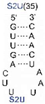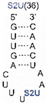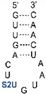1. Introduction
Transfer RNAs (tRNAs) are present in three domains of life as a universal component of cellular protein synthesis machinery. To become fully functional, tRNAs, which were originally transcribed in the form of longer precursors, undergo a well-defined multistep maturation process, in which an important step that influences tRNA structure and functionality is the incorporation of numerous post-transcriptional chemical modifications. The four canonical RNA bases (A, C, G, and U) are modified in different ways by specific enzymes [
1,
2,
3,
4].
Natural tRNAs contain a highest proportion of modified nucleosides compared to other RNA types [
5,
6]. The post-transcriptional modifications in tRNA can be divided into two groups based on their complexity. The first group of modifications is introduced by a single enzymatic reaction, such as methylation, and is found at numerous locations within the tRNA core region, in which tertiary interactions between the D and T arms stabilize the three-dimensional structure. The second group includes complex modifications whose syntheses require the sequential activity of multiple enzymes. These hyper-modifications are mainly found in the anticodon loop, in which they maintain the structure as a prerequisite for efficient translation [
7]. The anticodon stem-loop (ASL) is the most frequently modified part of the tRNA molecule, particularly nucleosides in the wobble position (which is also referred to as the 1st position in the anticodon or the 34th position in the tRNA chain), which interact with the third base of the codon in the mRNA. The second commonly modified position in the ASL is the 37th position, which is located directly after the last base of the anticodon. The modification of the nucleosides in these positions play a crucial role in the extension or restriction of the decoding properties of a particular tRNA molecule [
8]. Positions outside the ASL usually play a more structural role, but they have also been shown to influence decoding properties [
7,
9].
Hypermodified uridines that contain sulfur or selenium atom are widely distributed in a variety of organisms and have been identified in the wobble position of the anticodon sequence of tRNA
Lys, tRNA
Glu, and tRNA
Gln. Moreover, in 2012, Dumelin et al. discovered a new type of modification with a hydrophobic geranyl group conjugated to the sulfur atom of the 2-thiouridine nucleoside in bacterial tRNAs [
10].
Figure 1 shows the structure of 5-methylaminomethyl-2-thiouridine (mnm5S2U) and its counterparts: 5-methylaminomethyl-
S-geranyl-2-thiouridine (mnm5geS2U) and 5-methylaminomethyl-2-selenouridine (mnm5Se2U). These counterparts are specific to the bacterial tRNAs for lysine (tRNA
Lys) and glutamic acid (tRNA
Glu). The bacterial tRNA specific to glutamine (tRNA
Gln) contains 5-carboxymethylaminomethyl-2-thiouridine (cmnm5S2U) and its counterparts: 5-carboxymethylaminomethyl-
S-geranyl-2-thiouridine (cmnm5geS2U) and 5-carboxymethylaminomethyl-2-selenouridine (cmnm5Se2U) [
5,
6,
10,
11].
Sulfur and selenium modifications in the tRNA wobble uridines play important role in the precise reading of genetic information and the tuning of the protein synthesis [
12,
13]. We and other researchers have attempted to explain why nature has introduced such a complex system of modifications into the wobble position of bacterial tRNAs [
14,
15,
16,
17,
18,
19]. Uridines in the RNA chain preferentially recognize the A-complement through Watson–Crick interactions and, with a lower affinity, the G-complement through the wobble hydrogen bonding pattern [
16]. The introduction of sulfur or selenium atoms into the C2 position of uridine increases the thermodynamic stability of RNA duplexes that contain S2U-A or Se2U-A base pairs and restricts the formation of S2U-G or Se2U-G base pairs [
17]. However, the situation changes when the side chain is present in the C5 position of 2-thiouridines and 2-selenouridines [
14]. Under physiological conditions (pH 7.4), R5Se2Us preferentially adopt the zwitterionic form (ZI, about 90%) with the positive charge on the aminoalkyl side chain and the negative charge on the Se2-N3-O4 edge. The tRNA anticodons with the R5Se2Us wobble can also read the synonymous 5′-NNG-3′ codons in mRNA in contrast to their 2-oxo precursors, which preferentially read the 5′-NNA-3′ codons [
14], and to their 2-thio precursors, which recognize both synonymous codons depending on the character of the substituent in the C5 position. Moreover, the presence of sulfur and selenium in biological entities, such as proteins or nucleic acids, can be expected to protect cellular elements from oxidative damage. Selenium-containing compounds are superior ROS scavengers because, unlike sulfur analogs, they can react reversibly with oxidizing species [
18,
19].
The enzyme that is responsible for the subsequent conversion of S2U→Se2U is the subject of our current research [
11,
20]. The enzymatic activity in extracts of
Salmonella enterica serovar
Typhimurium, which catalyzes the conversion of mnm5S2U into mnm5Se2U in the presence of selenide and ATP, was first described by Verez et al. in 1992 [
21]. Two years later, Veres and Stadtman isolated and purified the enzyme that is responsible for this conversion and introduced the name tRNA 2-selenouridine synthase (SelU) [
22]. In 2012, Dumelin et al. demonstrated that SelU is responsible for the introduction of the geranyl group into two types of R5-substituted thio-nucleosides (mnm5S2U and cmnm5S2U) in bacterial tRNAs [
10]. The tRNA 2-selenouridine synthase (SelU, MnmH, and YbbB) is a 41.1 kDa protein that contains a 364-amino acid chain, which is divided into two structural domains: an N-terminal domain with rhodanese homology, with a -Cys-X-X-Cys- active site, and a C-terminal P-loop domain, which contains a Walker A motif and an isoleucine–tRNA synthetase (IleS)-like helical region [
23,
24]. The P-loop domain is found in proteins that bind ATP or GTP [
25,
26]. The intact Walker A motif is required for the geranylation activity of the enzyme, which could mean it is also the binding site for geranyl pyrophosphate (GePP), a donor molecule in the geranyl transfer reaction in vitro [
23]. This information was confirmed by the validation of molecular dynamics in our recently published study [
27]. SelU synthase, under the natural conditions in the bacterial cells, contains a tightly bound specific tRNA fraction. Therefore, the purified protein has an unusual absorption spectrum with a maximum at 260 nm, as in nucleic acids, and no peak at 280 nm, which is characteristic of proteins. Wolfe et al. estimated that one protein molecule binds two tRNA molecules [
24]. To date, the crystal structure of the SelU protein has not been determined. The putative 3D structure of the SelU protein was recently predicted based on its amino acid sequence using the AlphaFold v2.0 system. The structure is available for analysis on the AlphaFold Protein Structure Database website (
https://alphafold.ebi.ac.uk/entry/P33667, accessed on 1 July 2021) [
28].
