardioprotection. Cardiovasc. Res. 2019, 115, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Zuurbier, C.J.; Jong, W.M.; Eerbeek, O.; Koeman, A.; Pulskens, W.P.; Butter, L.M.; Leemans, J.C.; Hollmann, M.W. Deletion of the innate immune NLRP3 receptor abolishes cardiac ischemic preconditioning and is associated with decreased Il-6/STAT3 signaling. PLoS ONE 2012, 7, e40643. [Google Scholar] [CrossRef]
- Jong, W.M.; Leemans, J.C.; Weber, N.C.; Juffermans, N.P.; Schultz, M.J.; Hollmann, M.W.; Zuurbier, C.J. Nlrp3 plays no role in acute cardiac infarction due to low cardiac expression. Int. J. Cardiol. 2014, 177, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome Activation of Cardiac Fibroblasts Is Essential for Myocardial Ischemia/Reperfusion Injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef]
- Toldo, S.; Marchetti, C.; Mauro, A.G.; Chojnacki, J.; Mezzaroma, E.; Carbone, S.; Zhang, S.; Van Tassell, B.; Salloum, F.N.; Abbate, A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia–reperfusion in the mouse. Int. J. Cardiol. 2016, 209, 215–220. [Google Scholar] [CrossRef]
- Van Hout, G.P.; Bosch, L.; Ellenbroek, G.H.; de Haan, J.J.; van Solinge, W.W.; Cooper, M.A.; Arslan, F.; de Jager, S.C.; Robertson, A.A.; Pasterkamp, G.; et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 2017, 38, 828–836. [Google Scholar] [CrossRef]
- Silvis, M.J.M.; Demkes, E.J.; Timmers, L.; Arslan, F.; de Jager, S.C.A.; Sluijter, J.P.G.; Mosterd, A.; de Kleijn, D.P.V.; Bosch, L.; van Hout, G.P.J. NLRP3-Inflammasome Inhibition with IZD334 Does Not Reduce Cardiac Damage in a Pig Model of Myocardial Infarction. Biomedicines 2022, 10, 3056. [Google Scholar] [CrossRef]
- Mewton, N.; Roubille, F.; Bresson, D.; Prieur, C.; Bouleti, C.; Bochaton, T.; Ivanes, F.; Dubreuil, O.; Biere, L.; Hayek, A.; et al. Effect of Colchicine on Myocardial Injury in Acute Myocardial Infarction. Circulation 2021, 144, 859–869. [Google Scholar] [CrossRef]
- Shah, B.; Pillinger, M.; Zhong, H.; Cronstein, B.; Xia, Y.; Lorin, J.D.; Smilowitz, N.R.; Feit, F.; Ratnapala, N.; Keller, N.M.; et al. Effects of Acute Colchicine Administration Prior to Percutaneous Coronary Intervention: COLCHICINE-PCI Randomized Trial. Circ. Cardiovasc. Interv. 2020, 13, e008717. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a008656. [Google Scholar] [CrossRef] [PubMed]
- Bahi, N.; Zhang, J.; Llovera, M.; Ballester, M.; Comella, J.X.; Sanchis, D. Switch from caspase-dependent to caspase-independent death during heart development: Essential role of endonuclease G in ischemia-induced DNA processing of differentiated cardiomyocytes. J. Biol. Chem. 2006, 281, 22943–22952. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Cardona, M.; Poncelas-Nozal, M.; Hernando, V.; Vilardosa, Ú.; Aluja, D.; Parra, V.M.; Sanchis, D.; Garcia-Dorado, D. Studies on the role of apoptosis after transient myocardial ischemia: Genetic deletion of the executioner caspases-3 and -7 does not limit infarct size and ventricular remodeling. Basic Res. Cardiol. 2016, 111, 18. [Google Scholar] [CrossRef]
- Cardona, M.; Lopez, J.A.; Serafín, A.; Rongvaux, A.; Inserte, J.; García-Dorado, D.; Flavell, R.; Llovera, M.; Cañas, X.; Vázquez, J.; et al. Executioner Caspase-3 and 7 Deficiency Reduces Myocyte Number in the Developing Mouse Heart. PLoS ONE 2015, 10, e0131411. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Wakiyama, H.; Hsieh, Y.-J.; Jones, M.; Levitsky, S. Differential contribution of necrosis and apoptosis in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1923–H1935. [Google Scholar] [CrossRef]
- Mocanu, M.M.; Baxter, G.F.; Yellon, D.M. Caspase inhibition and limitation of myocardial infarct size: Protection against lethal reperfusion injury. Br. J. Pharmacol. 2000, 130, 197–200. [Google Scholar] [CrossRef]
- Kovacs, P.; Bak, I.; Szendrei, L.; Vecsernyes, M.; Varga, E.; Blasig, I.E.; Tosaki, A. Non-specific caspase inhibition reduces infarct size and improves post-ischaemic recovery in isolated ischaemic/reperfused rat hearts. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2001, 364, 501–507. [Google Scholar] [CrossRef]
- He, Z.; Davidson, S.M.; Yellon, D.M. The importance of clinically relevant background therapy in cardioprotective studies. Basic Res. Cardiol. 2020, 115, 69. [Google Scholar] [CrossRef]
- Audia, J.P.; Yang, X.-M.; Crockett, E.S.; Housley, N.; Haq, E.U.; O’donnell, K.; Cohen, M.V.; Downey, J.M.; Alvarez, D.F. Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y12 receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function. Basic Res. Cardiol. 2018, 113, 32. [Google Scholar] [CrossRef]
- Yue, R.-C.; Lu, S.-Z.; Luo, Y.; Wang, T.; Liang, H.; Zeng, J.; Liu, J.; Hu, H.-X. Calpain silencing alleviates myocardial ischemia-reperfusion injury through the NLRP3/ASC/Caspase-1 axis in mice. Life Sci. 2019, 233, 116631. [Google Scholar] [CrossRef]
- Zhang, Y.; Rong, H.; Zhang, F.-X.; Wu, K.; Mu, L.; Meng, J.; Xiao, B.; Zamponi, G.W.; Shi, Y. A Membrane Potential- and Calpain-Dependent Reversal of Caspase-1 Inhibition Regulates Canonical NLRP3 Inflammasome. Cell Rep. 2018, 24, 2356–2369.e5. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Gao, Y.; Dong, Z.; Yang, J.; Gao, R.; Li, X.; Zhang, S.; Ma, L.; Sun, X.; Wang, Z.; et al. GSDMD-Mediated Cardiomyocyte Pyroptosis Promotes Myocardial I/R Injury. Circ. Res. 2021, 129, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Andreadou, I.; Aragno, M.; Beauloye, C.; Bertrand, L.; Lazou, A.; Falcão-Pires, I.; Bell, R.; Zuurbier, C.J.; Pagliaro, P. Effect of hyperglycaemia and diabetes on acute myocardial ischaemia–reperfusion injury and cardioprotection by ischaemic conditioning protocols. Br. J. Pharmacol. 2020, 177, 5312–5335. [Google Scholar] [CrossRef]
- Meng, Z.; Zhang, Z.; Zhao, J.; Liu, C.; Yao, P.; Zhang, L.; Xie, D.; Lau, W.B.; Tsukuda, J.; Christopher, T.A.; et al. Nitrative Modification of Caveolin-3: A Novel Mechanism of Cardiac Insulin Resistance and a Potential Therapeutic Target Against Ischemic Heart Failure in Prediabetic Animals. Circulation 2023, 147, 1162–1179. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Preckel, B.; Hermanides, J.; Hollmann, M.W.; Zuurbier, C.J.; Weber, N.C. Amelioration of endothelial dysfunction by sodium glucose co-transporter 2 inhibitors: Pieces of the puzzle explaining their cardiovascular protection. Br. J. Pharmacol. 2022, 179, 4047–4062. [Google Scholar] [CrossRef]
