
Surface Hydration Protects Cystic Fibrosis Airways from Infection by Restoring Junctional Networks

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Apical Surface Liquid Manipulation
2.3. Bacteria and Infection
2.4. Epithelium Integrity
2.5. Western Blot
2.6. Confocal Microscopy
2.7. Short-Circuit Current
2.8. RNA Extraction, RT-PCR, and qPCR
2.9. NanoString Gene Expression
2.10. Statistical Analysis
3. Results
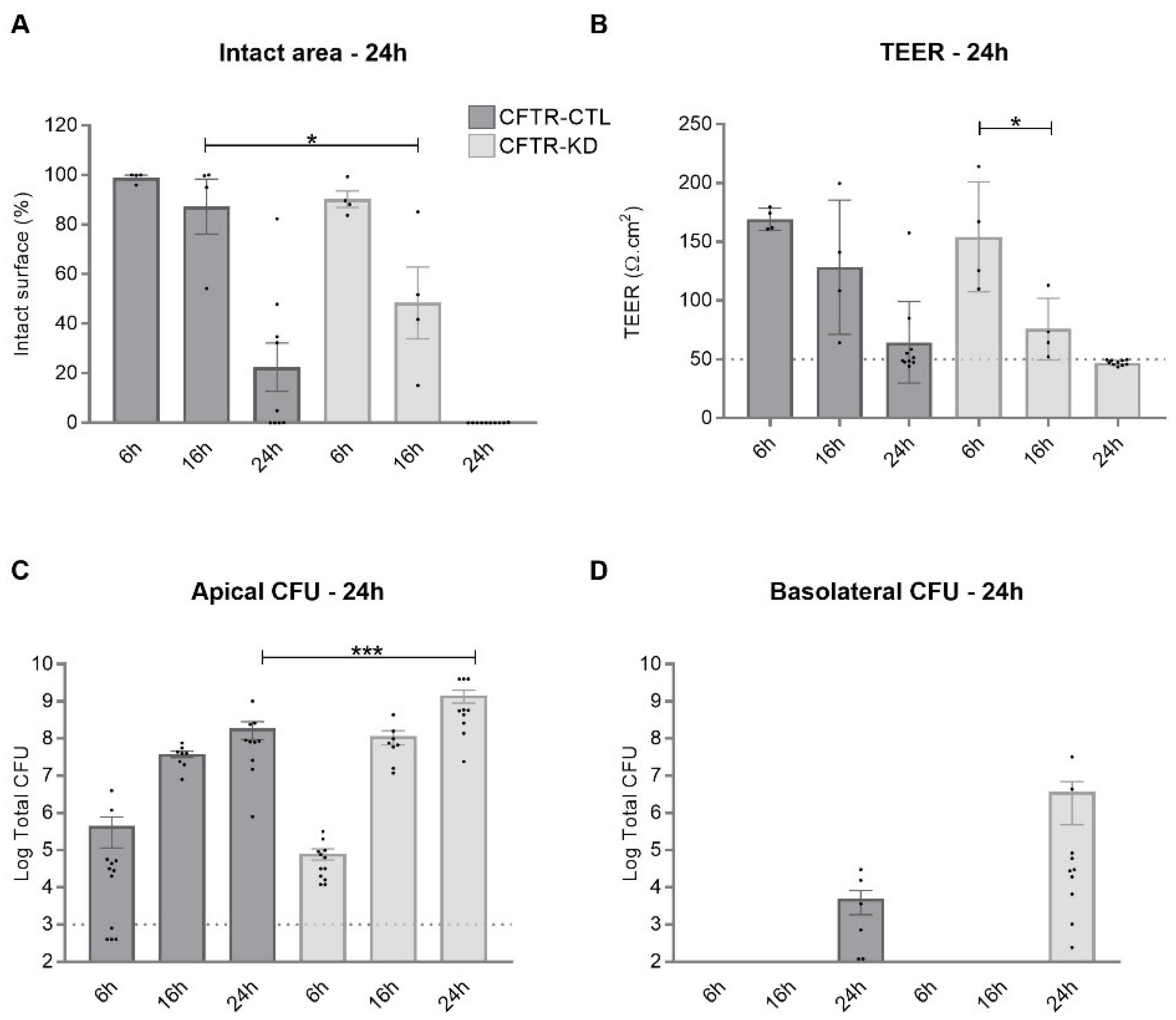
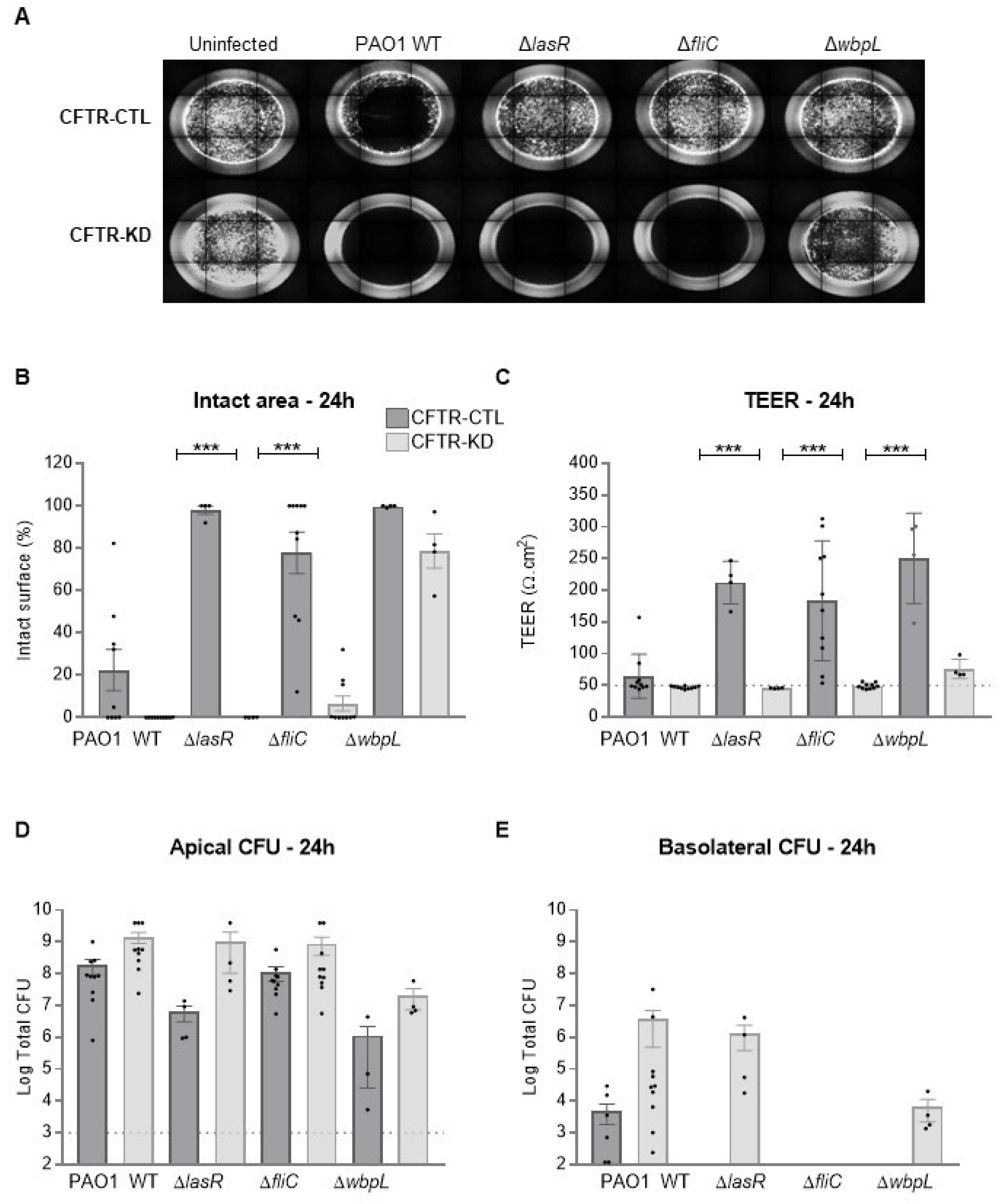
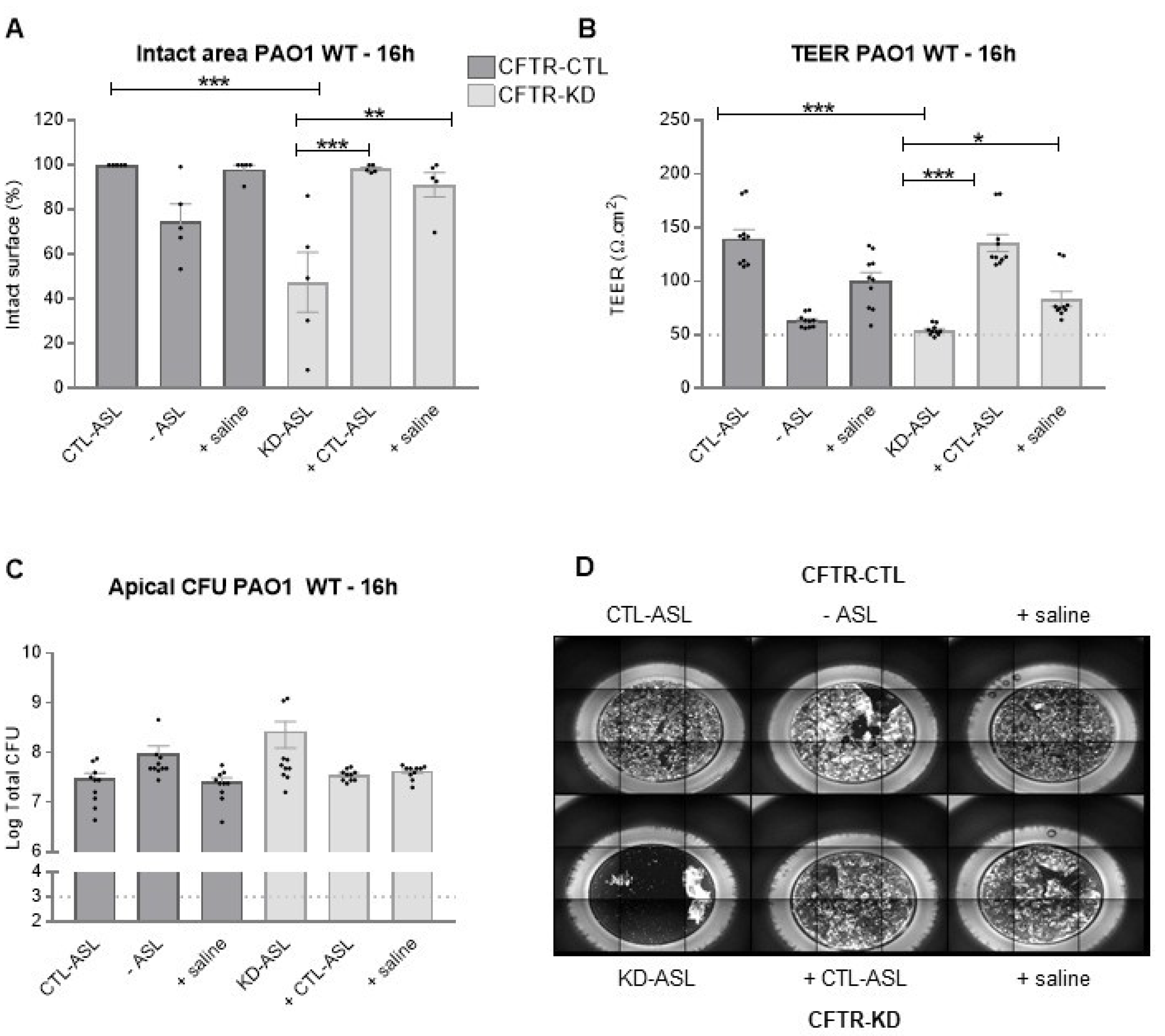
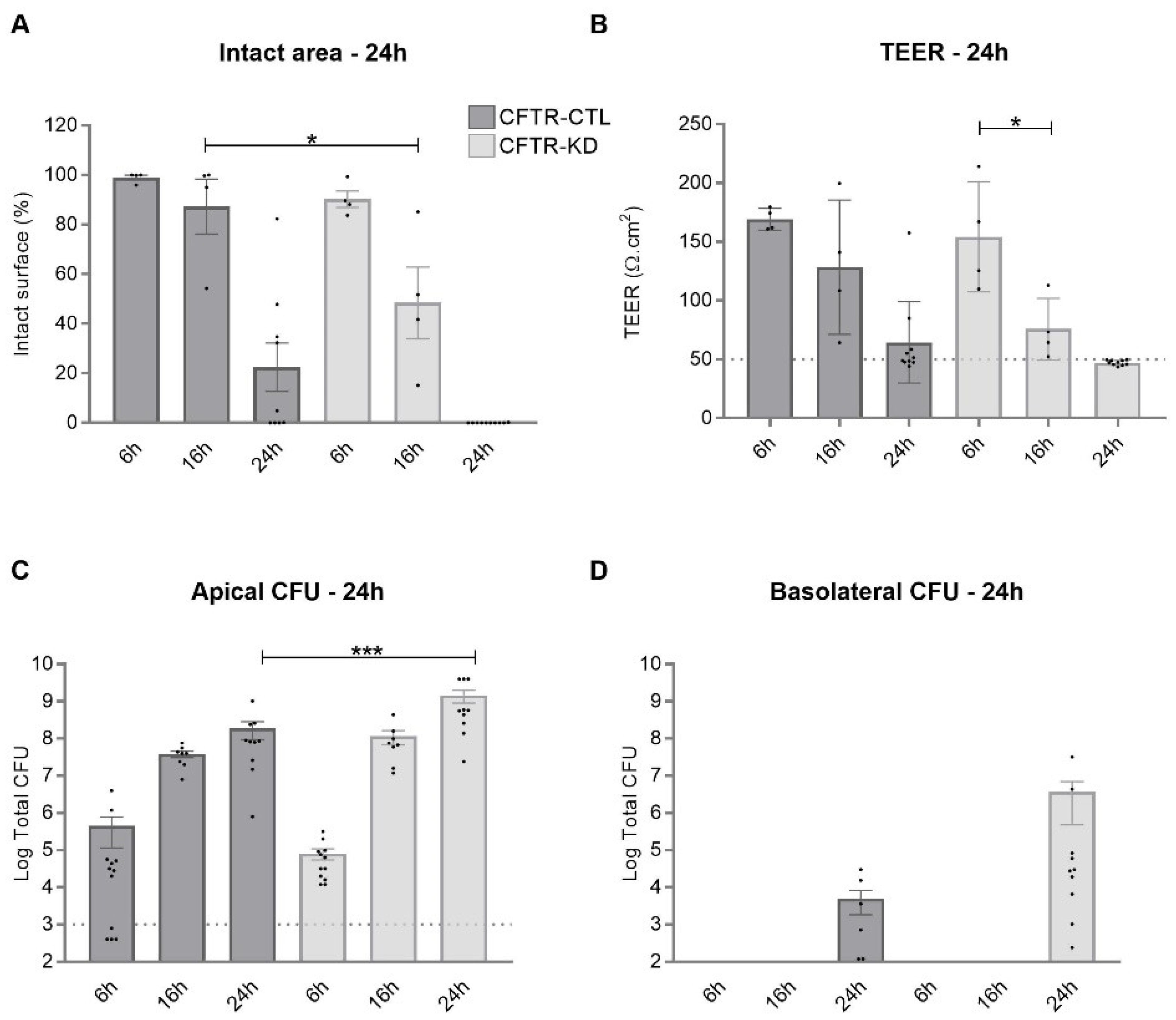
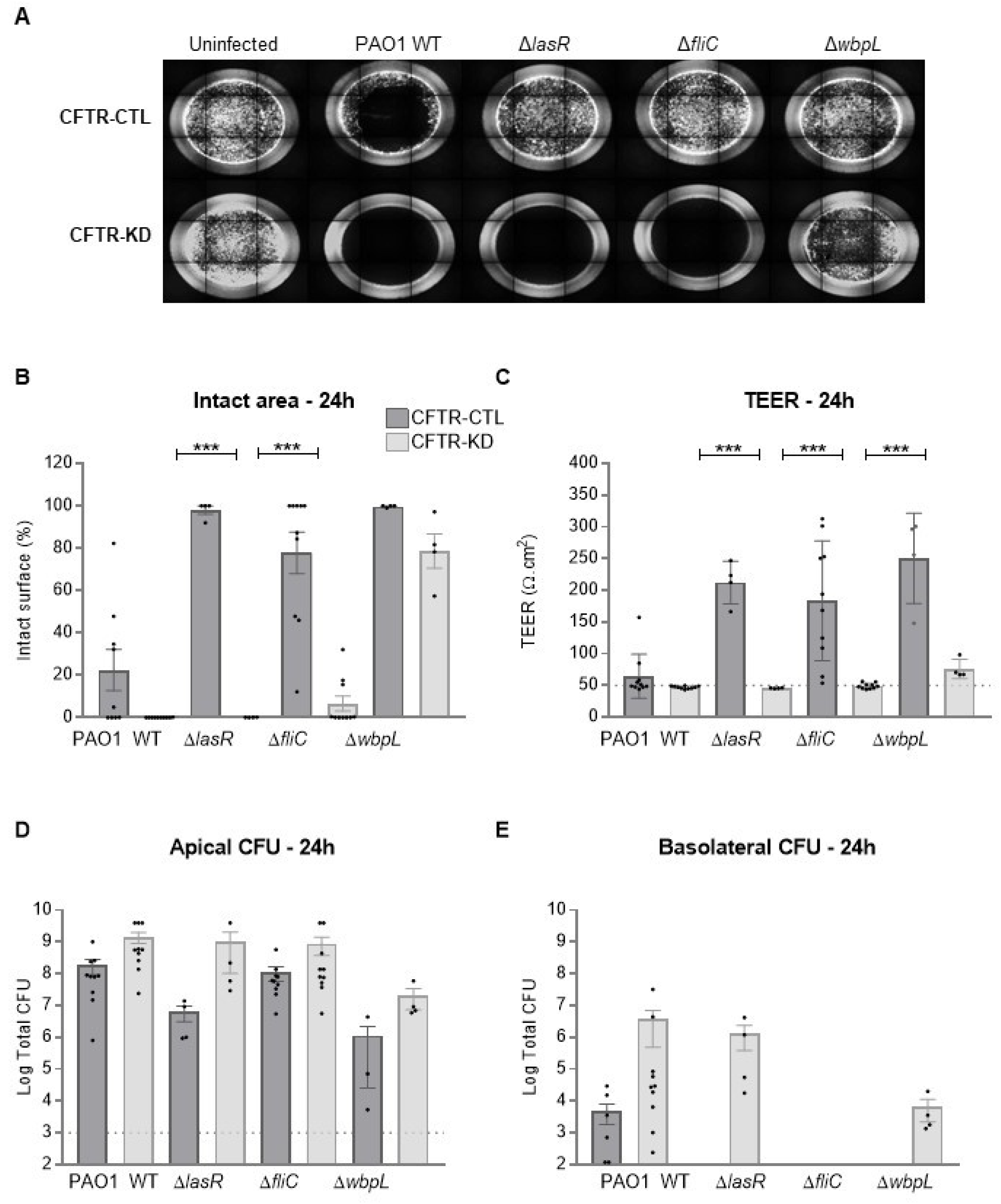
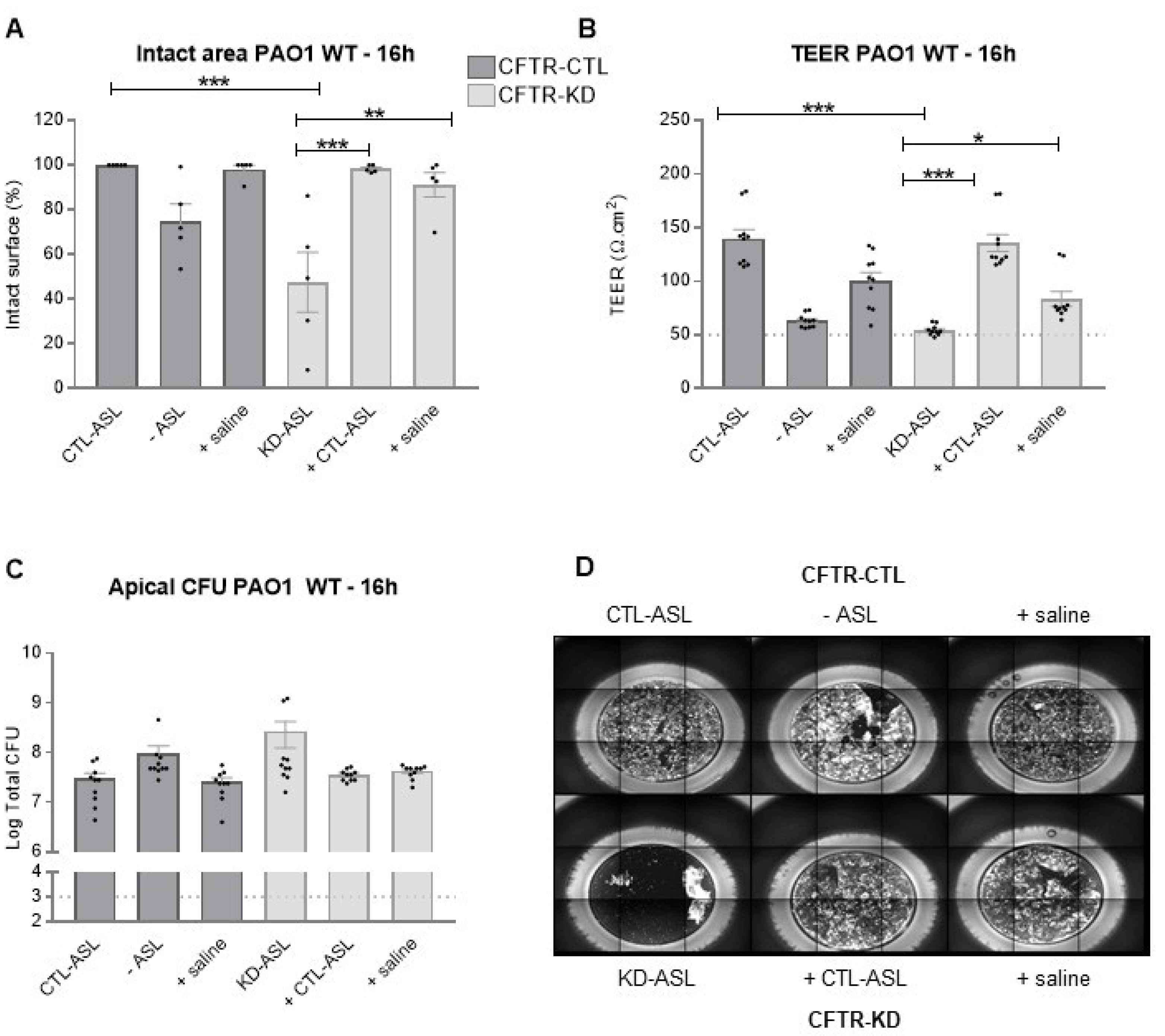
3.1. CFTR-KD Calu-3 Cells Exhibit Enhanced Susceptibility to Pa Virulence
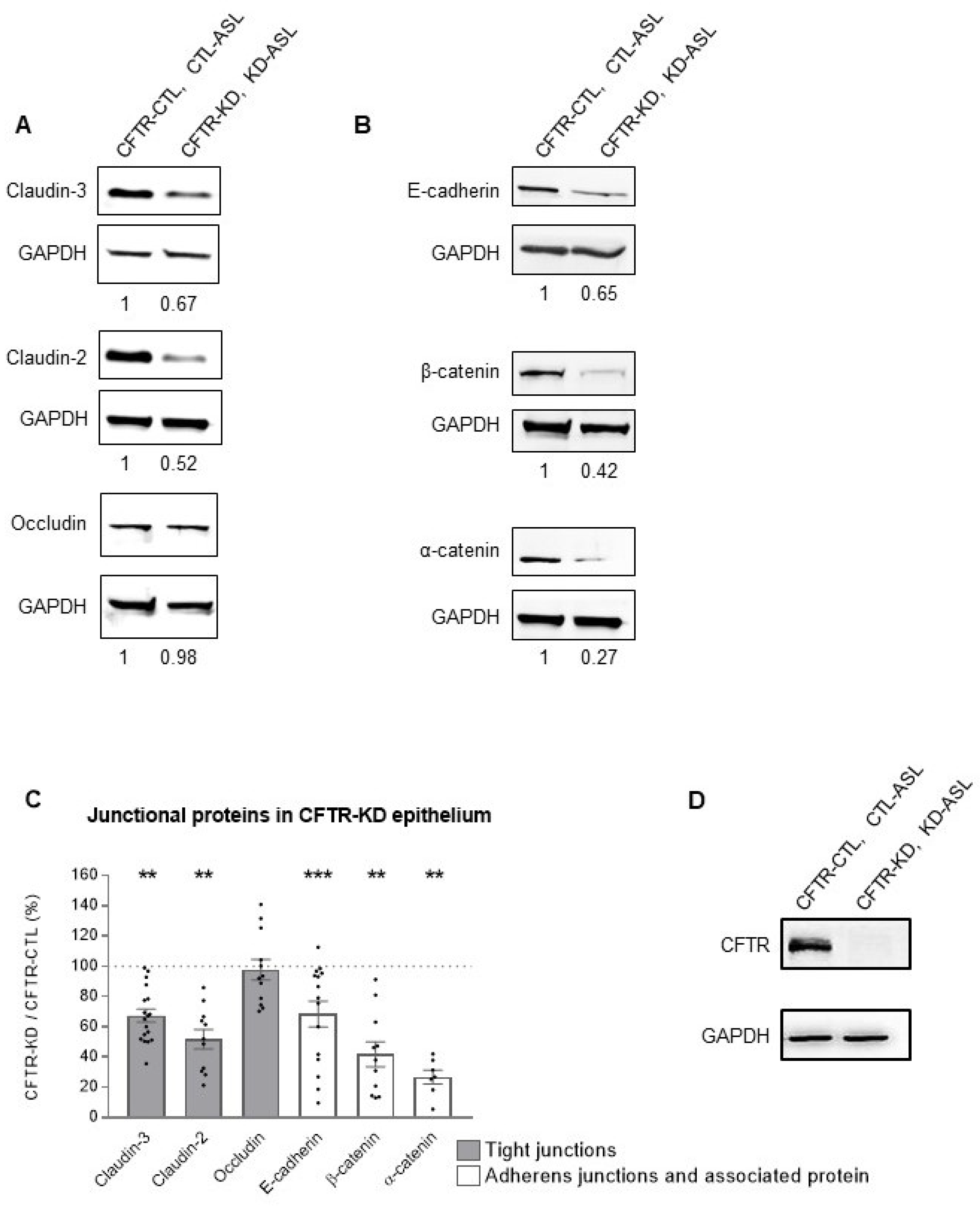
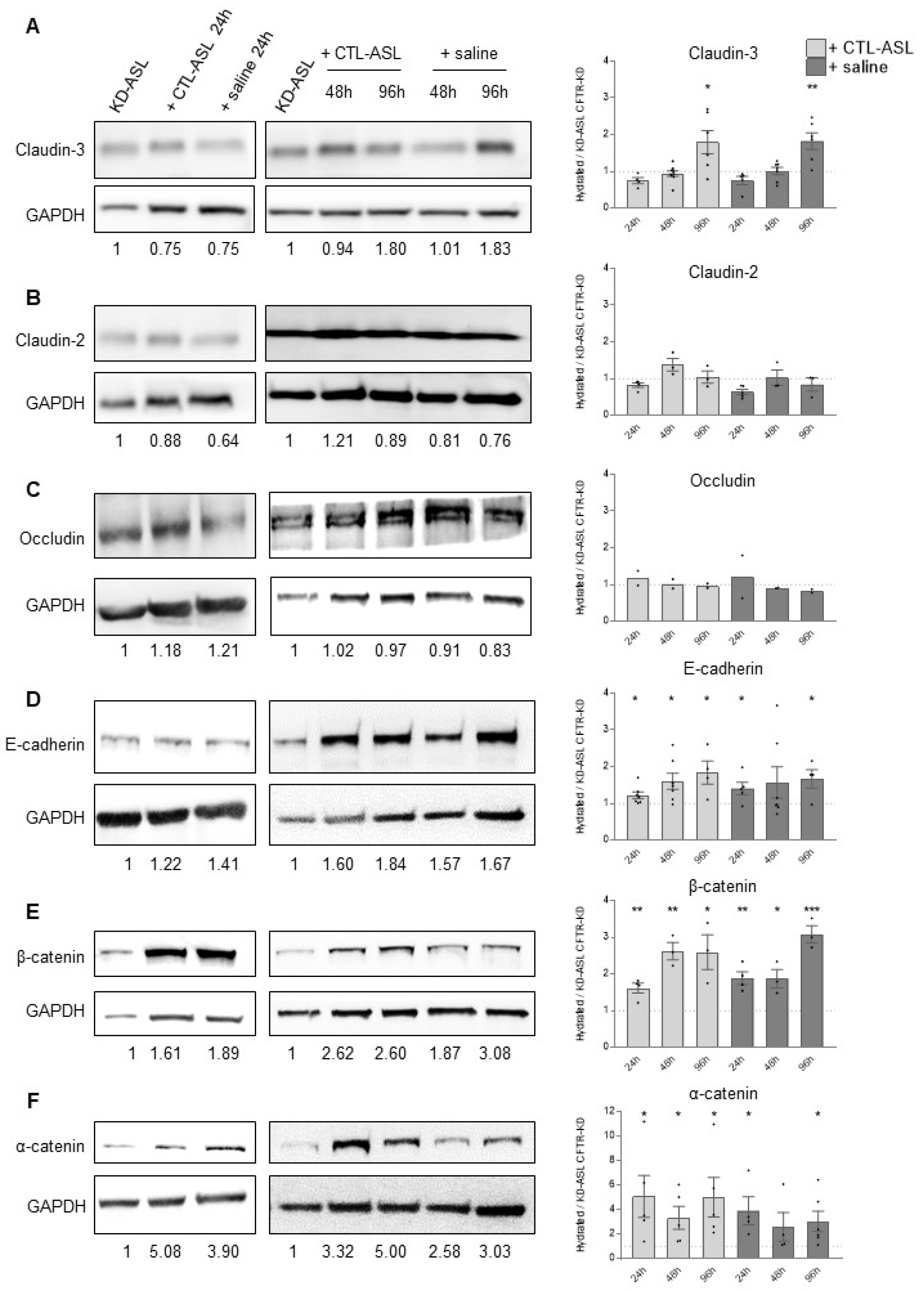
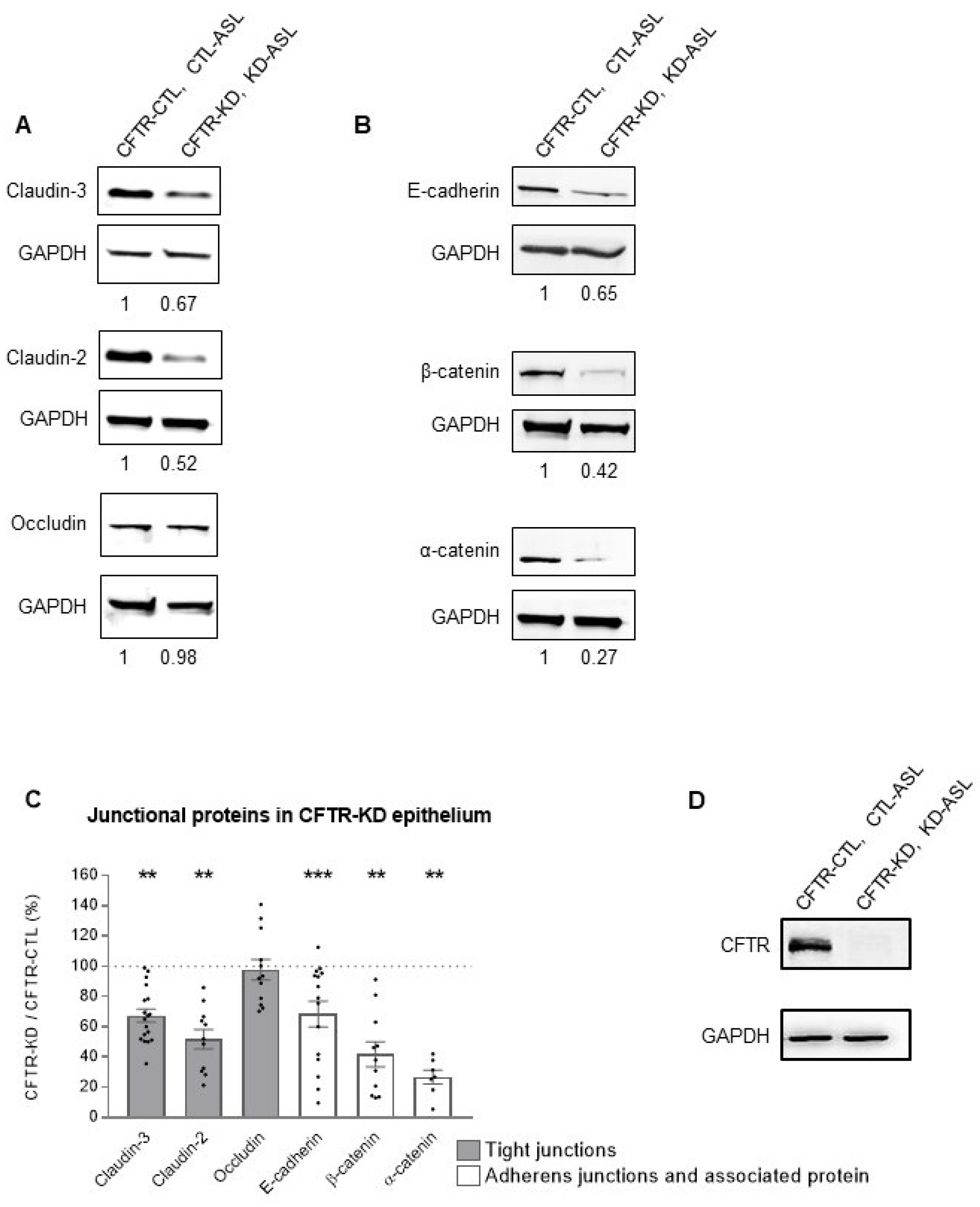
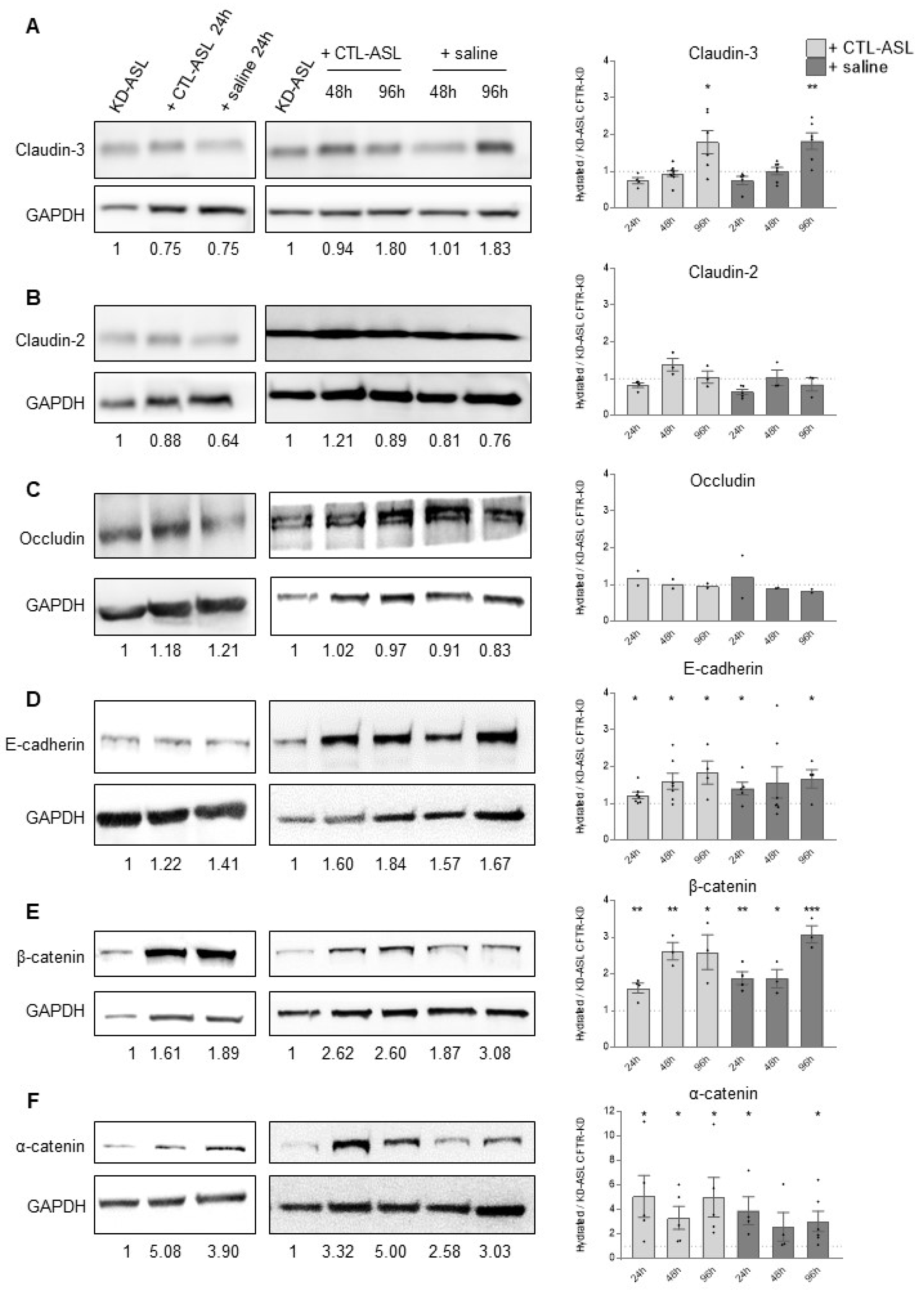
3.2. CFTR-KD Calu-3 Cells Exhibit Altered Expression of Junctional Proteins
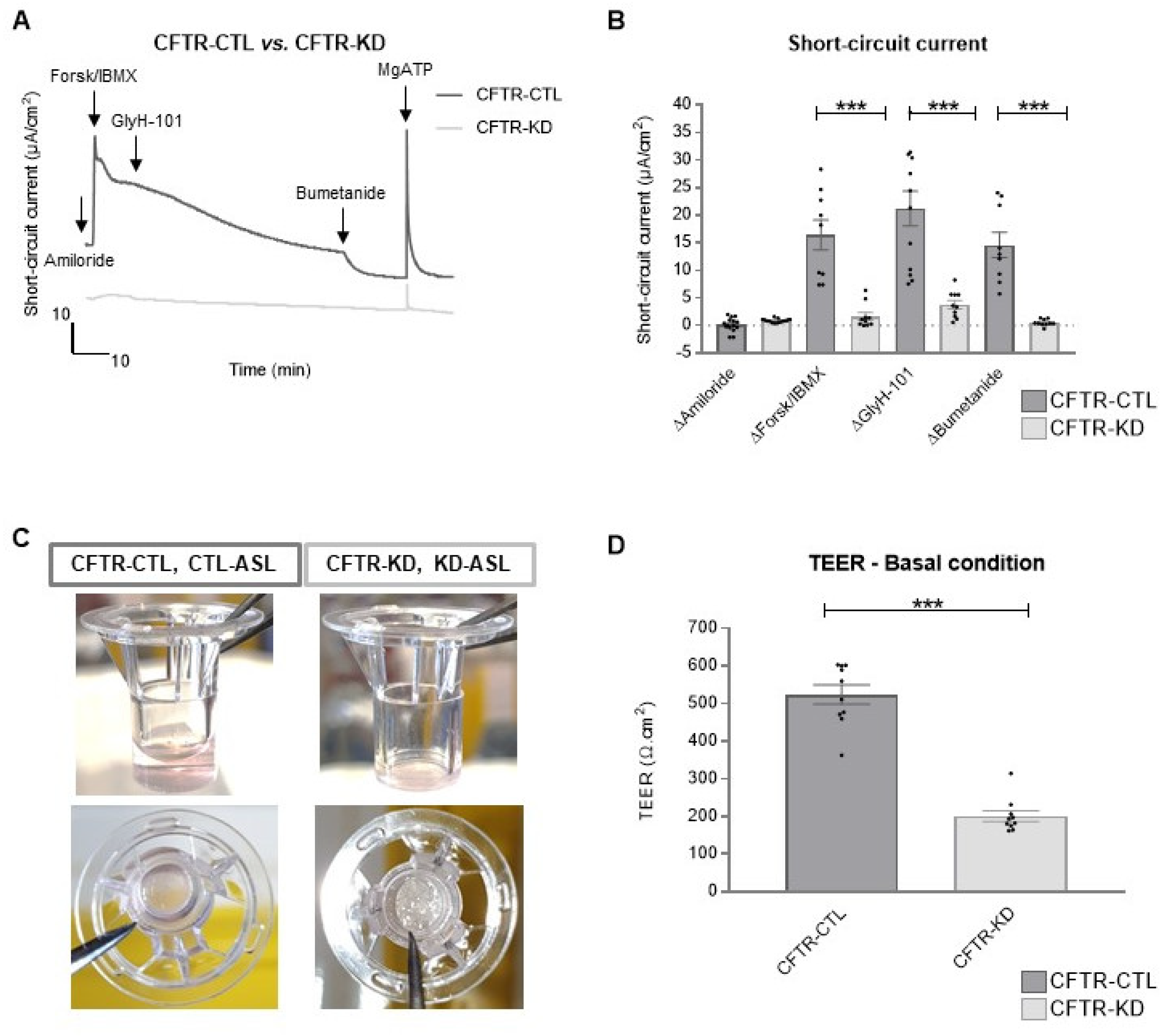
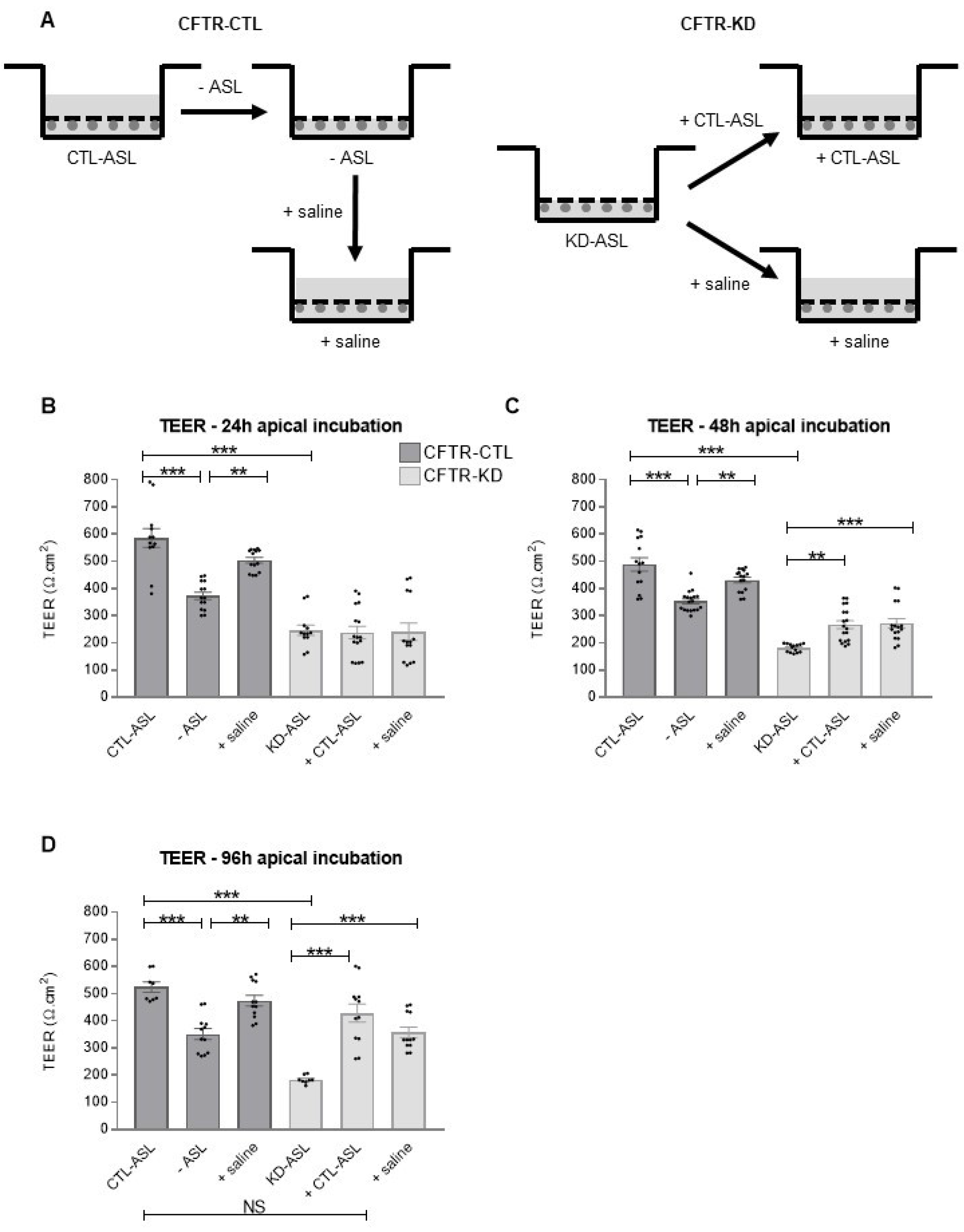
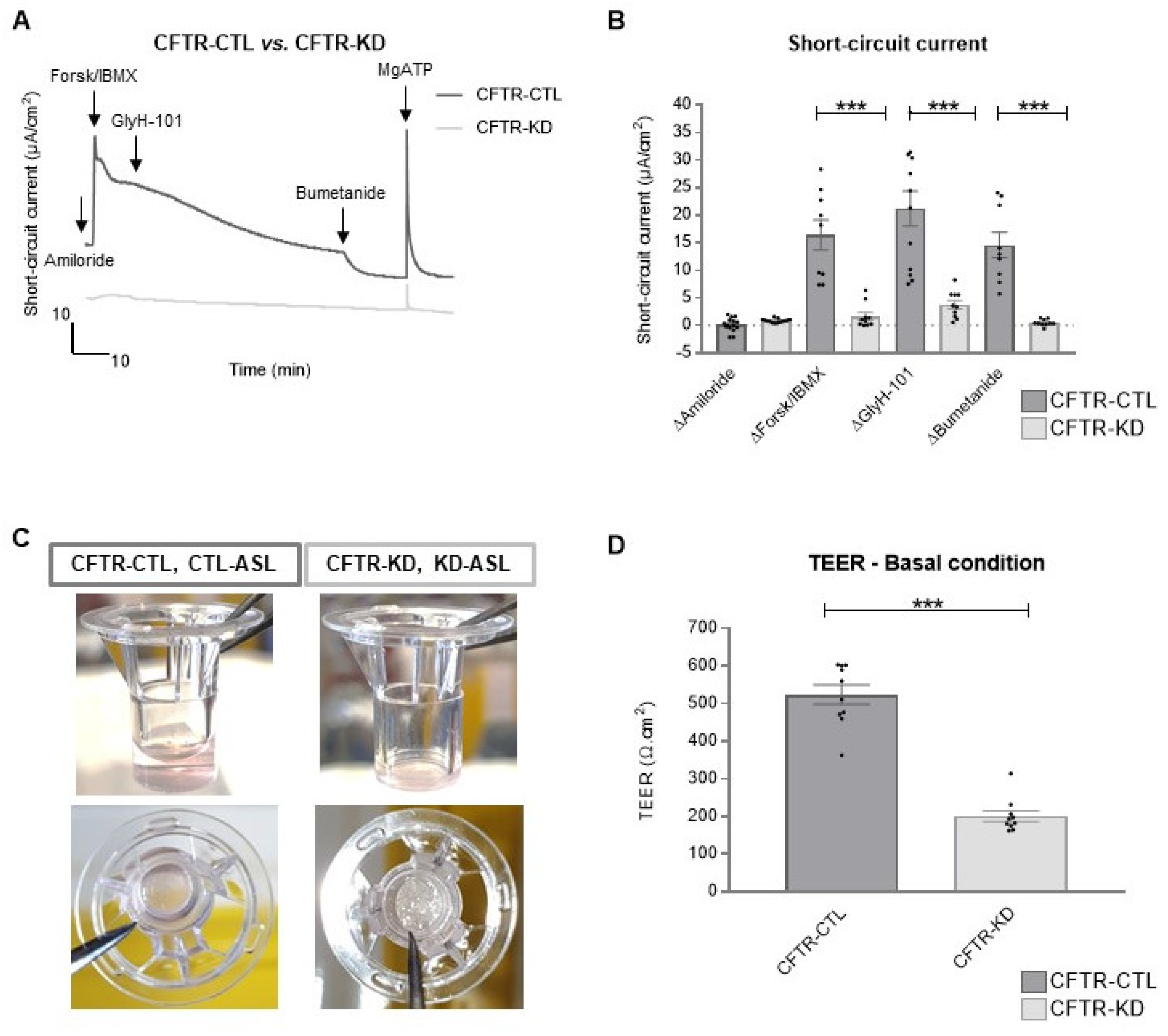
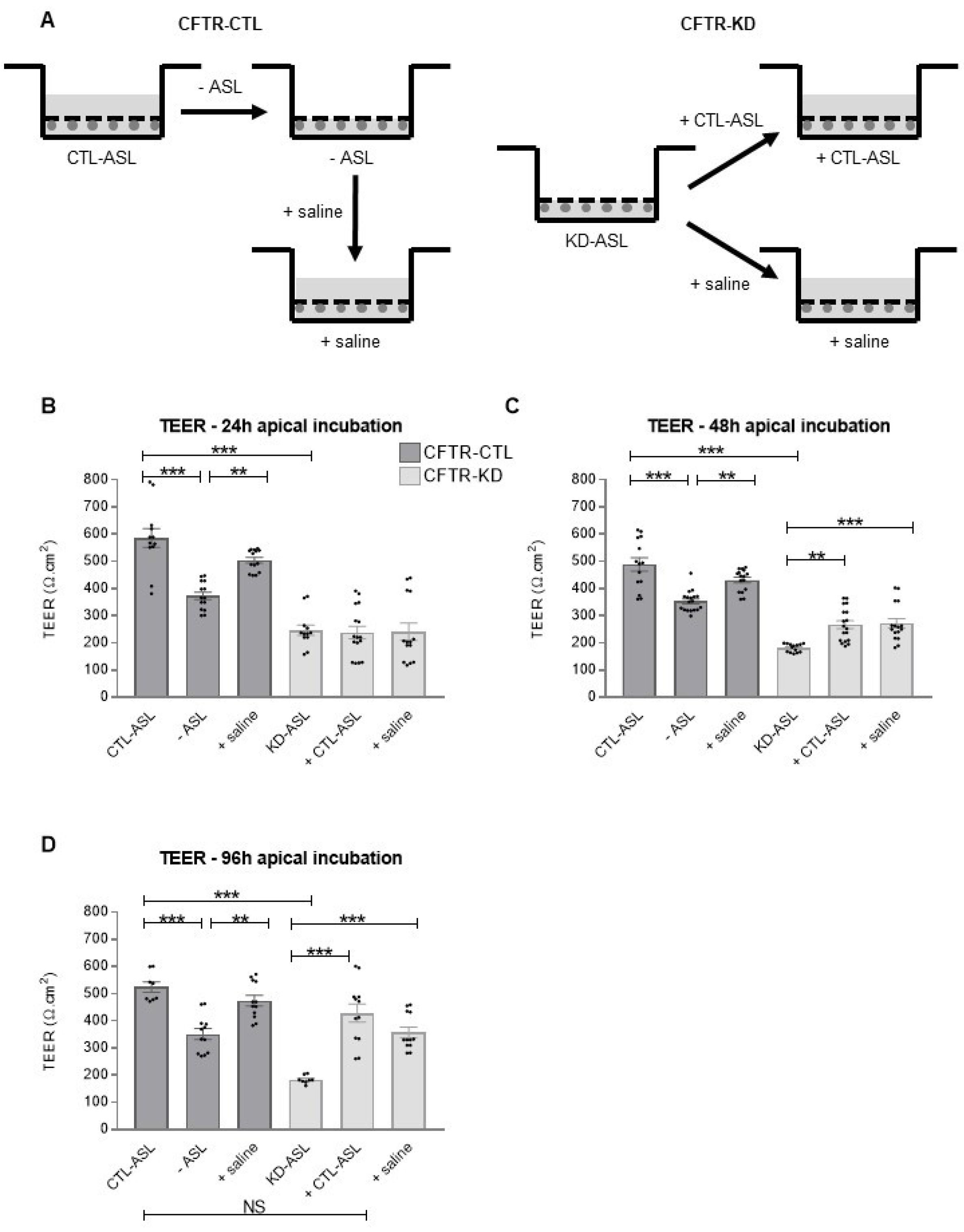
3.3. Rehydration of the CFTR-KD Calu-3 Cell Surface Restored Barrier Integrity
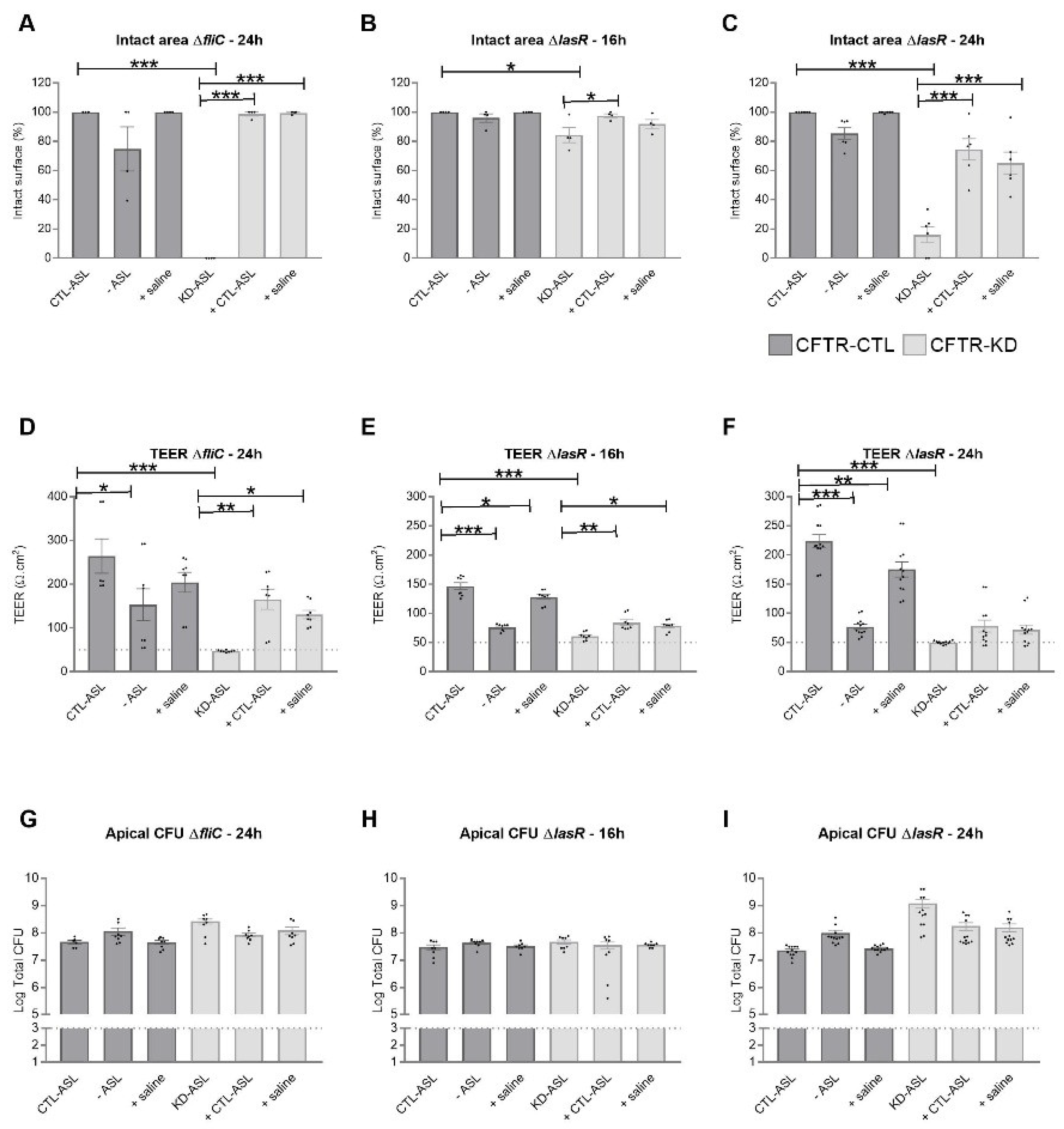
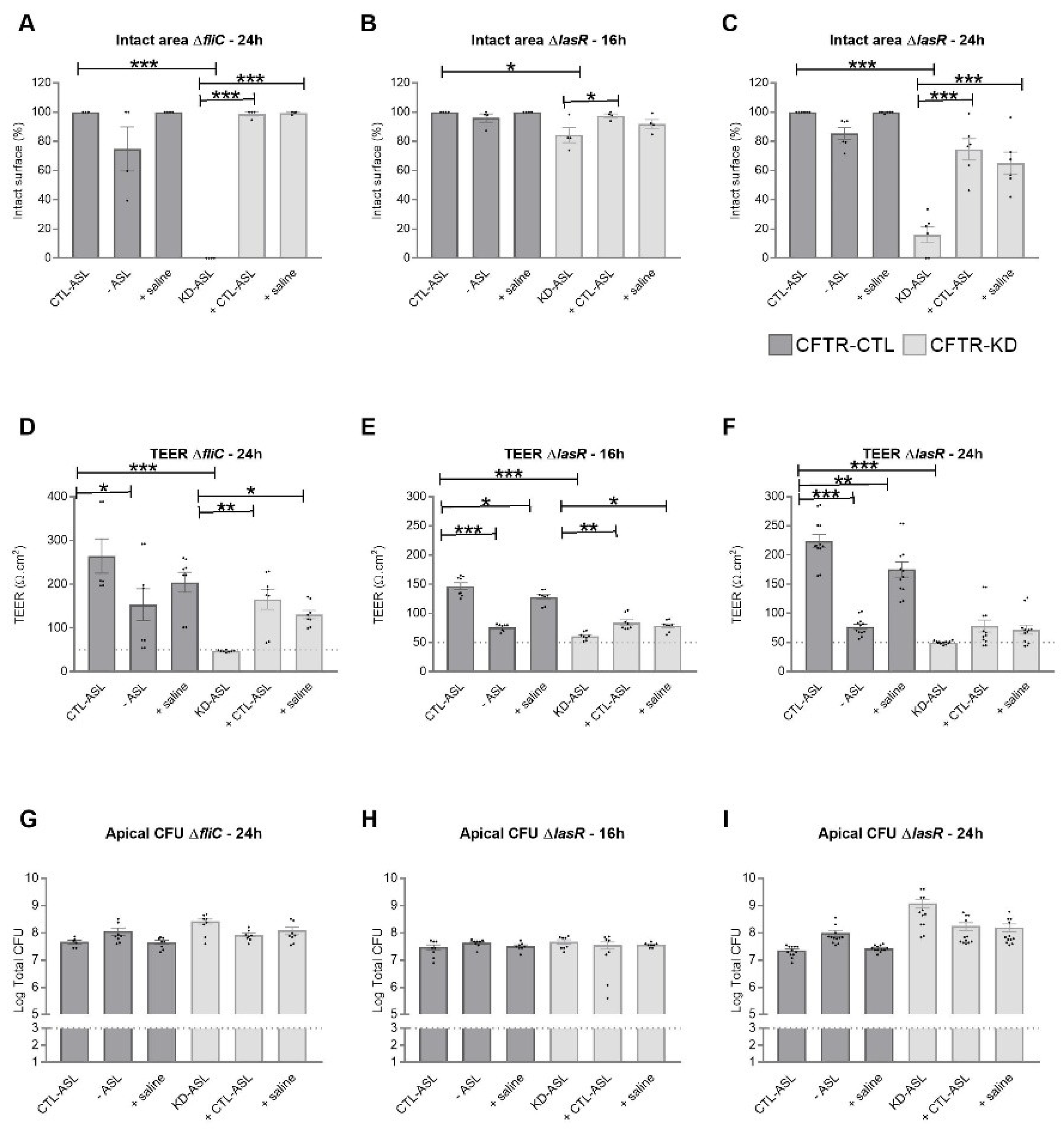
3.4. Rehydration of the CFTR-KD Calu-3 Cell Surface Reduced the Epithelium Vulnerability to Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Boeck, K. Cystic Fibrosis in the Year 2020: A Disease with a New Face. Acta Paediatr. 2020, 109, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic Fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in Epithelial Physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Webster, M.J.; Tarran, R. Slippery When Wet: Airway Surface Liquid Homeostasis and Mucus Hydration. Curr. Top. Membr. 2018, 81, 293–335. [Google Scholar] [CrossRef]