Originally, the SelU enzyme was thought to be responsible for the direct conversion of R5S2U→R5Se2U and, additionally, for the
S-geranylation of the R5S2U-tRNA as a second independent modification pathway. Dumelin et al. claimed that the
S-geranylation of tRNA is an alternative to the selenation process, which is prolonged at low selenium concentrations [
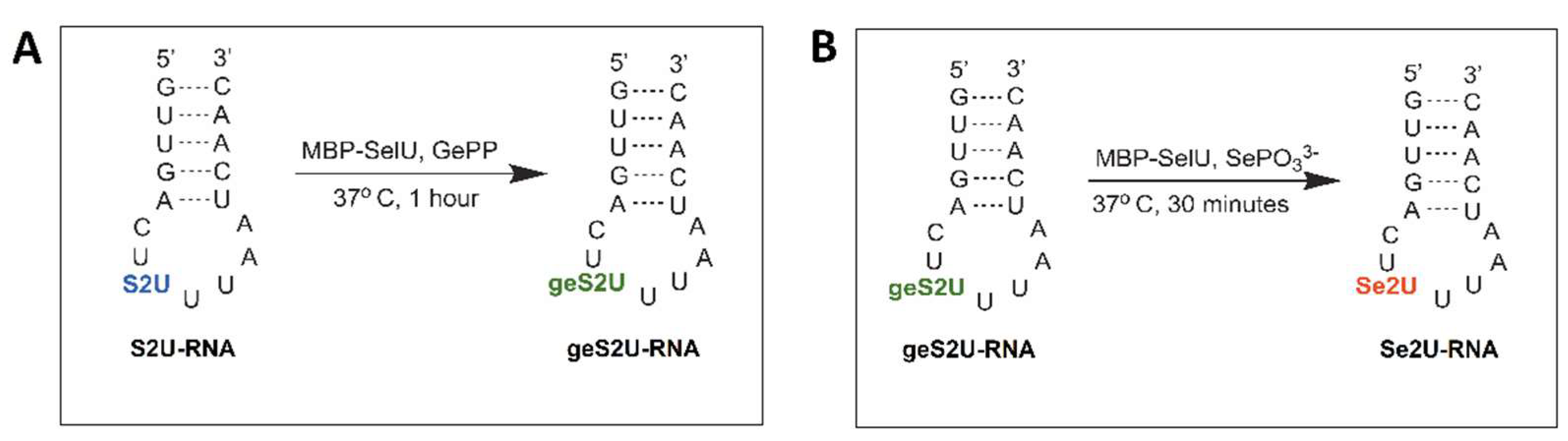
10]. For several years, the selenation and geranylation of the R5S2U-tRNA were thought to occur independently and in parallel. Since these two reactions (S2U→Se2U and S2U→geS2U) are mechanistically distinct, we attempted to elucidate the reaction pathway, first using chemical conversions and then using enzymatic conversions [
11,
20,
29]. In 2018, we demonstrated using 17-mer model ASL–RNAs that the conversion of the S2U-RNA into the Se2U-RNA occurs via the geS2U-RNA intermediate product, which corresponds to the two subsequent reactions cycle: S2U-RNA→geS2U-RNA→Se2U-RNA [
11].
In previous studies, we used the wild type SelU protein, whose gene was isolated from the
E. coli RNA through reverse transcription followed by PCR with specific primers, and cloned it into the DNA expression plasmid pET28c, which also encoded a short His
6 tag. [
11,
20]. The recombinant SelU–His
6 protein was not stable under the conditions applied, so the yield of geranylation was only ~40% for a freshly prepared sample and decreased to ~10% for the protein stored at −70 °C for several weeks [
11,
20]. This enzyme efficiency was not sufficient for us to explain the properties and differences of the interactions with the substrate that were studied. Therefore, we decided to change the system and constructed an MBP–SelU fusion protein with an MBP tag (42 kDa) at the N-terminus of the SelU protein. In our case, the MBP tag acted as a stabilizing factor in the MBP–SelU fusion protein [
30]. The MBP-modified SelU retained the substrate preferences, tRNA-binding properties, and catalytic ability of SelU synthase but showed much higher stability and activity in the specific reactions: geranylation (>90%) and selenation (100%). Thanks to these parameters of the MBP–SelU fusion protein, we obtained a molecular tool that can be used to characterize and evaluate the properties of SelU synthase.
2. Materials and Methods
2.1. The Chemical Synthesis of Modified Nucleosiede Standards
The chemical syntheses of an R5-substituted 2-oxo-, 2-thio-, S-geranyl-2-thio-, and 2-selenouridines was performed at the Lodz University of Technology and we used the standards for LC-MS analysis. The procedures for the synthesis of nm5U, mnm5U, cmnm5U, S2U, nm5S2U, mnm5S2U, cmnm5S2U, Se2U, mnm5Se2U, cmnm5Se2U, geS2U, mnm5geS2U, and cmnm5geS2U have been previously described [
14,
17,
31,
32,
33]. The procedure for the synthesis of the nm5geS2U and nm5Se2U standards is presented in the Synthetic Procedures in the
Supplementary Materials.