- Chen, S.; Coronel, R.; Hollmann, M.W.; Weber, N.C.; Zuurbier, C.J. Direct cardiac effects of SGLT2 inhibitors. Cardiovasc. Diabetol. 2022, 21, 45. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, Q.; Zuurbier, C.J. Efficacy of cardioprotective interventions depends critically on duration of ischemia. Cond. Med. 2022, 5, 93–99. [Google Scholar]
- Hausenloy, D.J.; Kharbanda, R.K.; Møller, U.K.; Ramlall, M.; Aarøe, J.; Butler, R.; Bulluck, H.; Clayton, T.; Dana, A.; Dodd, M.; et al. Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): A single-blind randomised controlled trial. Lancet 2019, 394, 1415–1424. [Google Scholar] [CrossRef]
- Park, J.; Choi, K.H.; Lee, J.M.; Kim, H.K.; Hwang, D.; Rhee, T.M.; Kim, J.; Park, T.K.; Yang, J.H.; Song, Y.B.; et al. Prognostic Implications of Door-to-Balloon Time and Onset-to-Door Time on Mortality in Patients with ST -Segment-Elevation Myocardial Infarction Treated with Primary Percutaneous Coronary Intervention. J. Am. Heart Assoc. 2019, 8, e012188. [Google Scholar] [CrossRef]
- Zahler, D.; Rozenfeld, K.-L.; Pasternak, Y.; Itach, T.; Lupu, L.; Banai, S.; Shacham, Y. Relation of Pain-to-Balloon Time and Mortality in Patients with ST-Segment Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention. Am. J. Cardiol. 2021, 163, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Sabia, P.J.; Powers, E.R.; Ragosta, M.; Sarembock, I.J.; Burwell, L.R.; Kaul, S. An Association between Collateral Blood Flow and Myocardial Viability in Patients with Recent Myocardial Infarction. N. Engl. J. Med. 1992, 327, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef]
- Lecour, S.; Andreadou, I.; Bøtker, H.E.; Davidson, S.M.; Heusch, G.; Ruiz-Meana, M.; Schulz, R.; Zuurbier, C.J.; Ferdinandy, P.; Hausenloy, D.J.; et al. Improving Preclinical Assessment of Cardioprotective Therapies (IMPACT) criteria: Guidelines of the EU-CARDIOPROTECTION COST Action. Basic Res. Cardiol. 2021, 116, 52. [Google Scholar] [CrossRef]
- Warltier, D.C.; Al-Wathiqui, M.H.; Kampine, J.P.; Schmeling, W.T. Recovery of Contractile Function of Stunned Myocardium in Chronically Instrumented Dogs is Enhanced by Halothane or Isoflurane. Anesthesiology 1988, 69, 552–565. [Google Scholar] [CrossRef]
- Weber, N.C.; Goletz, C.; Huhn, R.; Grueber, Y.; Preckel, B.; Schlack, W.; Ebel, D. Blockade of anaesthetic-induced preconditioning in the hyperglycaemic myocardium: The regulation of different mitogen-activated protein kinases. Eur. J. Pharmacol. 2008, 592, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.D.; Pravdic, D.; Bienengraeber, M.; Pratt, P.F., Jr.; Auchampach, J.A.; Gross, G.J.; Kersten, J.R.; Warltier, D.C. Isoflurane postconditioning protects against reperfusion injury by preventing mitochondrial permeability transition by an endothelial nitric oxide synthase-dependent mechanism. Anesthesiology 2010, 112, 73–85. [Google Scholar] [CrossRef]
- Lotz, C.; Kehl, F. Volatile anesthetic-induced cardiac protection: Molecular mechanisms, clinical aspects, and interactions with nonvolatile agents. J. Cardiothorac. Vasc. Anesth. 2015, 29, 749–760. [Google Scholar] [CrossRef]
- Ma, L.; Zhu, J.; Gao, Q.; Rebecchi, M.J.; Wang, Q.; Liu, L. Restoring Pharmacologic Preconditioning in the Aging Heart: Role of Mitophagy/Autophagy. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 489–498. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, K.; Hu, P. The Role of Autophagy in Acute Myocardial Infarction. Front. Pharmacol. 2019, 10, 551. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, Å.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Jiao, Y.; Yan, N.; Wu, B.; Ren, Y.; Li, H.; Sun, J.; Gao, J. NOD2 mediates isoflurane preconditioning-induced protection of myocardial injury. Neurosci. Lett. 2017, 637, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Qiao, S.; Li, H.; Deng, Y.; Wang, C.; An, J. The Effect of Mitochondrial Complex I-Linked Respiration by Isoflurane Is Independent of Mitochondrial Nitric Oxide Production. Cardiorenal Med. 2018, 8, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Jang, S.; Parodi-Rullán, R.; Khuchua, Z.; Kuznetsov, A.V. Mitochondrial permeability transition in cardiac ischemia-reperfusion: Whether cyclophilin D is a viable target for cardioprotection? Cell. Mol. Life Sci. 2017, 74, 2795–2813. [Google Scholar] [CrossRef] [PubMed]
- Harisseh, R.; Chiari, P.; Villedieu, C.; Sueur, P.; Abrial, M.; Fellahi, J.-L.; Ovize, M.; Gharib, A. Cyclophilin D Modulates the Cardiac Mitochondrial Target of Isoflurane, Sevoflurane, and Desflurane. J. Cardiovasc. Pharmacol. 2017, 69, 326–334. [Google Scholar] [CrossRef]
- Qiao, S.; Olson, J.M.; Paterson, M.; Yan, Y.; Zaja, I.; Liu, Y.; Riess, M.L.; Kersten, J.R.; Liang, M.; Warltier, D.C.; et al. MicroRNA-21 Mediates Isoflurane-induced Cardioprotection against Ischemia–Reperfusion Injury via Akt/Nitric Oxide Synthase/Mitochondrial Permeability Transition Pore Pathway. Anesthesiology 2015, 123, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.M.; Yan, Y.; Bai, X.; Ge, Z.D.; Liang, M.; Kriegel, A.J.; Twaroski, D.M.; Bosnjak, Z.J. Up-regulation of microRNA-21 mediates isoflurane-induced protection of cardiomyocytes. Anesthesiology 2015, 122, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.J.; Liu, B. Inhibition of MicroRNA-23 Contributes to the Isoflurane-Mediated Cardioprotection against Oxidative Stress. Cardiovasc. Toxicol. 2018, 18, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, S.; Tritapepe, L.; Hanouz, J.L.; Puddu, P.E. The mechanisms of cardio-protective effects of desflurane and sevoflurane at the time of reperfusion: Anaesthetic post-conditioning potentially translatable to humans? Br. J. Anaesth. 2016, 116, 456–475. [Google Scholar] [CrossRef]
- Chen, S.; Lotz, C.; Roewer, N.; Broscheit, J.-A. Comparison of volatile anesthetic-induced preconditioning in cardiac and cerebral system: Molecular mechanisms and clinical aspects. Eur. J. Med. Res. 2018, 23, 10. [Google Scholar] [CrossRef]