- Button, B.; Cai, L.-H.; Ehre, C.; Kesimer, M.; Hill, D.B.; Sheehan, J.K.; Boucher, R.C.; Rubinstein, M. A Periciliary Brush Promotes the Lung Health by Separating the Mucus Layer from Airway Epithelia. Science 2012, 337, 937–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermund, A.; Trillo-Muyo, S.; Hansson, G.C. Assembly, Release, and Transport of Airway Mucins in Pigs and Humans. Ann. Am. Thorac. Soc. 2018, 15, S159–S163. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.B.; Markovetz, M.R.; Ehre, C. Mucus, Mucins, and Cystic Fibrosis. Pediatr. Pulmonol. 2019, 54 (Suppl. 3), S84–S96. [Google Scholar] [CrossRef] [Green Version]
- Matsui, H.; Wagner, V.E.; Hill, D.B.; Schwab, U.E.; Rogers, T.D.; Button, B.; Taylor, R.M.; Superfine, R.; Rubinstein, M.; Iglewski, B.H.; et al. A Physical Linkage between Cystic Fibrosis Airway Surface Dehydration and Pseudomonas aeruginosa Biofilms. Proc. Natl. Acad. Sci. USA 2006, 103, 18131–18136. [Google Scholar] [CrossRef] [Green Version]
- Whitsett, J.A.; Alenghat, T. Respiratory Epithelial Cells Orchestrate Pulmonary Innate Immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.; Lillehoj, E.P.; Park, Y.; Kyo, Y.; Kim, K.C. Analysis of the Proteome of Human Airway Epithelial Secretions. Proteome Sci. 2011, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Peters-Hall, J.R.; Brown, K.J.; Pillai, D.K.; Tomney, A.; Garvin, L.M.; Wu, X.; Rose, M.C. Quantitative Proteomics Reveals an Altered Cystic Fibrosis in Vitro Bronchial Epithelial Secretome. Am. J. Respir. Cell Mol. Biol. 2015, 53, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced Airway Surface PH Impairs Bacterial Killing in the Porcine Cystic Fibrosis Lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef]
- Simonin, J.; Bille, E.; Crambert, G.; Noel, S.; Dreano, E.; Edwards, A.; Hatton, A.; Pranke, I.; Villeret, B.; Cottart, C.-H.; et al. Airway Surface Liquid Acidification Initiates Host Defense Abnormalities in Cystic Fibrosis. Sci. Rep. 2019, 9, 6516. [Google Scholar] [CrossRef]