2.2. The Chemical Synthesis of S2U and geS2U Phosphoramidites and Model RNAs Oligonucleotides
The chemical syntheses of the S2U and geS2U phosphoramidites and the model RNA oligonucleotides were performed in-house according to previously described procedures [
11,
20,
34]. The detailed procedures are presented in the Synthetic Procedures in the
Supplementary Materials. The list of prepared RNA models, their sequences, and the mass spectrometry analysis data are presented in
Table S1A,B.
2.3. The Chemical Synthesis of Selenophosphate
The synthesis of selenophosphate SePO
33− was performed according to the procedure previously described [
20,
35,
36]. The detailed procedure is presented in the Synthetic Procedures in the
Supplementary Materials.
2.4. Protein Procedures
The detailed procedure for the synthesis and purification of the MBP–SelU fusion protein is presented in the Protein Procedures in the
Supplementary Materials. The SDS-PAGE validation of the protein overexpression level and the purification of MBP–SelU are presented in
Figures S1 and S2.
2.5. Geranylation Reaction
The geranylation reaction conditions are described in
Table S2A: 21 µg (0.249 nmol) of purified MBP–SelU was incubated with 6.11 µg (1.113 nmol) of the S2U-RNA in the presence of a 5-fold excess of geranyl pyrophosphate ammonium salt (GePP 5 × H
2O, Axon Medchem, Reston, VA, USA) (5.56 nmol, 2.03 µg) in 100 μL of the reaction buffer (10 mM Tricine–KOH, pH 7.2, 10 mM MgCl
2) at 37 °C for 1 h. The reaction products were analyzed using RP-HPLC (Shimadzu, Kyoto, Japan) with the Kinetex C-18 column, (5 µm, 100 A, and 250 × 4.60 mm) (Phenomenex, Warsaw, Poland) and buffers (A: 0.1 M CH
3COONH
4 and B: 0.1 M CH
3COONH
4/40% CH
3CN). The reaction products were separated in a linear acetonitrile gradient: 0–5 min for 0% B, 5–21 min for 0–80% B, and 21–26 min for 80–0% B. The reaction products present in the collected fractions were identified by ESI-MS.
2.6. Selenation Reaction
The selenation reaction conditions are described in
Table S2B: 21 µg (0.249 nmol) of purified MBP–SelU was incubated with 6.11 µg (1.113 nmol) of the geS2U-RNA in the presence of 18 equivalents of SePO
33− (20 nmol, 3.16 µg) in 100 μL of the reaction buffer (10 mM Tricine–KOH, pH 7.2, 10 mM MgCl
2), at 37 °C for 30 min, under anaerobic conditions. The Se2U-RNA product formation was monitored using RP-HPLC (Shimadzu, Kyoto, Japan) with the Kinetex C-18 column (5 µm, 100 A, and 250 × 4.60 mm), (Phenomenex, Warsaw, Poland) and buffers (A: 0.1 M CH
3COONH
4 and B: 0.1 M CH
3COONH
4 / 40% CH
3CN). The reaction products were separated in a linear acetonitrile gradient: 0–5 min for 0% B, 5–21 min for 0–80% B, and 21–26 min for 80–0% B. The reaction products present in the collected fractions were identified by ESI-MS.
2.7. Kinetics of Geranylation and Selenation Reactions
The kinetic parameters of the S2U-RNA geranylation catalyzed by the MBP–SelU enzyme were determined in the reactions, in which the initial rates were maintained until approximately 10% of the S2U-RNA was converted into geS2U-RNA. The reactions were performed in the 200 µL reaction buffer containing 10 mM Tricine–KOH, pH 7.2, 10 mM MgCl2, with the constant GePP concentration (30 µM) and variable concentrations of the S2U-RNA oligonucleotide: 0, 0.625, 1.25, 2.5, 5, 7.5, 15, 20, and 25 µM. The enzyme was used in two concentrations: 8.3 pmol (0.041 µM) for lower concentrations of the oligonucleotide (0.625–7.5 μM) and 16.6 pmol (0.083 µM) for higher concentrations of the oligonucleotide (15–25 μM). The reaction products were separated using RP-HPLC (Shimadzu, Kyoto, Japan) with the Kinetex C-18 column (5 µm, 100 A, and 250 × 4.60 mm) (Phenomenex, Warsaw, Poland) in a linear acetonitrile gradient: 0–5 min for 0% B, 5–21 min for 0–80% B, and 21–26 min for 80–0% B (A: 0.1 M CH3COONH4 and B: 0.1 M CH3COONH4/40% CH3CN). Each reaction was repeated at least three times. The obtained results were analyzed using GraphPad Prism 4.0 software to plot the V(S) dependence curve, were V means reaction rate and S is the substrate concentration. The kinetic constants KM and kcat were determined.
To determine the kinetic parameters for the selenation of the geS2U-RNA catalyzed by the MBP–SelU enzyme, the initial rates were maintained until approximately 10% of the geS2U-RNA was converted into Se2U-RNA. The reactions were performed in the 200 µL reaction buffer containing 10 mM Tricine–KOH, pH 7.2, 10 mM MgCl2, with a constant concentration of SePO33− (30 µM) and variable concentrations of the geS2U-RNA oligonucleotide: 0, 0.3125, 0.625, 1.25, 2.5, and 5 μM. The enzyme concentration varied from 0.0386 pmol (0.193 nM) for the lower concentrations of the geS2U-RNA to 0.622 pmol (3.11 nM) for the higher concentrations of oligonucleotide. The reaction products were separated using RP-HPLC (Shimadzu, Kyoto, Japan) with a Kinetex C-18 column (5 µm, 100 A, and 250 × 4.60 mm) (Phenomenex, Warsaw, Poland) in the linear acetonitrile gradient: 0–5 min for 0% B, 5–21 min for 0–80% B, and 21–26 min for 80–0% B (A: 0.1 M CH3COONH4 and B: 0.1 M CH3COONH4/40% CH3CN). Each reaction was repeated three times. The obtained results were analyzed using GraphPad Prism 4.0 software to plot the V(S) dependence curve and to determine the kinetic constants KM and kcat.