- Torregroza, C.; Raupach, A.; Feige, K.; Weber, N.C.; Hollmann, M.W.; Huhn, R. Perioperative Cardioprotection: General Mechanisms and Pharmacological Approaches. Anesth. Analg. 2020, 131, 1765–1780. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, N.R.; Krafft, P.R.; Leitzke, A.S.; Applegate, R.L., 2nd; Tang, J.; Zhang, J.H. The role of Volatile Anesthetics in Cardioprotection: A systematic review. Med. Gas Res. 2012, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yu, J.; Xie, P.; Maimaitili, Y.; Wang, J.; Yang, L.; Ma, H.; Zhang, X.; Yang, Y.; Zheng, H. Sevoflurane postconditioning protects the myocardium against ischemia/reperfusion injury via activation of the JAK2–STAT3 pathway. PeerJ 2017, 5, e3196. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Bu, M.; Yun, H. Sevoflurane prevents hypoxia/reoxygenation-induced cardiomyocyte apoptosis by inhibiting PI3KC3-mediated autophagy. Hum. Cell 2019, 32, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.; Sun, Y.; Sun, B.; Wang, A.; Qiu, J.; Hong, L.; An, J.-Z.; Wang, C.; Zhang, H.-L. Sevoflurane postconditioning protects against myocardial ischemia/reperfusion injury by restoring autophagic flux via an NO-dependent mechanism. Acta Pharmacol. Sin. 2018, 40, 35–45. [Google Scholar] [CrossRef]
- Qian, B.; Yang, Y.; Yao, Y.; Liao, Y.; Lin, Y. Upregulation of vascular endothelial growth factor receptor-1 contributes to sevoflurane preconditioning–mediated cardioprotection. Drug Des. Dev. Ther. 2018, 12, 769–776. [Google Scholar] [CrossRef]
- Huang, G.; Hao, F.; Hu, X. Downregulation of microRNA-155 stimulates sevoflurane-mediated cardioprotection against myocardial ischemia/reperfusion injury by binding to SIRT1 in mice. J. Cell. Biochem. 2019, 120, 15494–15505. [Google Scholar] [CrossRef]
- Xie, D.; Zhao, J.; Guo, R.; Jiao, L.; Zhang, Y.; Lau, W.B.; Lopez, B.; Christopher, T.; Gao, E.; Cao, J.; et al. Sevoflurane Pre-conditioning Ameliorates Diabetic Myocardial Ischemia/Reperfusion Injury Via Differential Regulation of p38 and ERK. Sci. Rep. 2020, 10, 23. [Google Scholar] [CrossRef]
- Zhong, C.Y.; Qiu, H.; Chen, J.; Liu, H. Effects of volatile anesthetic preconditioning on expression of NFkB-regulated genes in aged rat myocardium. J. Biomed. Res. 2017, 33, 264–270. [Google Scholar] [CrossRef]
- Smit, K.F.; Weber, N.C.; Hollmann, M.W.; Preckel, B. Noble gases as cardioprotectants-translatability and mechanism. Br. J. Pharmacol. 2015, 172, 2062–2073. [Google Scholar] [CrossRef]
- Weber, N.C.; Preckel, B. Gaseous mediators: An updated review on the effects of helium beyond blowing up balloons. Intensive Care Med. Exp. 2019, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Anna, R.; Rolf, R.; Mark, C. Update of the organoprotective properties of xenon and argon: From bench to beside. Intensive Care Med. Exp. 2020, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.; Torregroza, C.; Feige, K.; Preckel, B.; Hollmann, M.W.; Weber, N.C.; Huhn, R. Pharmacological Conditioning of the Heart: An Update on Experimental Developments and Clinical Implications. Int. J. Mol. Sci. 2021, 22, 2519. [Google Scholar] [CrossRef] [PubMed]
- De Deken, J.; Rex, S.; Monbaliu, D.; Pirenne, J.; Jochmans, I. The Efficacy of Noble Gases in the Attenuation of Ischemia Reperfusion Injury: A Systematic Review and Meta-Analyses. Crit. Care Med. 2016, 44, e886–e896. [Google Scholar] [CrossRef] [PubMed]
- Pagel, P.S.; Krolikowski, J.G.; Shim, Y.H.; Venkatapuram, S.; Kersten, J.R.; Weihrauch, D.; Warltier, D.C.; Pratt, P.F., Jr. Noble gases without anesthetic properties protect myocardium against infarction by activating prosurvival signaling kinases and inhibiting mitochondrial permeability transition in vivo. Anesth. Analg. 2007, 105, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.C.; Toma, O.; Wolter, J.I.; Obal, D.; Müllenheim, J.; Preckel, B.; Schlack, W. The noble gas xenon induces pharmacological preconditioning in the rat heart in vivo via induction of PKC-epsilon and p38 MAPK. Br. J. Pharmacol. 2005, 144, 123–132. [Google Scholar] [CrossRef]
- Weber, N.C.; Toma, O.; Wolter, J.I.; Wirthle, N.M.; Schlack, W.; Preckel, B. Mechanisms of xenon- and isoflurane-induced preconditioning-a potential link to the cytoskeleton via the MAPKAPK-2/HSP27 pathway. Br. J. Pharmacol. 2005, 146, 445–455. [Google Scholar] [CrossRef]
- Weber, N.C.; Stursberg, J.; Wirthle, N.M.; Toma, O.; Schlack, W.; Preckel, B. Xenon preconditioning differently regulates p44/42 MAPK (ERK 1/2) and p46/54 MAPK (JNK 1/2 and 3) in vivo. Br. J. Anaesth. 2006, 97, 298–306. [Google Scholar] [CrossRef]
- Li, Q.; Lian, C.; Zhou, R.; Li, T.; Xiang, X.; Liu, B. Pretreatment With Xenon Protected Immature Rabbit Heart From Ischaemia/Reperfusion Injury by Opening of the mitoKATP Channel. Heart Lung Circ. 2013, 22, 276–283. [Google Scholar] [CrossRef]
- Roehl, A.B.; Funcke, S.; Becker, M.M.; Goetzenich, A.; Bleilevens, C.; Rossaint, R.; Steendijk, P.; Hein, M. Xenon and Isoflurane Reduce Left Ventricular Remodeling after Myocardial Infarction in the Rat. Anesthesiology 2013, 118, 1385–1394. [Google Scholar] [CrossRef]
- Hein, M.; Roehl, A.B.; Baumert, J.H.; Bleilevens, C.; Fischer, S.; Steendijk, P.; Rossaint, R. Xenon and isoflurane improved biventricular function during right ventricular ischemia and reperfusion. Acta Anaesthesiol. Scand. 2010, 54, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.C.; Frässdorf, J.; Ratajczak, C.; Grueber, Y.; Schlack, W.; Hollmann, M.W.; Preckel, B. Xenon induces late cardiac preconditioning in vivo: A role for cyclooxygenase 2? Anesth. Analg. 2008, 107, 1807–1813. [Google Scholar] [CrossRef] [PubMed]
- Pagel, P.S.; Krolikowski, J.G.; Pratt, P.F.; Shim, Y.H.; Amour, J.; Warltier, D.C.; Weihrauch, D. The Mechanism of Helium-Induced Preconditioning: A Direct Role for Nitric Oxide in Rabbits. Anesth. Analg. 2008, 107, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Pagel, P.S.; Krolikowski, J.G.; Pratt, P.F.; Shim, Y.H.; Amour, J.; Warltier, D.C.; Weihrauch, D. Reactive Oxygen Species and Mitochondrial Adenosine Triphosphate–Regulated Potassium Channels Mediate Helium-Induced Preconditioning against Myocardial Infarction In Vivo. J. Cardiothorac. Vasc. Anesth. 2008, 22, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Pagel, P.S.; Krolikowski, J.G.; Pratt, P.F.; Shim, Y.H.; Amour, J.; Warltier, D.C.; Weihrauch, D. Inhibition of Glycogen Synthase Kinase or the Apoptotic Protein p53 Lowers the Threshold of Helium Cardioprotection In Vivo: The Role of Mitochondrial Permeability Transition. Anesth. Analg. 2008, 107, 769–775. [Google Scholar] [CrossRef]