- Teichgräber, V.; Ulrich, M.; Endlich, N.; Riethmüller, J.; Wilker, B.; De Oliveira-Munding, C.C.; van Heeckeren, A.M.; Barr, M.L.; von Kürthy, G.; Schmid, K.W.; et al. Ceramide Accumulation Mediates Inflammation, Cell Death and Infection Susceptibility in Cystic Fibrosis. Nat. Med. 2008, 14, 382–391. [Google Scholar] [CrossRef]
- Grassmé, H.; Henry, B.; Ziobro, R.; Becker, K.A.; Riethmüller, J.; Gardner, A.; Seitz, A.P.; Steinmann, J.; Lang, S.; Ward, C.; et al. Β1-Integrin Accumulates in Cystic Fibrosis Luminal Airway Epithelial Membranes and Decreases Sphingosine, Promoting Bacterial Infections. Cell Host Microbe 2017, 21, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Golovkine, G.; Reboud, E.; Huber, P. Pseudomonas aeruginosa Takes a Multi-Target Approach to Achieve Junction Breach. Front. Cell. Infect. Microbiol. 2017, 7, 532. [Google Scholar] [CrossRef]
- Popoff, M.R. Bacterial Factors Exploit Eukaryotic Rho GTPase Signaling Cascades to Promote Invasion and Proliferation within Their Host. Small GTPases 2014, 5, e28209. [Google Scholar] [CrossRef] [Green Version]
- Yuksel, H.; Turkeli, A. Airway Epithelial Barrier Dysfunction in the Pathogenesis and Prognosis of Respiratory Tract Diseases in Childhood and Adulthood. Tissue Barriers 2017, 5, e1367458. [Google Scholar] [CrossRef]
- Murphy, J.; Ramezanpour, M.; Drilling, A.; Roscioli, E.; Psaltis, A.J.; Wormald, P.-J.; Vreugde, S. In Vitro Characteristics of an Airway Barrier-Disrupting Factor Secreted by Staphylococcus aureus. Int. Forum. Allergy Rhinol. 2019, 9, 187–196. [Google Scholar] [CrossRef]
- Tam, K.; Torres, V.J. Staphylococcus Aureus Secreted Toxins and Extracellular Enzymes. Microbiol. Spectr. 2019, 7, 2–17. [Google Scholar] [CrossRef]
- Zihni, C.; Balda, M.S.; Matter, K. Signalling at Tight Junctions during Epithelial Differentiation and Microbial Pathogenesis. J. Cell Sci. 2014, 127, 3401–3413. [Google Scholar] [CrossRef] [Green Version]