2.8. Microscale Thermophoresis (MST) Experiments
The relative binding affinity between the MBP–SelU protein and the Cy3-labeled ASL–RNA oligonucleotides, containing the unmodified uridine (U-) or C2 modified uridine (S2U-, geS2U- or Se2U-, respectively) in the position that mimicked the wobble position in tRNA (sequences are given in
Table S1), was investigated using microscale thermophoresis (MST) with the Monolith NT.115 system (NanoTemper Technologies, München, Germany). The 20 µL samples prepared for the MST measurements contained fluorescently labeled RNA oligonucleotide (target) at the constant concentration and the unlabeled protein (ligand) at sixteen serial dilutions (1:1). The samples were prepared according to the protocol described in the User Starting Guide for the Monolith NT.115 (NanoTemper Technologies, München, Germany). The working concentration of the Cy3-RNA oligonucleotides was chosen based on the fluorescence intensity, which should range from 200 to 1500 counts. The type of capillaries used for the MST measurements was chosen based on the shape of the fluorescence peaks, which were tested through Capillary Scan before the start of the experiments. The MBP–SelU ligand (452 µM) was subjected to the series of dilutions (from 226 µM to 0.0069 µM) in 2-times concentrated assay buffer (20 mM Tricine, pH 7.2, 20 mM MgCl
2, 0.1% Tween-20) and mixed with the Cy3-RNA and incubated for 30 min at room temperature. Then, the mixtures were centrifuged (15,000 rpm for 5 min) and the samples were loaded into the Monolith NT.115 Standard Treated Capillary (Nanotemper Technologies, München, Germany) and measured with 40% MST power, 20% LED excitation power (excitation type: green) at a constant temperature of 22 °C. The measurements were supported by the MO Control v2.2.4 software (NanoTemper Technologies, München, Germany). The experiments were repeated at least three times for each pair of interacting molecules: MBP–SelU:U-RNA, MBP–SelU:S2U-RNA, MBP–SelU:geS2U-RNA, and MBP–SelU:Se2U-RNA. As a control, the interactions between the MBP protein and the tested oligonucleotides were examined. The initial fluorescence signals were analyzed using MO Affinity Analysis v2.2.4 software (NanoTemper Technologies, München, Germany) and the interaction affinity and dissociation constant (K
d) were determined for each target–ligand pair using the K
d fitting model. The protein–RNA binding specificity was confirmed by the SD assay based on the denaturation of the protein in the samples (capillaries: 1, 2, and 3 vs. 14, 15, and 16) using a mixture of 4% SDS and 40 mM DTT in combination with heating to 95 °C, followed by the measurement of fluorescence intensity using a Monolith NT.115 instrument. When the fluorescence intensities of the samples were identical after denaturation, it could be concluded that the fluorescence changes observed before the denaturation were triggered by a binding event.
2.9. Isolation of tRNA Associated with MBP-SelU Protein
The fraction of the purified MBP–SelU protein (purity ~99%) was dissolved in the 400 µL of storage buffer (20 mM Tris-HCl, pH 7.4, 25 mM NaCl) and mixed with an equal volume of phenol solution, equilibrated with 10 mM Tris-HCl, pH 7.4 (Sigma-Aldrich, Poznan, Poland), vortexed and centrifuged at maximum speed for 15 min. Then, the clear aqueous phase was transferred into a new eppendorf tube, treated with 400 µL of a phenol–chloroform mixture (1:1), and centrifuged as above. The aqueous phase was transferred into the new tube and washed twice with chloroform (400 µL) to remove the phenol residues. The nucleic acid present in the aqueous phase was precipitated with 2.5 volumes of 99% ethanol with the addition of 0.1 volume of 3 M sodium acetate, pH 5.0. The obtained pellet was washed with 70% ethanol and air dried. The same procedure was repeated for the isolation of tRNA from the pure MBP protein to check whether the MBP tag bound the nucleic acids (control isolation). The amount of isolated tRNA was assessed spectrophotometrically after measuring the Abs260.
For the analysis of the full-length tRNAs associated with the MBP–SelU, the protein sample (200 µg) in the storage buffer was thermally denatured (5 min at 95 °C) and centrifuged at maximum speed for 15 min at 4 °C. The supernatant was collected and analyzed using UPLC-PDA-ESI(-)-MS.
2.10. Isolation of tRNALys, tRNAGlu and tRNAGln from E. coliΔSelU
The Escherichia coli, strain BW25113 with the SelU knockout (E. coli Genetic Stock Center Yale College, New Haven, CT, USA) was cultured in 12 L of LB medium (with 1% inoculum from the overnight culture) at 37 °C for several hours (~4–5 h) until the Abs600 reached 0.6–0.8 OD. Then, the culture was centrifuged (4000 rpm for 30 min at 4 °C) and the cells were lysed in TriReagent (ThermoFisher Sci., Waltham, MA, USA). The total cellular RNA was isolated according to the manufacturer’s procedure for TriReagent. The individual tRNAs were isolated from the total cellular RNA mixture using a specific DNA probes labeled with biotin at the 5′ end. The probe specific for tRNALys was 5′-biotin-TGCGACCAATTGATTAAAAGTCAACTGCTC-3′. The probe specific for tRNAGlu was 5′-biotin-CCTGTTACCGCCGTGAAAGGGCGGTGTCC-3′. The probespecific for tRNAGln was 5′-biotin-AGGGAATGCCGGTATCAAAAACCGGTGCCT-3′. They were all bound to Streptavidin Agarose resin (ThermoFisher Sci.). The total cellular RNA resuspended in 10 mM Tris-HCl, pH 7.4, and 150 mM NaCl buffer was added to the agarose beads with bound the appropriate probe and incubated at 85 °C for 5 min. Then, the beads were cooled to room temperature for 10 min and incubated overnight at 4 °C with vigorous mixing (2000 rpm). The next day, the samples were centrifuged (3000 rpm for 2 min) to remove unbound RNA and washed with 10 mM Tris-HCl pH 7.4 buffer until the Abs260 of the solution reached zero. The bound tRNA was eluted by the successive addition of 200 μL of deionized water, incubation at 85 °C for 3 min, and centrifugation. The harvested tRNA-containing solution was evaporated in a vacuum centrifuge (Savant). The obtained full-length tRNAs were subjected to UPLC-PDA-ESI(-)-MS analysis and, in the final stage of the experiments, geranylation and selenation reactions.