- Pagel, P.S.; Krolikowski, J.G. Transient metabolic alkalosis during early reperfusion abolishes helium preconditioning against myocardial infarction: Restoration of cardioprotection by cyclosporin A in rabbits. Anesth. Analg. 2009, 108, 1076–1082. [Google Scholar] [CrossRef]
- Pagel, P.S.; Krolikowski, J.G.; Amour, J.; Warltier, D.C.; Weihrauch, D. Morphine reduces the threshold of helium preconditioning against myocardial infarction: The role of opioid receptors in rabbits. J. Cardiothorac. Vasc. Anesth. 2009, 23, 619–624. [Google Scholar] [CrossRef]
- Huhn, R.; Heinen, A.; Weber, N.C.; Hieber, S.; Hollmann, M.W.; Schlack, W.; Preckel, B. Helium-induced late preconditioning in the rat heart in vivo. Br. J. Anaesth. 2009, 102, 614–619. [Google Scholar] [CrossRef]
- Oei, G.T.M.L.; Aslami, H.; Kerindongo, R.P.; Steenstra, R.J.; Beurskens, C.J.P.; Boer, A.M.T.-D.; Juffermans, N.P.; Hollmann, M.W.; Preckel, B.; Weber, N.C. Prolonged Helium Postconditioning Protocols during Early Reperfusion Do Not Induce Cardioprotection in the Rat Heart In Vivo: Role of Inflammatory Cytokines. J. Immunol. Res. 2015, 2015, 216798. [Google Scholar] [CrossRef]
- Oei, G.T.; Heger, M.; van Golen, R.F.; Alles, L.K.; Flick, M.; van der Wal, A.C.; van Gulik, T.M.; Hollmann, M.W.; Preckel, B.; Weber, N.C. Reduction of cardiac cell death after helium postconditioning in rats: Transcriptional analysis of cell death and survival pathways. Mol. Med. 2015, 20, 516–526. [Google Scholar] [CrossRef]
- Heinen, A.; Huhn, R.; Smeele, K.M.; Zuurbier, C.J.; Schlack, W.; Preckel, B.; Weber, N.C.; Hollmann, M.W. Helium-induced preconditioning in young and old rat heart: Impact of mitochondrial Ca(2+) -sensitive potassium channel activation. Anesthesiology 2008, 109, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Huhn, R.; Weber, N.C.; Preckel, B.; Schlack, W.; Bauer, I.; Hollmann, M.W.; Heinen, A. Age-related loss of cardiac preconditioning: Impact of protein kinase A. Exp. Gerontol. 2012, 47, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Oei, G.T.; Huhn, R.; Heinen, A.; Hollmann, M.W.; Schlack, W.S.; Preckel, B.; Weber, N.C. Helium-induced cardioprotection of healthy and hypertensive rat myocardium in vivo. Eur. J. Pharmacol. 2012, 684, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Foster, C.R.; Satomi, S.; Kato, Y.; Patel, H.H. The caveolar-mitochondrial interface: Regulation of cellular metabolism in physiology and pathophysiology. Biochem. Soc. Trans. 2020, 48, 165–177. [Google Scholar] [CrossRef]
- Schilling, J.M.; Head, B.P.; Patel, H.H. Caveolins as Regulators of Stress Adaptation. Mol. Pharmacol. 2018, 93, 277–285. [Google Scholar] [CrossRef]
- Ohi, M.D.; Kenworthy, A.K. Emerging Insights into the Molecular Architecture of Caveolin-1. J. Membr. Biol. 2022, 255, 375–383. [Google Scholar] [CrossRef]
- Flick, M.; Albrecht, M.; Oei, G.T.; Steenstra, R.; Kerindongo, R.P.; Zuurbier, C.J.; Patel, H.H.; Hollmann, M.W.; Preckel, B.; Weber, N.C. Helium postconditioning regulates expression of caveolin-1 and -3 and induces RISK pathway activation after ischaemia/reperfusion in cardiac tissue of rats. Eur. J. Pharmacol. 2016, 791, 718–725. [Google Scholar] [CrossRef]
- Aehling, C.; Weber, N.C.; Zuurbier, C.J.; Preckel, B.; Galmbacher, R.; Stefan, K.; Hollmann, M.W.; Popp, E.; Knapp, J. Effects of combined helium pre/post-conditioning on the brain and heart in a rat resuscitation model. Acta Anaesthesiol. Scand. 2017, 62, 63–74. [Google Scholar] [CrossRef]
- Weber, N.C.; Schilling, J.M.; Warmbrunn, M.V.; Dhanani, M.; Kerindongo, R.; Siamwala, J.; Song, Y.; Zemljic-Harpf, A.E.; Fannon, M.J.; Hollmann, M.W.; et al. Helium-Induced Changes in Circulating Caveolin in Mice Suggest a Novel Mechanism of Cardiac Protection. Int. J. Mol. Sci. 2019, 20, 2640. [Google Scholar] [CrossRef]
- Jelemenský, M.; Kovácsházi, C.; Ferenczyová, K.; Hofbauerová, M.; Kiss, B.; Pállinger, É.; Kittel, Á.; Sayour, V.N.; Görbe, A.; Pelyhe, C.; et al. Helium Conditioning Increases Cardiac Fibroblast Migration Which Effect Is Not Propagated via Soluble Factors or Extracellular Vesicles. Int. J. Mol. Sci. 2021, 22, 10504. [Google Scholar] [CrossRef]
- Likhvantsev, V.V.; Landoni, G.; Levikov, D.I.; Grebenchikov, O.A.; Skripkin, Y.V.; Cherpakov, R.A. Sevoflurane Versus Total Intravenous Anesthesia for Isolated Coronary Artery Bypass Surgery with Cardiopulmonary Bypass: A Randomized Trial. J. Cardiothorac. Vasc. Anesth. 2016, 30, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Landoni, G.; Guarracino, F.; Cariello, C.; Franco, A.; Baldassarri, R.; Borghi, G.; Covello, R.D.; Gerli, C.; Crivellari, M.; Zangrillo, A. Volatile compared with total intravenous anaesthesia in patients undergoing high-risk cardiac surgery: A randomized multicentre study. Br. J. Anaesth. 2014, 113, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Landoni, G.; Lomivorotov, V.V.; Nigro Neto, C.; Monaco, F.; Pasyuga, V.V.; Bradic, N.; Lembo, R.; Gazivoda, G.; Likhvantsev, V.V.; Lei, C.; et al. Volatile Anesthetics versus Total Intravenous Anesthesia for Cardiac Surgery. N. Engl. J. Med. 2019, 380, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Lurati Buse, G.A.; Schumacher, P.; Seeberger, E.; Studer, W.; Schuman, R.M.; Fassl, J.; Kasper, J.; Filipovic, M.; Bolliger, D.; Seeberger, M.D. Randomized comparison of sevoflurane versus propofol to reduce perioperative myocardial ischemia in patients undergoing noncardiac surgery. Circulation 2012, 126, 2696–2704. [Google Scholar] [CrossRef]
- Devereaux, P.J.; Sessler, D.I.; Leslie, K.; Kurz, A.; Mrkobrada, M.; Alonso-Coello, P.; Villar, J.C.; Sigamani, A.; Biccard, B.M.; Meyhoff, C.S.; et al. Clonidine in patients undergoing noncardiac surgery. N. Engl. J. Med. 2014, 370, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Hofland, J.; Ouattara, A.; Fellahi, J.L.; Gruenewald, M.; Hazebroucq, J.; Ecoffey, C.; Joseph, P.; Heringlake, M.; Steib, A.; Coburn, M.; et al. Effect of Xenon Anesthesia Compared to Sevoflurane and Total Intravenous Anesthesia for Coronary Artery Bypass Graft Surgery on Postoperative Cardiac Troponin Release: An International, Multicenter, Phase 3, Single-blinded, Randomized Noninferiority Trial. Anesthesiology 2017, 127, 918–933. [Google Scholar] [CrossRef]