- Losa, D.; Köhler, T.; Bacchetta, M.; Saab, J.B.; Frieden, M.; van Delden, C.; Chanson, M. Airway Epithelial Cell Integrity Protects from Cytotoxicity of Pseudomonas aeruginosa Quorum-Sensing Signals. Am. J. Respir. Cell Mol. Biol. 2015, 53, 265–275. [Google Scholar] [CrossRef]
- Zulianello, L.; Canard, C.; Köhler, T.; Caille, D.; Lacroix, J.-S.; Meda, P. Rhamnolipids Are Virulence Factors That Promote Early Infiltration of Primary Human Airway Epithelia by Pseudomonas aeruginosa. Infect. Immun. 2006, 74, 3134–3147. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Y.C.; Wang, Y.; Da Silva, N.; Kim, B.; Diao, R.Y.; Hill, E.; Brown, D.; Chan, H.C.; Breton, S. CFTR Interacts with ZO-1 to Regulate Tight Junction Assembly and Epithelial Differentiation through the ZONAB Pathway. J. Cell. Sci. 2014, 127, 4396–4408. [Google Scholar] [CrossRef] [Green Version]
- LeSimple, P.; Liao, J.; Robert, R.; Gruenert, D.C.; Hanrahan, J.W. Cystic Fibrosis Transmembrane Conductance Regulator Trafficking Modulates the Barrier Function of Airway Epithelial Cell Monolayers. J. Physiol. 2010, 588, 1195–1209. [Google Scholar] [CrossRef]
- Nilsson, H.E.; Dragomir, A.; Lazorova, L.; Johannesson, M.; Roomans, G.M. CFTR and Tight Junctions in Cultured Bronchial Epithelial Cells. Exp. Mol. Pathol. 2010, 88, 118–127. [Google Scholar] [CrossRef]
- Castellani, S.; Guerra, L.; Favia, M.; Di Gioia, S.; Casavola, V.; Conese, M. NHERF1 and CFTR Restore Tight Junction Organisation and Function in Cystic Fibrosis Airway Epithelial Cells: Role of Ezrin and the RhoA/ROCK Pathway. Lab. Investig. 2012, 92, 1527–1540. [Google Scholar] [CrossRef] [Green Version]
- Lochbaum, R.; Schilpp, C.; Nonnenmacher, L.; Frick, M.; Dietl, P.; Wittekindt, O.H. Retinoic Acid Signalling Adjusts Tight Junction Permeability in Response to Air-Liquid Interface Conditions. Cell Signal. 2020, 65, 109421. [Google Scholar] [CrossRef]