2.11. The tRNA Nucleolytic Hydrolysis
Tthe natural tRNA sample (10 µg) was hydrolyzed into single nucleosides using a combination of two nucleases: Benzonase from Serratia marcescens (Sigma-Aldrich, Poznan, Poland), 20 units per reaction in 50 mM Tris-HCl, pH 8.0, 1 mM MgCl2 buffer for 4 h at 37 °C; Phosphodiesterase I from Crotalus adamanteus venom (Sigma-Aldrich), 0.8 units per reaction in 50 mM Tris-HCl.0, pH 8.0, and 20 mM of MgCl2 for 16 h at 37 °C; and finally Alkaline Phosphatase (EURx, Gdansk, Poland), 10 units per reaction in the manufacturer’s buffer (1 M diethanolamine, 10 mM p-nitrophenylophosphate, and 0.25 mM MgCl2, pH 9.8) for 1 h at 37 °C. Each sample after hydrolysis was filtered using a 10 000-MW cut-off spin filter (Merck, Poznan, Poland) and dried in a vacuum centrifuge. The obtained mixture of nucleosides was analyzed using UPLC-PDA-ESI(-)-HRMS.
2.12. The In Vitro Transformation of the R5S2U-tRNA into R5Se2U-tRNA
The natural (c)mnm5S2U-tRNAs (tRNALys, tRNAGln, and tRNAGlu) isolated from the E. coliΔSelU were used to perform the geranylation and selenation reactions. For each type of tRNA, three reaction mixtures were prepared, each containing 5 µg of tRNA (0.2 nmol) in 10 mM Tricine-HCl buffer (pH 7.2) and 10 mM MgCl2. For the geranylation reaction (i), the R5S2U-tRNA was incubated with 5-fold molar excess (1 nmol) of the geranyl pyrophosphate (GePP) and MBP–SelU enzyme (2 µg = 0.0236 nmol), at 37 °C for 2 h. For the geranylation and subsequent selenation reactions (ii), the R5S2U-tRNA was incubated with 5-fold molar excess (1 nmol) of the geranyl pyrophosphate (GePP) and MBP–SelU enzyme (2 µg = 0.0236 nmol), at 37 °C for 2 h. After the geranylation step, the SePO33− (~4 nmol) and the additional portion of MBP–SelU (1 µg = 0.0118 nmol) were added into the mixture and the reaction was continued for the next 1 h at 37 °C. For the direct selenation reaction (iii), the R5S2U-tRNA was incubated with the MBP–SelU enzyme (3 µg and 0.0355 nmol, respectively) in the presence of the SePO33− (~4 nmol), at 37 °C for 1 h. The total amount of enzyme used in the second and third reactions (ii and iii) was 3 µg = 0.0355 nmol, which corresponded to approximately 0.8 µg (0.035 nmol) of the protein-bound tRNA fraction being added into the reaction mixture, according to our calculation of the amount of tRNA that was isolated from the pure protein. This amount of specific tRNAs that was present in the reaction was taken as background tRNA. All reactions were stopped by the thermal inactivation of the enzyme (95 °C for 3 min) and then the mixtures were centrifuged for the separation of the denatured protein pellets. The identification of the reaction products that were in the form of full-length tRNAs, was performed using the UPLC-PDA-ESI(-)-MS analysis or, after nucleolytic hydrolysis and dephosphorylation, using the UPLC-PDA-ESI(-)-HRMS analysis of the nucleosides that were derived from the tRNA products.
2.13. Ultra-Performance Liquid Chromatography Coupled with a Mass Spectrometry and Photodiode Array Detection
The LC-MS analysis was carried out on ACQUITY UPLC I Class chromatography system equipped with a photodiode array detector with a binary solvent manager (Waters Corp., Milford, MA, USA) coupled with an SYNAPT G2-Si mass spectrometer equipped with an electrospray source and quadrupole, Time-of-Flight mass analyzer (Waters Corp., Milford, MA, USA).