- Mehta, R.H.; Leimberger, J.D.; van Diepen, S.; Meza, J.; Wang, A.; Jankowich, R.; Harrison, R.W.; Hay, D.; Fremes, S.; Duncan, A.; et al. Levosimendan in Patients with Left Ventricular Dysfunction Undergoing Cardiac Surgery. N. Engl. J. Med. 2017, 376, 2032–2042. [Google Scholar] [CrossRef]
- Uhlig, C.; Bluth, T.; Schwarz, K.; Deckert, S.; Heinrich, L.; De Hert, S.; Landoni, G.; Serpa Neto, A.; Schultz, M.J.; Pelosi, P.; et al. Effects of Volatile Anesthetics on Mortality and Postoperative Pulmonary and Other Complications in Patients Undergoing Surgery: A Systematic Review and Meta-analysis. Anesthesiology 2016, 124, 1230–1245. [Google Scholar] [CrossRef]
- Smit, K.F.; Oei, G.T.M.L.; Brevoord, D.; Stroes, E.S.; Nieuwland, R.; Schlack, W.S.; Hollmann, M.W.; Weber, N.C.; Preckel, B. Helium Induces Preconditioning in Human Endothelium In Vivo. Anesthesiology 2013, 118, 95–104. [Google Scholar] [CrossRef]
- Smit, K.F.; Brevoord, D.; de Hert, S.; de Mol, B.A.; Kerindongo, R.P.; Van Dieren, S.; Schlack, W.S.; Hollmann, M.W.; Weber, N.C.; Preckel, B. Effect of helium pre- or postconditioning on signal transduction kinases in patients undergoing coronary artery bypass graft surgery. J. Transl. Med. 2016, 14, 294. [Google Scholar] [CrossRef]
- Arola, O.; Saraste, A.; Laitio, R.; Airaksinen, J.; Hynninen, M.; Bäcklund, M.; Ylikoski, E.; Wennervirta, J.; Pietilä, M.; Roine, R.O.; et al. Inhaled Xenon Attenuates Myocardial Damage in Comatose Survivors of Out-of-Hospital Cardiac Arrest: The Xe-Hypotheca Trial. J. Am. Coll. Cardiol. 2017, 70, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Saraste, A.; Ballo, H.; Arola, O.; Laitio, R.; Airaksinen, J.; Hynninen, M.; Bäcklund, M.; Ylikoski, E.; Wennervirta, J.; Pietilä, M.; et al. Effect of Inhaled Xenon on Cardiac Function in Comatose Survivors of Out-of-Hospital Cardiac Arrest—A Substudy of the Xenon in Combination With Hypothermia After Cardiac Arrest Trial. Crit. Care Explor. 2021, 3, e0502. [Google Scholar] [CrossRef]
- Derwall, M.; Ebeling, A.; Nolte, K.W.; Weis, J.; Rossaint, R.; Ichinose, F.; Nix, C.; Fries, M.; Brücken, A. Inhaled nitric oxide improves transpulmonary blood flow and clinical outcomes after prolonged cardiac arrest: A large animal study. Crit. Care 2015, 19, 328. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, Y.; Pokreisz, P.; Vermeersch, P.; Marsboom, G.; Swinnen, M.; Verbeken, E.; Santos, J.; Pellens, M.; Gillijns, H.; et al. Nitric Oxide Inhalation Improves Microvascular Flow and Decreases Infarction Size After Myocardial Ischemia and Reperfusion. J. Am. Coll. Cardiol. 2007, 50, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Minamishima, S.; Kida, K.; Tokuda, K.; Wang, H.; Sips, P.Y.; Kosugi, S.; Mandeville, J.B.; Buys, E.S.; Brouckaert, P.; Liu, P.K.; et al. Inhaled Nitric Oxide Improves Outcomes After Successful Cardiopulmonary Resuscitation in Mice. Circulation 2011, 124, 1645–1653. [Google Scholar] [CrossRef]
- Russo, I.; Barale, C.; Melchionda, E.; Penna, C.; Pagliaro, P. Platelets and Cardioprotection: The Role of Nitric Oxide and Carbon Oxide. Int. J. Mol. Sci. 2023, 24, 6107. [Google Scholar] [CrossRef]
- Hayashida, K.; Sano, M.; Ohsawa, I.; Shinmura, K.; Tamaki, K.; Kimura, K.; Endo, J.; Katayama, T.; Kawamura, A.; Kohsaka, S.; et al. Inhalation of hydrogen gas reduces infarct size in the rat model of myocardial ischemia–reperfusion injury. Biochem. Biophys. Res. Commun. 2008, 373, 30–35. [Google Scholar] [CrossRef]
- Janssens, S.P.; Bogaert, J.; Zalewski, J.; Toth, A.; Adriaenssens, T.; Belmans, A.; Bennett, J.; Claus, P.; Desmet, W.; Dubois, C.; et al. Nitric oxide for inhalation in ST-elevation myocardial infarction (NOMI): A multicentre, double-blind, randomized controlled trial. Eur. Heart J. 2018, 39, 2717–2725. [Google Scholar] [CrossRef]
- Yuan, T.; Zhao, J.N.; Bao, N.R. Hydrogen applications: Advances in the field of medical therapy. Med. Gas Res. 2023, 13, 99–107. [Google Scholar]
- Ohsawa, I.; Ishikawa, M.; Takahashi, K.; Watanabe, M.; Nishimaki, K.; Yamagata, K.; Katsura, K.; Katayama, Y.; Asoh, S.; Ohta, S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med. 2007, 13, 688–694. [Google Scholar] [CrossRef]
- Chambers, D.J.; Fallouh, H.B. Cardioplegia and cardiac surgery: Pharmacological arrest and cardioprotection during global ischemia and reperfusion. Pharmacol. Ther. 2010, 127, 41–52. [Google Scholar] [CrossRef]
- Francica, A.; Tonelli, F.; Rossetti, C.; Tropea, I.; Luciani, G.B.; Faggian, G.; Dobson, G.P.; Onorati, F. Cardioplegia between Evolution and Revolution: From Depolarized to Polarized Cardiac Arrest in Adult Cardiac Surgery. J. Clin. Med. 2021, 10, 4485. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef]
- Colliva, A.; Braga, L.; Giacca, M.; Zacchigna, S. Endothelial cell–cardiomyocyte crosstalk in heart development and disease. J. Physiol. 2019, 598, 2923–2939. [Google Scholar] [CrossRef] [PubMed]
- Os, M.M.D.; Brom, C.E.V.D.; van Leeuwen, A.L.I.; Dekker, N.A.M. Microcirculatory perfusion disturbances following cardiopulmonary bypass: A systematic review. Crit. Care 2020, 24, 218. [Google Scholar] [CrossRef]
- Dekker, N.A.M.; van Leeuwen, A.L.I.; van de Ven, P.M.; de Vries, R.; Hordijk, P.L.; Boer, C.; van den Brom, C.E. Pharmacological interventions to reduce edema following cardiopulmonary bypass: A systematic review and meta-analysis. J. Crit. Care 2020, 56, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Tan, Z.; Zhong, X.; Tian, Y.; Wang, X.; Yu, B.; Ramirez-Correa, G.; Murphy, A.; Gabrielson, K.; Paolocci, N.; et al. Endocardial endothelium is a key determinant of force-frequency relationship in rat ventricular myocardium. J. Appl. Physiol. 2013, 115, 383–393. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Cote, G.M.; Guo, X.; Lebrasseur, N.K.; Cui, L.; Liao, R.; Sawyer, D.B. Cardiac endothelial cells regulate reactive oxygen species-induced cardiomyocyte apoptosis through neuregulin-1beta/erbB4 signaling. J. Biol. Chem. 2004, 279, 51141–51147. [Google Scholar] [CrossRef]
- Hernández-Reséndiz, S.; Muñoz-Vega, M.; E Contreras, W.; E Crespo-Avilan, G.; Rodriguez-Montesinos, J.; Arias-Carrión, O.; Pérez-Méndez, O.; A Boisvert, W.; Preissner, K.T.; A Cabrera-Fuentes, H. Responses of Endothelial Cells Towards Ischemic Conditioning Following Acute Myocardial Infarction. Cond. Med. 2018, 1, 247–258. [Google Scholar] [PubMed]