- Vermeer, P.D.; Panko, L.; Welsh, M.J.; Zabner, J. ErbB1 Functions as a Sensor of Airway Epithelial Integrity by Regulation of Protein Phosphatase 2A Activity. J. Biol. Chem. 2006, 281, 1725–1730. [Google Scholar] [CrossRef] [Green Version]
- Amaral, M.D.; Quaresma, M.C.; Pankonien, I. What Role Does CFTR Play in Development, Differentiation, Regeneration and Cancer? Int. J. Mol. Sci. 2020, 21, 3133. [Google Scholar] [CrossRef]
- Higgins, G.; Fustero Torre, C.; Tyrrell, J.; McNally, P.; Harvey, B.J.; Urbach, V. Lipoxin A4 Prevents Tight Junction Disruption and Delays the Colonization of Cystic Fibrosis Bronchial Epithelial Cells by Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L1053–L1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellec, J.; Bacchetta, M.; Losa, D.; Anegon, I.; Chanson, M.; Nguyen, T.H. CFTR Inactivation by Lentiviral Vector-Mediated RNA Interference and CRISPR-Cas9 Genome Editing in Human Airway Epithelial Cells. Curr. Gene Ther. 2015, 15, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Köhler, T.; Curty, L.K.; Barja, F.; van Delden, C.; Pechère, J.C. Swarming of Pseudomonas aeruginosa Is Dependent on Cell-to-Cell Signaling and Requires Flagella and Pili. J. Bacteriol. 2000, 182, 5990–5996. [Google Scholar] [CrossRef] [Green Version]
- Fleiszig, S.M.; Arora, S.K.; Van, R.; Ramphal, R. FlhA, a Component of the Flagellum Assembly Apparatus of Pseudomonas aeruginosa, Plays a Role in Internalization by Corneal Epithelial Cells. Infect. Immun. 2001, 69, 4931–4937. [Google Scholar] [CrossRef] [Green Version]
- Rocchetta, H.L.; Burrows, L.L.; Pacan, J.C.; Lam, J.S. Three Rhamnosyltransferases Responsible for Assembly of the A-Band D-Rhamnan Polysaccharide in Pseudomonas aeruginosa: A Fourth Transferase, WbpL, Is Required for the Initiation of Both A-Band and B-Band Lipopolysaccharide Synthesis. Mol. Microbiol. 1998, 28, 1103–1119. [Google Scholar] [CrossRef]