The conditions for the nucleoside mixture analysis using the UPLC-PDA-ESI(-)-HRMS method: the ACQUITY HSS T3 1.8 μm column (100 × 2.1 mm, 1.7 µm) (Waters Corp., Milford, MA, USA) and HSS T3 guard column (2.1 × 5 mm, 1.8 µm), which was at 25 °C, were used for the chromatographic separation of the analyte. A gradient program was employed for the mobile phase, which combined solvent A (0.01% formic acid in water) and solvent B (0.01% formic acid in acetonitrile) as follows: 0% B (0–1.0 min), 0–0.2% B (1.0–2.4 min), 0.2–0.8% B (2.4–3.8 min), 0.8–1.8% B (3.8–5.2 min), 1.8–3.2% B (5.2–6.6 min), 3.2–5% B (6.6–10.0 min), 5–8% B (10.0–13.5 min), 8.0–30% B (13.5–15.5 min), 30–100% B (15.5–19.5 min), 100–100% B (19.5–20.5 min), 100–0% B (20.5–21.0 min), and 0% B (21.0–25.0 min). The flow rate was 0.2 mL/min and the injection volume was 5 μL. For the mass spectrometric detection, the electrospray source was operated in negative high-resolution mode at 30,000 FWHM, which resolved the power of the TOF analyzer. To ensure accurate mass measurements, the data were collected in centroid mode and the mass was corrected during acquisition using leucine encephalin solution as an external reference, Lock-Spray TM (Waters Corp., Milford, MA, USA), which generated a reference ion at m/z 554.2615 [M-H]− in negative ESI mode. The optimized source parameters were: capillary voltage, 2.8 kV for negative mode; cone voltage, 20 V; source temperature, 110 °C; desolvation gas (nitrogen) flow rate, 600 L/h; temperature, 350 °C; nebulizer gas pressure, 6.5 bar. The mass spectrometer conditions were optimized using the direct infusion of the standard solution. The mass spectra were recorded over a range of m/z 100 to 1200. The PDA spectra were measured over the wavelength range of 210–400 nm in steps of 1.2 nm. The results of the measurements were processed using the MassLynx 4.1 software (Waters Corp., Milford, MA, USA), which was incorporated into the instrument.
The conditions for the full-length tRNA analysis using the UPLC-PDA-ESI(-)-MS method: the ACQUITY UPLC® Oligonucleotides BEH C18 column (50 × 2.1 mm, 1.7 μm), which was maintained at 60 °C, was used for the chromatographic separation of analyte. A gradient program was employed for the mobile phase, which combined solvent A (15 mM of triethylamine and 400 mM of 1,1,1,3,3,3- hexafluoro-2-propanol in water) and solvent B (50% methanol and 50% solvent A, v/v) as follows: 42.5% B (0–1.0 min), 42.5–52% B (1.0–22.0 min), 52–52% B (22.0–24.0 min), 52–42.5% B (24.0–24.1 min), and 42.5–42.5% B (24.1–28 min). Water was used as the weak wash solvent and 10% methanol in water was used as the strong wash solvent. The flow rate was 0.2 mL/min and the injection volume was 10 μL. For the mass spectrometric detection, the electrospray source was operated in a negative resolution mode. The optimized source parameters were: capillary voltage, 2.7 kV; cone voltage, 40 V; desolvation gas flow rate, 600 L/h; temperature, 400 °C; nebulizer gas pressure, 6.5 bar; source temperature, 120 °C. The mass spectra were recorded over the range of m/z 500 to 2000. The mass spectrometer conditions were optimized using the direct infusion of the standard solution. The system was controlled using the MassLynx software v 4.1. The raw ESI mass spectra were deconvoluted to a zero-charge state mass using the MaxEnt1 algorithm.
4. Discussion
In the study presented here, we discussed the requirements for the substrates of the tRNA 2-selenouridine synthase (SelU), the composition of the tRNA molecules bound to this nucleoprotein, in particular the pattern of nucleoside modifications, and we provided unequivocal evidence that confirmed the postulated cellular pathway of the R5S2U→R5Se2U transformation in bacterial tRNAs. In the presented study, we used the fusion protein with an MBP tag at the N-terminus of SelU and proved that the presence of the MBP tag significantly stabilized the SelU protein, as reflected by its high activity in the specific reactions of geranylation and selenation. We do not know the mechanism through which the MBP interacted with the SelU. At this stage, we can only speculate that the MBP appeared to act as a “molecular chaperone”, which promoted the stability of the proteins that were fused to it, and that its maintenance function likely relied on direct stabilizing contact with the attached protein [
30,
41]. The catalytic efficiency of the studied MBP–SelU variant was assessed, under conditions that were described previously [
11], as being 1000-fold higher than that of SelU–His
6 during geranylation and 50-fold higher than that during selenation (
Figure S4,
Table S3). The increased stability improved the catalytic performance of the enzyme, which could be consistent with the stability and efficacy of SelU under native conditions, i.e., in bacterial cells. Thanks to these parameters of MBP–SelU, we had the necessary molecular tool to characterize and evaluate the properties of this enzyme.
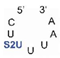
We found that SelU recognized the position of the S2U modification in the anticodon loop, the S2U flanking sequence, and the length of the RNA substrate. The enzyme recognized the simplest chemically synthesized ASL–RNA model substrate (17-mer), which mimicked the native anticodon loop of tRNA
Lys with a 7-nt loop sequence (5′-CU
S2UUUAA-3′) during the geranylation and selenation reactions and achieved the high efficiency in the applied conditions (4.5-fold excess of ASL–RNA substrate for 1 h of incubation at 37 °C). Shifting the S2U position in the ASL loop structure (to the 33
rd, 35
th or 36
th position) resulted in a much poorer recognition by the enzyme and impaired the efficiency of the geranylation reaction (
Table 1). S2U(34)–RNA substrates that contained purine residues A or G in the 35
th position were barely accepted by the enzyme, whereas natural U(35) was the better substrate compared to the C(35) variant (
Table 2). The length of the oligonucleotide substrate that mimicked the structural elements of tRNA
Lys also affected recognition by the enzyme. The oligonucleotide that truncated to 7-nt, which mimicked the anticodon loop (AL) of tRNA
Lys, and the oligonucleotide truncated to 3-nt, which mimicked only the anticodon (A), were less accepted and the efficiency of the enzyme in the geranylation reaction decreased to 25% and 14%, respectively. This indicated that the enzyme required the fully structured loop to exert its catalytic activity. The single nucleosides (S2U, mnm5S2U, and geS2U) did not serve as substrates for the MBP–SelU enzyme. Interestingly, the enzyme was less demanding in terms of substrate specificity during the selenation reaction, as all geS2U–ASL models tested were converted quantitatively (geS2U in the 33rd, 34th, and 35th position of ASL) or still produced the high yield (66% for geS2U in the 36
th position of ASL) for their selenouridine analogs. The MBP–SelU enzyme also accepted the geS2U–RNA
Arg ASL in the selenation reaction, which proceeded with a 94% yield. These results suggested that the recognition of tRNA substrates by the enzyme occurred through different mechanisms in the two reactions that were tested (i.e., alkylation at the sulfur atom in geranylation and nucleophilic substitution of selenophosphate at C2 in selenation) [
11].