- Zakkar, M.; Guida, G.; Suleiman, M.-S.; Angelini, G.D. Cardiopulmonary Bypass and Oxidative Stress. Oxidative Med. Cell. Longev. 2015, 2015, 189863. [Google Scholar] [CrossRef]
- Kiziltepe, U.; Tunçtan, B.; Eyileten, Z.B.; Sirlak, M.; Arikbuku, M.; Tasoz, R.; Uysalel, A.; Ozyurda, U. Efficiency of l-arginine enriched cardioplegia and non-cardioplegic reperfusion in ischemic hearts. Int. J. Cardiol. 2004, 97, 93–100. [Google Scholar] [CrossRef]
- Orhan, G.; Yapici, N.; Yuksel, M.; Sargin, M.; Şenay, S.; Yalçin, A.S.; Aykaç, Z.; Aka, S.A. Effects of N-acetylcysteine on myocardial ischemia–reperfusion injury in bypass surgery. Heart Vessel. 2006, 21, 42–47. [Google Scholar] [CrossRef]
- Prabhu, A.; Sujatha, D.; Kanagarajan, N.; Vijayalakshmi, M.; Ninan, B. Effect of N-Acetylcysteine in Attenuating Ischemic Reperfusion Injury in Patients Undergoing Coronary Artery Bypass Grafting with Cardiopulmonary Bypass. Ann. Vasc. Surg. 2009, 23, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Hallström, S.; Franz, M.; Gasser, H.; Vodrazka, M.; Semsroth, S.; Losert, U.M.; Haisjackl, M.; Podesser, B.K.; Malinski, T. S-nitroso human serum albumin reduces ischaemia/reperfusion injury in the pig heart after unprotected warm ischaemia. Cardiovasc. Res. 2007, 77, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-W.; Major, T.; Deatrick, K.B.; Mohammed, A.; Jeakle, M.; Charpie, J.R. Nicorandil attenuates ventricular dysfunction and organ injury after cardiopulmonary bypass. Int. J. Cardiol. 2022, 368, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yamada, T.; Kotake, Y.; Takeda, J. Cardioprotective Effects of Nicorandil in Patients Undergoing On-Pump Coronary Artery Bypass Surgery. J. Cardiothorac. Vasc. Anesth. 2008, 22, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Kadosaki, M.; Nara, N.; Wei, J.; Endo, S.; Inada, K. Nicorandil attenuates NF-kappaB activation, adhesion molecule expression, and cytokine production in patients with coronary artery bypass surgery. Shock 2005, 24, 103–108. [Google Scholar] [CrossRef]
- Yavuz, T.; Altuntas, I.; Odabasi, D.; Delibas, N.; Ocal, A.; Ibrisim, E.; Kutsal, A. Beneficial Effect of the Addition of Nitroglycerin to the Cardioplegic Solution on Lipid Peroxidation during Coronary Artery Bypass Surgery. Int. Heart J. 2005, 46, 45–55. [Google Scholar] [CrossRef]
- Hamarneh, A.; Ho, A.F.W.; Bulluck, H.; Sivaraman, V.; Ricciardi, F.; Nicholas, J.; Shanahan, H.; Hardman, E.A.; Wicks, P.; Ramlall, M.; et al. Negative interaction between nitrates and remote ischemic preconditioning in patients undergoing cardiac surgery: The ERIC-GTN and ERICCA studies. Basic Res. Cardiol. 2022, 117, 31. [Google Scholar] [CrossRef]
- Clark, D., 3rd; Tesseneer, S.; Tribble, C.G. Nitroglycerin and sodium nitroprusside: Potential contributors to postoperative bleeding? Heart Surg. Forum 2012, 15, E92–E96. [Google Scholar] [CrossRef]
- Rubio-Gayosso, I.; Platts, S.H.; Duling, B.R. Reactive oxygen species mediate modification of glycocalyx during ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2247–H2256. [Google Scholar] [CrossRef]
- Mulivor, A.W.; Lipowsky, H.H. Inflammation- and ischemia-induced shedding of venular glycocalyx. Am. J. Physiol. Circ. Physiol. 2004, 286, H1672–H1680. [Google Scholar] [CrossRef] [PubMed]
- Bonfig, N.L.; Soukup, C.R.; Shah, A.A.; Olet, S.; Davidson, S.J.; Schmidt, C.W.; Peterson, R.; Henry, T.D.; Traverse, J.H. Increasing myocardial edema is associated with greater microvascular obstruction in ST-segment elevation myocardial infarction. Am. J. Physiol. Circ. Physiol. 2022, 323, H818–H824. [Google Scholar] [CrossRef]
- Palmier, M.; Cornet, E.; Renet, S.; Dumesnil, A.; Perzo, N.; Cohen, Q.; Richard, V.; Plissonnier, D. A Supraceliac Aortic Cross Clamping Model to Explore Remote Lung Injury and the Endothelial Glycocalyx. Ann. Vasc. Surg. 2022, S0890-5096(22)00906-2. [Google Scholar] [CrossRef] [PubMed]
- Dekker, N.A.M.; Veerhoek, D.; Koning, N.J.; van Leeuwen, A.L.I.; Elbers, P.W.G.; Brom, C.E.V.D.; Vonk, A.B.A.; Boer, C. Postoperative microcirculatory perfusion and endothelial glycocalyx shedding following cardiac surgery with cardiopulmonary bypass. Anaesthesia 2019, 74, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Passov, A.; Schramko, A.; Salminen, U.-S.; Aittomäki, J.; Andersson, S.; Pesonen, E. Endothelial glycocalyx during early reperfusion in patients undergoing cardiac surgery. PLoS ONE 2021, 16, e0251747. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Vlastos, D.; Andreadou, I.; Gazouli, M.; Efentakis, P.; Varoudi, M.; Makavos, G.; Kapelouzou, A.; Lekakis, J.; Parissis, J.; et al. Vascular conditioning prevents adverse left ventricular remodelling after acute myocardial infarction: A randomised remote conditioning study. Basic Res. Cardiol. 2021, 116, 9. [Google Scholar] [CrossRef]
- Annecke, T.; Chappell, D.; Chen, C.; Jacob, M.; Welsch, U.; Sommerhoff, C.P.; Rehm, M.; Conzen, P.F.; Becker, B.F. Sevoflurane preserves the endothelial glycocalyx against ischaemia-reperfusion injury. Br. J. Anaesth. 2010, 104, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.Q.; Sun, J.H.; Wu, Q.L.; Feng, L.Y.; Fan, Y.X.; Ye, J.X.; Gao, W.; He, G.L.; Wang, W.J. Protective effect of sevoflurane on vascular endothelial glycocalyx in patients undergoing heart valve surgery: A randomised controlled trial. Eur. J. Anaesthesiol. 2021, 38, 477–486. [Google Scholar] [CrossRef]
- Flameng, W.; Borgers, M.; Van Der Vusse, G.J.; Demeyere, R.; Vandermeersch, E.; Thoné, F.; Suy, R. Cardioprotective effects of lidoflazine in extensive aorta-coronary bypass grafting. J. Thorac. Cardiovasc. Surg. 1983, 85, 758–768. [Google Scholar] [CrossRef]
- Shen, A.C.; Jennings, R.B. Myocardial calcium and magnesium in acute ischemic injury. Am. J. Pathol. 1972, 67, 417–440. [Google Scholar] [PubMed]
- Serraino, G.F.; Jiritano, F.; Costa, D.; Ielapi, N.; Battaglia, D.; Bracale, U.M.; Mastroroberto, P.; Andreucci, M.; Serra, R. Metalloproteinases in Cardiac Surgery: A Systematic Review. Biomolecules 2023, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Fang, F.; Xia, T.J.; Zhang, Y.; Sun, J.; Wu, Q.; Wang, W. Doxycycline can reduce glycocalyx shedding by inhibiting matrix metalloproteinases in patients undergoing cardiopulmonary bypass: A randomized controlled trial. Microvasc. Res. 2022, 142, 104381. [Google Scholar] [CrossRef] [PubMed]