- Losa, D.; Köhler, T.; Bellec, J.; Dudez, T.; Crespin, S.; Bacchetta, M.; Boulanger, P.; Hong, S.S.; Morel, S.; Nguyen, T.H.; et al. Pseudomonas aeruginosa-Induced Apoptosis in Airway Epithelial Cells Is Mediated by Gap Junctional Communication in a JNK-Dependent Manner. J. Immunol. 2014, 192, 4804–4812. [Google Scholar] [CrossRef] [Green Version]
- Hogardt, M.; Heesemann, J. Adaptation of Pseudomonas aeruginosa during Persistence in the Cystic Fibrosis Lung. Int. J. Med. Microbiol. 2010, 300, 557–562. [Google Scholar] [CrossRef]
- Cattoir, V.; Narasimhan, G.; Skurnik, D.; Aschard, H.; Roux, D.; Ramphal, R.; Jyot, J.; Lory, S. Transcriptional Response of Mucoid Pseudomonas aeruginosa to Human Respiratory Mucus. mBio 2013, 3, e00410-12. [Google Scholar] [CrossRef] [Green Version]
- Schwagerus, E.; Sladek, S.; Buckley, S.T.; Armas-Capote, N.; Alvarez de la Rosa, D.; Harvey, B.J.; Fischer, H.; Illek, B.; Huwer, H.; Schneider-Daum, N.; et al. Expression and Function of the Epithelial Sodium Channel δ-Subunit in Human Respiratory Epithelial Cells in Vitro. Pflug. Arch. 2015, 467, 2257–2273. [Google Scholar] [CrossRef]
- Maiers, J.L.; Peng, X.; Fanning, A.S.; DeMali, K.A. ZO-1 Recruitment to α-Catenin—A Novel Mechanism for Coupling the Assembly of Tight Junctions to Adherens Junctions. J. Cell Sci. 2013, 126, 3904–3915. [Google Scholar] [CrossRef] [Green Version]
- Wolfgang, M.C.; Jyot, J.; Goodman, A.L.; Ramphal, R.; Lory, S. Pseudomonas aeruginosa Regulates Flagellin Expression as Part of a Global Response to Airway Fluid from Cystic Fibrosis Patients. Proc. Natl. Acad. Sci. USA 2004, 101, 6664–6668. [Google Scholar] [CrossRef] [Green Version]
- Vikström, E.; Bui, L.; Konradsson, P.; Magnusson, K.-E. The Junctional Integrity of Epithelial Cells Is Modulated by Pseudomonas aeruginosa Quorum Sensing Molecule through Phosphorylation-Dependent Mechanisms. Exp. Cell Res. 2009, 315, 313–326. [Google Scholar] [CrossRef]
- Schwarzer, C.; Fu, Z.; Patanwala, M.; Hum, L.; Lopez-Guzman, M.; Illek, B.; Kong, W.; Lynch, S.V.; Machen, T.E. Pseudomonas aeruginosa Biofilm-Associated Homoserine Lactone C12 Rapidly Activates Apoptosis in Airway Epithelia. Cell Microbiol. 2012, 14, 698–709. [Google Scholar] [CrossRef] [Green Version]