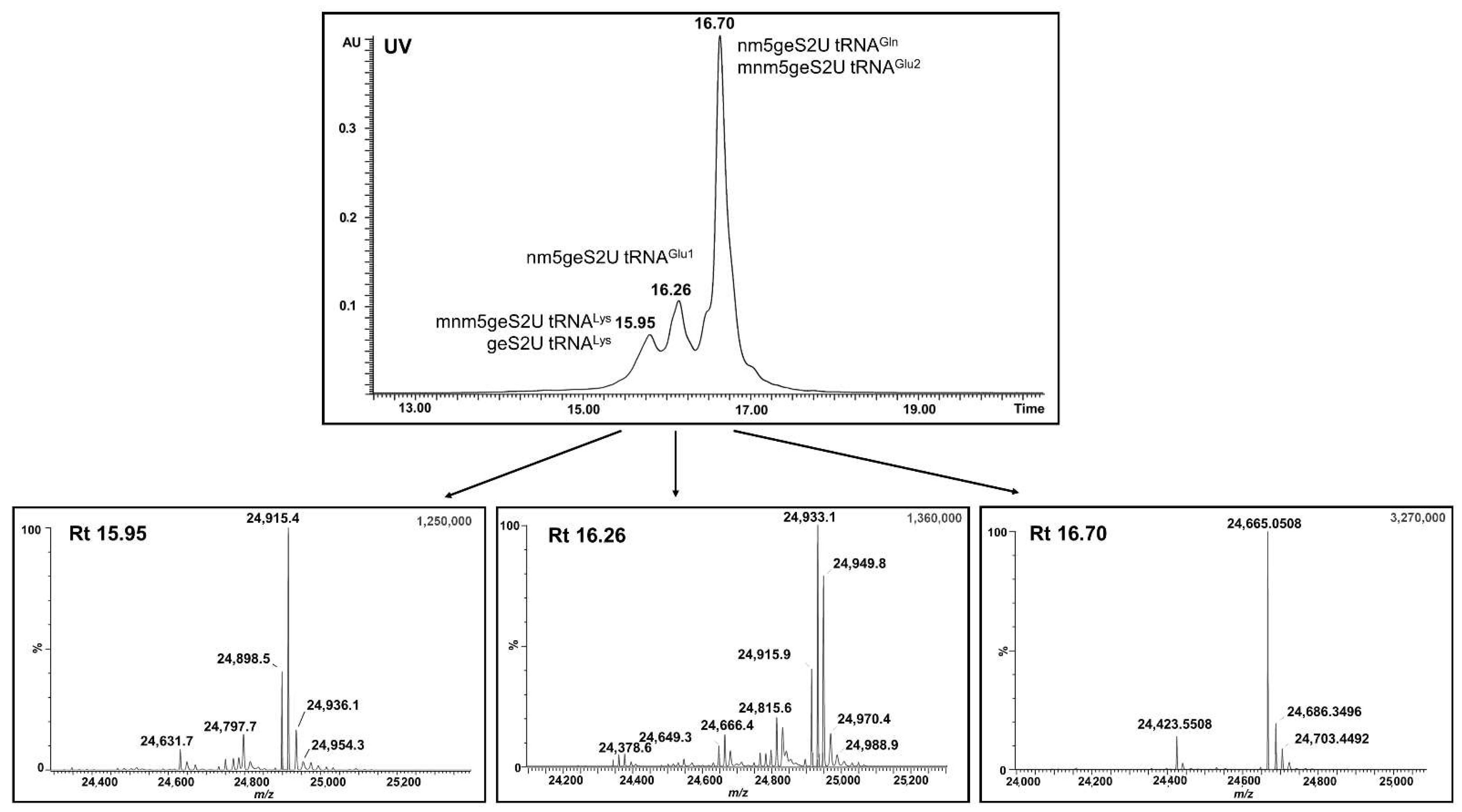
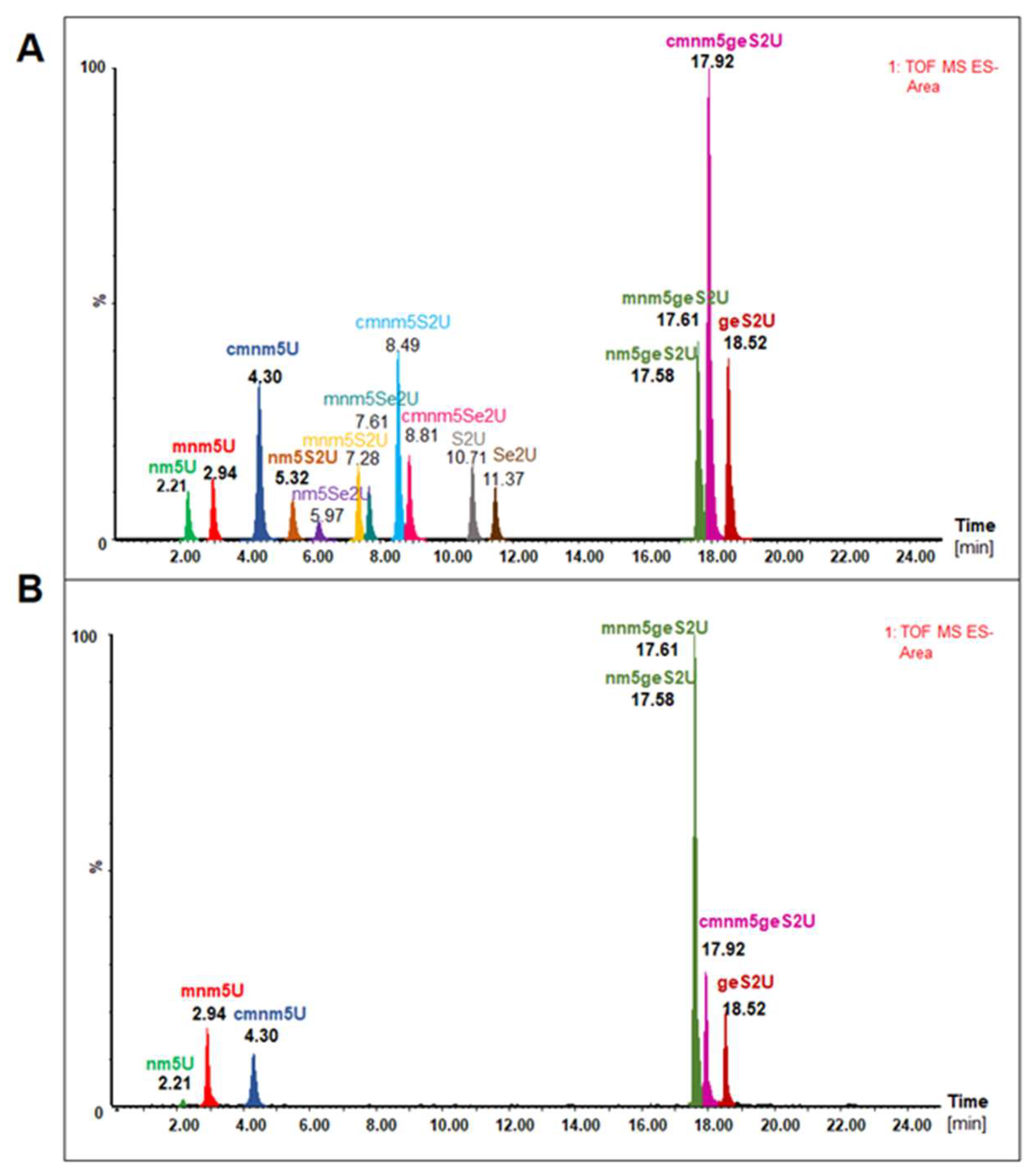
MBP–SelU and other fusion variants that were constructed in our studies (data not shown) had the tightly bound tRNA fraction that altered the spectral properties of the protein, which had an unusual absorption spectrum with a maximum at 260 nm, as in nucleic acids, and no peak at 280 nm, as in proteins. We roughly assessed the amount of tRNA associated with MBP–SelU as approximately one tRNA molecule per one protein molecule after the isolation of tRNA from the pure protein using the phenol–chloroform extraction method. Additionally, using the northern blot analysis, we found that three types of tRNAs (tRNA
Glu, tRNA
Lys, and tRNA
Gln) were associated with the MBP–SelU protein (data not shown). Then, using the UPLC-PDA-ESI(-)-MS technique, we identified the full-length tRNAs associated with the MBP–SelU protein (the results of the tRNA analysis are published for the first time) and assessed their nucleoside composition (
Figure 4 and
Figure 5,
Table 4,
Tables S6 and S7). The tRNAs that were associated with the MBP–SelU protein (exactly with SelU) were enriched in nucleosides that mainly contained the geranyl group (mnm5geS2U, geS2U, cmnm5geS2U, and nm5geS2U). The relatively high proportion of “naked” geS2U, without mnm- and cmnm- substituents in the C5 position, suggested that the biosynthesis of the final R5Se2U–tRNA occurred independently of the 5-substitution. Importantly, sulfur- and selenium-modified nucleosides were not detected in the hydrolysates of the tRNAs that were associated with the SelU protein. This result was confirmed by the microscale thermophoresis (MST) measurements, in which we assessed the relative binding affinity between the MBP–SelU protein and the synthetic RNAs, which contained a single modified uridine (S2U-, geS2U- or Se2U-) in the position that mimicked the wobble position in tRNA. Our results indicated that the geranylated RNA (Cy3–geS2U(34)–RNA