- Schulze, C.J.; Castro, M.M.; Kandasamy, A.D.; Cena, J.; Bryden, C.; Wang, S.H.; Koshal, A.; Tsuyuki, R.T.; Finegan, B.A.; Schulz, R. Doxycycline Reduces Cardiac Matrix Metalloproteinase-2 Activity but Does not Ameliorate Myocardial Dysfunction During Reperfusion in Coronary Artery Bypass Patients Undergoing Cardiopulmonary Bypass. Crit. Care Med. 2013, 41, 2512–2520. [Google Scholar] [CrossRef] [PubMed]
- Dekker, N.A.; Veerhoek, D.; van Leeuwen, A.L.; Vonk, A.B.; Brom, C.E.V.D.; Boer, C. Microvascular Alterations during Cardiac Surgery Using a Heparin or Phosphorylcholine-Coated Circuit. J. Cardiothorac. Vasc. Anesth. 2020, 34, 912–919. [Google Scholar] [CrossRef]
- Selim, J.; Hamzaoui, M.; Ghemired, A.; Djerada, Z.; Chevalier, L.; Piton, N.; Besnier, E.; Clavier, T.; Dumesnil, A.; Renet, S.; et al. Priming of Cardiopulmonary Bypass with Human Albumin Decreases Endothelial Dysfunction after Pulmonary Ischemia–Reperfusion in an Animal Model. Int. J. Mol. Sci. 2022, 23, 8938. [Google Scholar] [CrossRef]
- Vahldieck, C.; Cianflone, E.; Fels, B.; Löning, S.; Depelmann, P.; Sabatino, J.; Salerno, N.; Karsten, C.M.; Torella, D.; Weil, J.; et al. Endothelial Glycocalyx and Cardiomyocyte Damage Is Prevented by Recombinant Syndecan-1 in Acute Myocardial Infarction. Am. J. Pathol. 2023, 193, 474–492. [Google Scholar] [CrossRef]
- Juffermans, N.P.; van den Brom, C.E.; Kleinveld, D.J.B. Targeting Endothelial Dysfunction in Acute Critical Illness to Reduce Organ Failure. Anesth. Analg. 2020, 131, 1708–1720. [Google Scholar] [CrossRef]
- Addya, S.; Shiroto, K.; Turoczi, T.; Zhan, L.; Kaga, S.; Fukuda, S.; Surrey, S.; Duan, L.-J.; Fong, G.-H.; Yamamoto, F. Ischemic preconditioning-mediated cardioprotection is disrupted in heterozygous Flt-1 (VEGFR-1) knockout mice. J. Mol. Cell. Cardiol. 2005, 38, 345–351. [Google Scholar] [CrossRef]
- Thirunavukkarasu, M.; Juhasz, B.; Zhan, L.; Menon, V.P.; Tosaki, A.; Otani, H.; Maulik, N. VEGFR1 (Flt-1+/-) gene knockout leads to the disruption of VEGF-mediated signaling through the nitric oxide/heme oxygenase pathway in ischemic preconditioned myocardium. Free Radic. Biol. Med. 2007, 42, 1487–1495. [Google Scholar] [CrossRef]
- Gonca, S.; Kiliçkan, L.; Dalçik, C.; Dalçik, H.; Bayindir, O. The cardioprotective effects of thoracal epidural anestesia are induced by the expression of vascular endothelial growth factor and inducible nitric oxide synthase in cardiopulmonary bypass surgery. J. Cardiovasc. Surg. 2007, 48, 93–102. [Google Scholar]
- Braile, M.; Marcella, S.; Cristinziano, L.; Galdiero, M.R.; Modestino, L.; Ferrara, A.L.; Varricchi, G.; Marone, G.; Loffredo, S. VEGF-A in Cardiomyocytes and Heart Diseases. Int. J. Mol. Sci. 2020, 21, 5294. [Google Scholar] [CrossRef] [PubMed]
- Tofukuji, M.; Metais, C.; Li, J.; Franklin, A.; Simons, M.; Sellke, F.W. Myocardial VEGF expression after cardiopulmonary bypass and cardioplegia. Circulation 1998, 98((Suppl. 19)), II242-6, discussion II247–II248. [Google Scholar]
- Koning, N.J.; Overmars, M.A.H.; Brom, C.E.V.D.; van Bezu, J.; Simon, L.E.; Vonk, A.B.A.; Girbes, A.R.J.; Amerongen, G.P.V.N.; Boer, C. Endothelial hyperpermeability after cardiac surgery with cardiopulmonary bypass as assessed using an in vitro bioassay for endothelial barrier function. Br. J. Anaesth. 2016, 116, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhang, Y.; Yang, S.; Wu, M.; Fang, Y.; Feng, J.; Liu, B. Protective effect of vascular endothelial growth factor against cardiopulmonary bypass-associated acute kidney injury in beagles. Exp. Ther. Med. 2017, 15, 963–969. [Google Scholar] [CrossRef]
- Raza, Z.; Saleem, U.; Naureen, Z. Sphingosine 1-phosphate signaling in ischemia and reperfusion injury. Prostaglandins Other Lipid Mediat. 2020, 149, 106436. [Google Scholar] [CrossRef] [PubMed]
- Araibi, H.; van der Merwe, E.; Gwanyanya, A.; Kelly-Laubscher, R. The effect of sphingosine-1-phosphate on the endothelial glycocalyx during ischemia-reperfusion injury in the isolated rat heart. Microcirculation 2020, 27, e12612. [Google Scholar] [CrossRef]
- Samarska, I.V.; Bouma, H.R.; Buikema, H.; Mungroop, H.E.; Houwertjes, M.C.; Absalom, A.R.; Epema, A.H.; Henning, R.H. S1P1 Receptor Modulation Preserves Vascular Function in Mesenteric and Coronary Arteries after CPB in the Rat Independent of Depletion of Lymphocytes. PLoS ONE 2014, 9, e97196. [Google Scholar] [CrossRef]
- Greiwe, G.; Moritz, E.; Amschler, K.; Poppe, A.; Sarwari, H.; Nierhaus, A.; Kluge, S.; Reichenspurner, H.; Zoellner, C.; Schwedhelm, E.; et al. Dynamics of Vascular Protective and Immune Supportive Sphingosine-1-Phosphate During Cardiac Surgery. Front. Immunol. 2021, 12, 761475. [Google Scholar] [CrossRef]
- Clajus, C.; Lukasz, A.; David, S.; Hertel, B.; Lichtinghagen, R.; Parikh, S.M.; Simon, A.; Ismail, I.; Haller, H.; Kümpers, P. Angiopoietin-2 is a potential mediator of endothelial barrier dysfunction following cardiopulmonary bypass. Cytokine 2012, 60, 352–359. [Google Scholar] [CrossRef]
- Dekker, N.A.M.; Van Leeuwen, A.L.I.; Van Strien, W.W.J.; Majolée, J.; Szulcek, R.; Vonk, A.B.A.; Hordijk, P.L.; Boer, C.; van den Brom, C.E. Microcirculatory perfusion disturbances following cardiac surgery with cardiopulmonary bypass are associated with in vitro endothelial hyperpermeability and increased angiopoietin-2 levels. Crit. Care 2019, 23, 117. [Google Scholar] [CrossRef] [PubMed]
- Dekker, N.; van Meurs, M.; van Leeuwen, A.; Hofland, H.; van Slyke, P.; Vonk, A.; Boer, C.; Brom, C.V.D. Vasculotide, an angiopoietin-1 mimetic, reduces pulmonary vascular leakage and preserves microcirculatory perfusion during cardiopulmonary bypass in rats. Br. J. Anaesth. 2018, 121, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Lee, C.-K.; Kang, S.; Park, I.; Kim, Y.H.; Kim, S.K.; Hong, S.P.; Bae, H.; He, Y.; Kubota, Y.; et al. Angiopoietin-2 exacerbates cardiac hypoxia and inflammation after myocardial infarction. J. Clin. Investig. 2018, 128, 5018–5033. [Google Scholar] [CrossRef]
- Hu, S.; Cao, S.; Liu, J. Role of angiopoietin-2 in the cardioprotective effect of fibroblast growth factor 21 on ischemia/reperfusion-induced injury in H9c2 cardiomyocytes. Exp. Ther. Med. 2017, 14, 771–779. [Google Scholar] [CrossRef]