- Kojima, T.; Go, M.; Takano, K.; Kurose, M.; Ohkuni, T.; Koizumi, J.; Kamekura, R.; Ogasawara, N.; Masaki, T.; Fuchimoto, J.; et al. Regulation of Tight Junctions in Upper Airway Epithelium. Biomed. Res. Int. 2013, 2013, 947072. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, P.J.; Ernst, R.K.; Elborn, J.S.; Schock, B. Role of CFTR, Pseudomonas aeruginosa and Toll-like Receptors in Cystic Fibrosis Lung Inflammation. Biochem. Soc. Trans. 2009, 37, 863–867. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.J.; Travis, S.M.; Greenberg, E.P.; Welsh, M.J. Cystic Fibrosis Airway Epithelia Fail to Kill Bacteria Because of Abnormal Airway Surface Fluid. Cell 1996, 85, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Bals, R.; Weiner, D.J.; Wilson, J.M. The Innate Immune System in Cystic Fibrosis Lung Disease. J. Clin. Investig. 1999, 103, 303–307. [Google Scholar] [CrossRef] [Green Version]
- Guggino, W.B.; Stanton, B.A. New Insights into Cystic Fibrosis: Molecular Switches That Regulate CFTR. Nat. Rev. Mol. Cell Biol. 2006, 7, 426–436. [Google Scholar] [CrossRef]
- Favia, M.; Guerra, L.; Fanelli, T.; Cardone, R.A.; Monterisi, S.; Di Sole, F.; Castellani, S.; Chen, M.; Seidler, U.; Reshkin, S.J.; et al. Na+/H+ Exchanger Regulatory Factor 1 Overexpression-Dependent Increase of Cytoskeleton Organization Is Fundamental in the Rescue of F508del Cystic Fibrosis Transmembrane Conductance Regulator in Human Airway CFBE41o-Cells. Mol. Biol. Cell 2010, 21, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Morrison, C.B.; Shaffer, K.M.; Araba, K.C.; Markovetz, M.R.; Wykoff, J.A.; Quinney, N.L.; Hao, S.; Delion, M.F.; Flen, A.L.; Morton, L.C.; et al. Treatment of Cystic Fibrosis Airway Cells with CFTR Modulators Reverses Aberrant Mucus Properties via Hydration. Eur. Respir. J. 2021, 59, 2100185. [Google Scholar] [CrossRef]