Lys) had the highest affinity for MBP–SelU (K
d = 3.95 µM), whereas the other ASL–RNAs with 2-thio- and 2-seleno- modification bound to the enzyme in a similar range with a lower affinity. The Bjork group drew similar conclusions already in 2016, when they examined the modifications of tRNAs associated with wild type proteins, as well as mutants with the increased geranylation capacity, e.g., MnmH(G67E) [
23], and he was able to identify the cmnm5geS2UpU, mnm5geS2UpU, and nm5geS2UpU dimers. They concluded that the tRNAs bound to the enzymes were enriched in the geranylated derivatives and suggested that the MnmH type proteins appeared to have the strong affinity for such tRNAs. In the first report on the R5geS2U–tRNA, Dumelin et al. suggested that the geranylation of tRNA impaired cellular translation efficiency and was involved in the readout of GAG codons that were specific to Glu or AAG encoding Lys [
10]. If
S-geranyl–tRNA performed this function, it should be available to the translation apparatus in the cytoplasm of the bacterial cell. In both, this study (data not shown) and Bjork’s study, no free R5geS2U–tRNAs were detected in the cytoplasm [
23]. In our opinion, the total R5geS2U–tRNA is tightly associated with the SelU protein, which releases the final transformation product, i.e., the R5Se2U–tRNA, after the selenation of the R5geS2U–tRNA.
Finally, we demonstrated for the first time the pathway of conversion of native bacterial R5S2U–tRNA into R5Se2U–tRNA as the two-step process with the formation of R5geS2U–tRNA as the intermediate product, which is strongly associated with the SelU enzyme and plausibly is not involved in mRNA translation because the R5geS2U–tRNA content in the cellular fraction is negligible (data not shown). The reaction of direct R5S2U–tRNA selenation did not result in the R5Se2U–tRNA product.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}