- Aman, J.; van Bezu, J.; Damanafshan, A.; Huveneers, S.; Eringa, E.C.; Vogel, S.M.; Groeneveld, A.J.; Noordegraaf, A.V.; van Hinsbergh, V.W.; Amerongen, G.P.V.N. Effective Treatment of Edema and Endothelial Barrier Dysfunction with Imatinib. Circulation 2012, 126, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.N.; Aman, J.; Amerongen, G.P.V.N.; Dudek, S.M. Targeting Abl Kinases to Regulate Vascular Leak during Sepsis and Acute Respiratory Distress Syndrome. Arter. Thromb. Vasc. Biol. 2015, 35, 1071–1079. [Google Scholar] [CrossRef]
- Koning, N.; de Lange, F.; van Meurs, M.; Jongman, R.; Ahmed, Y.; Schwarte, L.; Amerongen, G.V.N.; Vonk, A.; Niessen, H.; Baufreton, C.; et al. Reduction of vascular leakage by imatinib is associated with preserved microcirculatory perfusion and reduced renal injury markers in a rat model of cardiopulmonary bypass. Br. J. Anaesth. 2018, 120, 1165–1175. [Google Scholar] [CrossRef]
- Konijnenberg, L.S.F.; Luiken, T.T.J.; Veltien, A.; Uthman, L.; Kuster, C.T.A.; Rodwell, L.; de Waard, G.A.; Lindert, M.K.-T.; Akiva, A.; Thijssen, D.H.J.; et al. Imatinib attenuates reperfusion injury in a rat model of acute myocardial infarction. Basic Res. Cardiol. 2023, 118, 2. [Google Scholar] [CrossRef]
- Boran, T.; Akyildiz, A.G.; Jannuzzi, A.T.; Alpertunga, B. Extended regorafenib treatment can be linked with mitochondrial damage leading to cardiotoxicity. Toxicol. Lett. 2020, 336, 39–49. [Google Scholar] [CrossRef]
- Raivio, P.; Lassila, R.; Petäjä, J. Thrombin in myocardial ischemia-reperfusion during cardiac surgery. Ann. Thorac. Surg. 2009, 88, 318–325. [Google Scholar] [CrossRef]
- Dekker, N.A.M.; van Leeuwen, A.L.I.; van Meurs, M.; Moser, J.; Pankras, J.E.; van der Wel, N.N.; Niessen, H.W.; Vervloet, M.G.; Vonk, A.B.A.; Hordijk, P.L.; et al. Preservation of renal endothelial integrity and reduction of renal edema by aprotinin does not preserve renal perfusion and function following experimental cardiopulmonary bypass. Intensiv. Care Med. Exp. 2021, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.A.; Bianchi, C.; Araujo, E.; Voisine, P.; Xu, S.-H.; Feng, J.; Li, J.; Sellke, F.W. Aprotinin Preserves Cellular Junctions and Reduces Myocardial Edema After Regional Ischemia and Cardioplegic Arrest. Circulation 2005, 112, I196–I201. [Google Scholar] [CrossRef] [PubMed]
- Karaca, P.; Konuralp, C.; Enc, Y.; Süzer, A.; Sokullu, O.; Ayoglu, U.; Cicek, S. Cardioprotective Effect of Aprotinin on Myocardial Ischemia/Reperfusion Injury during Cardiopulmonary Bypass. Circ. J. 2006, 70, 1432–1436. [Google Scholar] [CrossRef]
- Bert, C.; De Buck, F.; Sergeant, P.; Van Hemelrijck, J.; Kasran, A.; Van Duppen, V.; Ceuppens, J.; Meyns, B.; Delforge, M.; Wouters, P. Aprotinin Reduces Cardiac Troponin I Release and Inhibits Apoptosis of Polymorphonuclear Cells during Off-Pump Coronary Artery Bypass Surgery. J. Cardiothorac. Vasc. Anesth. 2008, 22, 16–22. [Google Scholar] [CrossRef]
- Mangano, D.T.; Tudor, I.C.; Dietzel, C. The risk associated with aprotinin in cardiac surgery. N. Engl. J. Med. 2006, 354, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, L.; He, L.; Yao, Y. The effect of tranexamic acid on myocardial injury in cardiac surgical patients: A systematic review and meta-analysis. Blood Coagul. Fibrinolysis 2022, 33, 429–437. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, H.; Li, J.; Bian, Y.; Song, Y.; Li, Z.; He, F.; Liu, S.; Tsai, Y. Hirudin protects against isoproternol-induced myocardial infraction by alleviating oxidative via an Nrf2 dependent manner. Int. J. Biol. Macromol. 2020, 162, 425–435. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
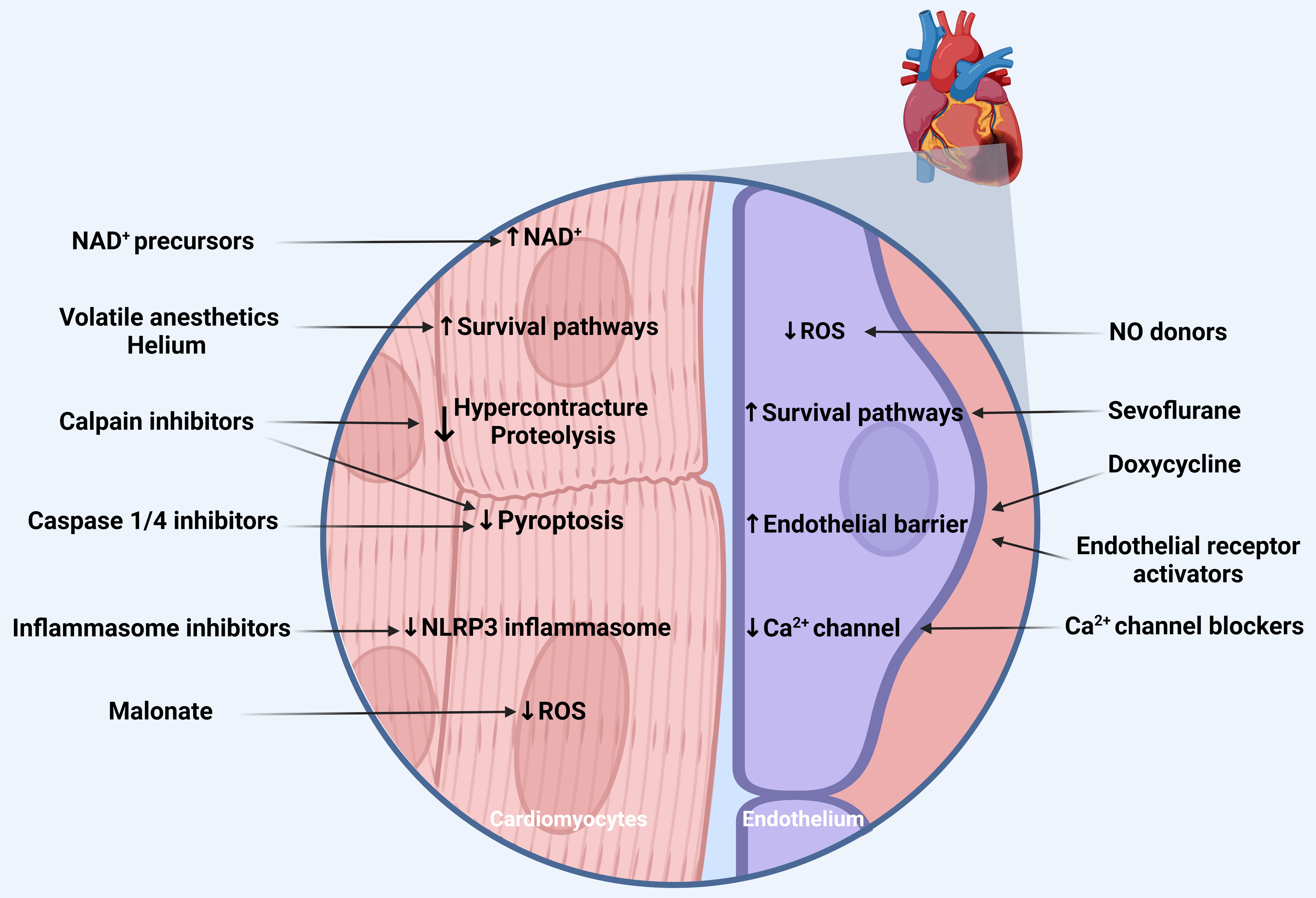
| Drugs/Compounds | Mechanism | Setting | = > Phase II Trial? |
|---|---|---|---|
| Heart | |||
| NAD+ precursors (NR, NMN) | restoring NAD levels | preclinical models of acute cardiac IRI | No |
| Malonate | reducing ROS at early reperfusion | preclinical models of acute cardiac IRI | No |
| NLRP3 inflammasome inhibitors | preventing inflammasome complex formation | preclinical models of cardiac IRI | No |
| Caspase-1,4 inhibitors (VRT, emricasan) | pyroptosis inhibition | preclinical models of acute cardiac IRI | No |
| Calpain inhibitors | proteolysis and pyroptosis inhibition | preclinical models of cardiac IRI | No |
| Volatile anesthetics/ Helium/Xenon | activation of Survival pathways (RISK/SAFE), Caveolin 1/3, mitophagy, and autophagy | preclinical models of cardiac IRI | Yes |
| Volatile anesthetics | reduced microRNAs (miRNA-155), induction of VEGFR1 | preclinical models of cardiac IRI | No |
| Endothelium | |||
| Sevoflurane | activation of survival pathways (RISK/SAFE) | preclinical CPB models cardiac surgery patients with CPB | Yes |
| Nitric oxide donors | reducing oxidative stress | preclinical CPB models cardiac surgery patients with CPB | Yes |
| Lidoflazine | calcium channel blocker | patients undergoing multiple aorta-coronary bypass grafting | Yes |
| Doxycycline | inhibition matrix metalloproteinases | cardiac surgery patients with CPB | Yes |
| (Indirect) Endothelial receptor activators | strengthening endothelial barrier | preclinical CPB models cardiac surgery patients with CPB | Yes, but not all |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Zuurbier, C.J.; Huhn, R.; Torregroza, C.; Hollmann, M.W.; Preckel, B.; van den Brom, C.E.; Weber, N.C. Pharmacological Cardioprotection against Ischemia Reperfusion Injury—The Search for a Clinical Effective Therapy. Cells 2023, 12, 1432. https://doi.org/10.3390/cells12101432
Wang Q, Zuurbier CJ, Huhn R, Torregroza C, Hollmann MW, Preckel B, van den Brom CE, Weber NC. Pharmacological Cardioprotection against Ischemia Reperfusion Injury—The Search for a Clinical Effective Therapy. Cells. 2023; 12(10):1432. https://doi.org/10.3390/cells12101432
Chicago/Turabian StyleWang, Qian, Coert J. Zuurbier, Ragnar Huhn, Carolin Torregroza, Markus W. Hollmann, Benedikt Preckel, Charissa E. van den Brom, and Nina C. Weber. 2023. "Pharmacological Cardioprotection against Ischemia Reperfusion Injury—The Search for a Clinical Effective Therapy" Cells 12, no. 10: 1432. https://doi.org/10.3390/cells12101432
APA StyleWang, Q., Zuurbier, C. J., Huhn, R., Torregroza, C., Hollmann, M. W., Preckel, B., van den Brom, C. E., & Weber, N. C. (2023). Pharmacological Cardioprotection against Ischemia Reperfusion Injury—The Search for a Clinical Effective Therapy. Cells, 12(10), 1432. https://doi.org/10.3390/cells12101432