- Citi, S. The Mechanobiology of Tight Junctions. Biophys. Rev. 2019, 11, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Vitzthum, C.; Clauss, W.G.; Fronius, M. Mechanosensitive Activation of CFTR by Increased Cell Volume and Hydrostatic Pressure but Not Shear Stress. Biochim. Biophys. Acta 2015, 1848, 2942–2951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.K.; Wang, D.; Duan, Y.; Loy, M.M.T.; Chan, H.C.; Huang, P. Mechanosensitive Gating of CFTR. Nat. Cell Biol. 2010, 12, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Cao, X.; Chen, X.; Wang, D.; Zhang, W.K.; Sun, Y.; Hu, W.; Zhou, Z.; Wang, Y.; Huang, P. Mechanosensitivity of Wild-Type and G551D Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Controls Regulatory Volume Decrease in Simple Epithelia. FASEB J. 2016, 30, 1579–1589. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simonin, J.L.; Luscher, A.; Losa, D.; Badaoui, M.; van Delden, C.; Köhler, T.; Chanson, M. Surface Hydration Protects Cystic Fibrosis Airways from Infection by Restoring Junctional Networks. Cells 2022, 11, 1587. https://doi.org/10.3390/cells11091587
Simonin JL, Luscher A, Losa D, Badaoui M, van Delden C, Köhler T, Chanson M. Surface Hydration Protects Cystic Fibrosis Airways from Infection by Restoring Junctional Networks. Cells. 2022; 11(9):1587. https://doi.org/10.3390/cells11091587
Chicago/Turabian StyleSimonin, Juliette L., Alexandre Luscher, Davide Losa, Mehdi Badaoui, Christian van Delden, Thilo Köhler, and Marc Chanson. 2022. "Surface Hydration Protects Cystic Fibrosis Airways from Infection by Restoring Junctional Networks" Cells 11, no. 9: 1587. https://doi.org/10.3390/cells11091587
APA StyleSimonin, J. L., Luscher, A., Losa, D., Badaoui, M., van Delden, C., Köhler, T., & Chanson, M. (2022). Surface Hydration Protects Cystic Fibrosis Airways from Infection by Restoring Junctional Networks. Cells, 11(9), 1587. https://doi.org/10.3390/cells11091587