
Mitochondria and the NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis

 ,
, 
Abstract
:1. Introduction
Overview of Mitochondria and NLRP3 Inflammasome Interplay
2. Alcoholic Steatohepatitis (ASH)
2.1. Epidemiology and Etiology
2.2. Progression and Stages
2.3. Pathogenesis
3. ASH, Mitochondria, and Inflammasome
4. Nonalcoholic Steatohepatitis (NASH)
4.1. Epidemiology
4.2. Etiology
4.3. Progression and Stages
5. NASH, Mitochondria, and Inflammasome
6. Sphingolipids and NLRP3 Inflammasome in NASH
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxynonenal |
| ASMase | acid sphingomyelinase |
| ADH | alcohol dehydrogenase |
| AH | alcoholic hepatitis |
| AIM2 | absent in melanoma 2 |
| ALD | alcoholic liver disease |
| ALDH | acetaldehyde dehydrogenase |
| AMPK | 5′-AMP-activated protein kinase |
| ASC | adaptor apoptosis speck protein |
| ASH | alcoholic steatohepatitis |
| ATP | adenosine triphosphate |
| CAT | catalase |
| CerSs | ceramide synthase |
| CYP2E1 | cytochrome P450 2E1 |
| DAG | diacylglycerol |
| DAMPS | danger-associated molecular patterns |
| ER | endoplasmic reticulum |
| FA | fatty acid |
| GSDMD | gasdermin D |
| GSH | glutathione |
| GSH-Px | glutathione peroxidase |
| GSSG-Rx | glutathione reductase |
| HCC | hepatocellular carcinoma |
| HSC | hepatic stellate cells |
| IL-18 | interleukin-18 |
| IL-1β | interleukin-1β |
| IL-6 | interleukin-6 |
| IR | insulin resistance |
| KC | Kupffer cells |
| LPS | lipopolysaccharide |
| MAFLD | metabolic dysfunction-associated fatty liver disease |
| MAM | mitochondria-associated membranes |
| MCD | methionine/choline-deficient diet |
| MDA | malondialdehyde |
| mGSH | mitochondrial glutathione |
| miRNAs | microRNAs |
| MMP | mitochondria membrane potential |
| mt-DNA | mitochondrial DNA |
| mtROS | mitochondrial ROS |
| NADH/NAD+ | oxidized and reduced nicotinamide adenine dinucleotide ratio |
| NAFL | nonalcoholic fatty liver |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRC4 | nucleotide-binding oligomerization domain-like receptors CARD domain-containing protein 4 |
| NLRP3 | nucleotide-binding oligomerization domain-like receptor protein 3 |
| ox-mtDNA | oxidized mitochondrial DNA |
| PAMPS | pathogen-associated molecular patterns |
| PKCδ | protein kinase Cδ |
| PPARα | peroxisome proliferator-activated receptor-α |
| ROS | reactive oxidative species |
| S1P | sphingosine-1-phosphate |
| S1PRs | sphingosine 1-phosphate receptors |
| S1PR4 | type 4 sphingosine-1-phosphate receptor |
| SM | sphingomyelin |
| SMases | sphingomyelinases |
| SMS1 | sphingomyelin synthase 1 |
| SOD | superoxide dismutase |
| SQSTM1/p62 | sequestosome-1 |
| SREBP1c | sterol regulatory element-binding protein 1c |
| T2DM | type 2 diabetes mellitus |
| TCA | tricarboxylic acid cycle |
| TLR | toll-like receptor |
| TNF-α | tumor necrosis factor alpha |
| TXNIP | thioredoxin-interacting protein |
| UPR | unfolded protein response |
| VDAC | voltage-dependent anion-selective channel |
| WD | Western diet |
| WHO | World Health Organization |
References
- Argemi, J.; Ventura-Cots, M.; Rachakonda, V.; Bataller, R. Alcoholic-Related Liver Disease: Pathogenesis, Management and Future Therapeutic Developments. Rev. Esp. Enferm. Dig. 2020, 112, 869–878. [Google Scholar] [CrossRef]
- Cariello, M.; Piccinin, E.; Moschetta, A. Transcriptional Regulation of Metabolic Pathways via Lipid-Sensing Nuclear Receptors PPARs, FXR, and LXR in NASH. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1519–1539. [Google Scholar] [CrossRef] [PubMed]
- Pelusi, S.; Cespiati, A.; Rametta, R.; Pennisi, G.; Mannisto, V.; Rosso, C.; Baselli, G.; Dongiovanni, P.; Fracanzani, A.L.; Badiali, S.; et al. Prevalence and Risk Factors of Significant Fibrosis in Patients With Nonalcoholic Fatty Liver Without Steatohepatitis. Clin. Gastroenterol. Hepatol. 2019, 17, 2310–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis Progression in Nonalcoholic Fatty Liver vs. Nonalcoholic Steatohepatitis: A Systematic Review and Meta-Analysis of Paired-Biopsy Studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, D.J.; Yang, V.W.; Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [Green Version]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and Mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–226. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Torres, S.; Brol, M.J.; Magdaleno, F.; Schierwagen, R.; Uschner, F.E.; Klein, S.; Ortiz, C.; Tyc, O.; Bachtler, N.; Stunden, J.; et al. The Specific NLRP3 Antagonist IFM-514 Decreases Fibrosis and Inflammation in Experimental Murine Non-Alcoholic Steatohepatitis. Front. Mol. Biosci. 2021, 8, 715765. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the Nlrp3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Dong, Z.; Song, W. NLRP3 Inflammasome as a Novel Therapeutic Target for Alzheimer’s Disease. Signal. Transduct. Target. Ther. 2020, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Antonio, I.; López-Sánchez, G.N.; Uribe, M.; Chávez-Tapia, N.C.; Nuño-Lámbarri, N. Role of the Inflammasome, Gasdermin D, and Pyroptosis in Non-Alcoholic Fatty Liver Disease. J. Gastroenterol. Hepatol. 2021, 36, 2720–2727. [Google Scholar] [CrossRef] [PubMed]
- Solsona-Vilarrasa, E.; Fucho, R.; Torres, S.; Nuñez, S.; Nuño-Lámbarri, N.; Enrich, C.; García-Ruiz, C.; Fernández-Checa, J.C. Cholesterol Enrichment in Liver Mitochondria Impairs Oxidative Phosphorylation and Disrupts the Assembly of Respiratory Supercomplexes. Redox Biol. 2019, 24, 101214. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [Green Version]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Hong, T.; Chen, Y.; Li, X.; Lu, Y. The Role and Mechanism of Oxidative Stress and Nuclear Receptors in the Development of NAFLD. Oxidative Med. Cell Longev. 2021, 2021, 6889533. [Google Scholar] [CrossRef]
- Quan, Y.; Xin, Y.; Tian, G.; Zhou, J.; Liu, X. Mitochondrial ROS-Modulated MtDNA: A Potential Target for Cardiac Aging. Oxidative Med. Cell Longev. 2020, 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than Just a Housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.W.; Lee, M.S. Mitochondria and the NLRP3 Inflammasome: Physiological and Pathological Relevance. Arch. Pharmacal. Res. 2016, 39, 1503–1518. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-ΚB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Hoque, R.; Vodovotz, Y.; Mehal, W. Therapeutic Strategies in Inflammasome Mediated Diseases of the Liver. J. Hepatol. 2013, 58, 1047–1052. [Google Scholar] [CrossRef] [Green Version]
- Rehm, J.; Samokhvalov, A.V.; Shield, K.D. Global Burden of Alcoholic Liver Diseases. J. Hepatol. 2013, 59, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Yang, Z.; Zhang, T.; Ma, J.; Chandler, K.; Liangpunsakul, S. Epidemiology of Alcohol-Associated Liver Disease. Clin. Liver Dis. 2021, 25, 483–492. [Google Scholar] [CrossRef]
- Becker, U.; Deis, A.; Sorensen, T.I.; Gronbaek, M.; Borch-Johnsen, K.; Muller, C.F.; Schnohr, P.; Jensen, G. Prediction of Risk of Liver Disease by Alcohol Intake, Sex, and Age: A Prospective Population Study. Hepatology 1996, 23, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic Liver Disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.O.; Bataller, R. Pathogenesis and management of alcoholic liver disease. Dig. Dis. 2016, 34, 347. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschke, R. Alcoholic Liver Disease: Alcohol Metabolism, Cascade of Molecular Mechanisms, Cellular Targets, and Clinical Aspects. Biomedicines 2018, 6, 106. [Google Scholar] [CrossRef] [Green Version]
- Pohl, K.; Moodley, P.; Dhanda, A.D. Alcohol’s Impact on the Gut and Liver. Nutrients 2021, 13, 3170. [Google Scholar] [CrossRef]
- Purohit, V.; Gao, B.; Song, B.J. Molecular Mechanisms of Alcoholic Fatty Liver. Alcohol. Clin. Exp. Res. 2009, 33, 191. [Google Scholar] [CrossRef] [Green Version]
- You, M.; Fischer, M.; Deeg, M.A.; Crabb, D.W. Ethanol Induces Fatty Acid Synthesis Pathways by Activation of Sterol Regulatory Element-Binding Protein (SREBP). J. Biol. Chem. 2002, 277, 29342–29347. [Google Scholar] [CrossRef] [Green Version]
- You, M.; Arteel, G.E. Effect of Ethanol on Lipid Metabolism. J. Hepatol. 2019, 70, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Liu, J. Ethanol and Liver: Recent Insights into the Mechanisms of Ethanol-Induced Fatty Liver. World J. Gastroenterol. WJG 2014, 20, 14672. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Checa, J.C.; Kaplowitz, N. Hepatic Mitochondrial Glutathione: Transport and Role in Disease and Toxicity. Toxicol. Appl. Pharm. 2005, 204, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef] [PubMed]
- Slevin, E.; Baiocchi, L.; Wu, N.; Ekser, B.; Sato, K.; Lin, E.; Ceci, L.; Chen, L.; Lorenzo, S.R.; Xu, W.; et al. Kupffer Cells: Inflammation Pathways and Cell-Cell Interactions in Alcohol-Associated Liver Disease. Am. J. Pathol. 2020, 190, 2185–2193. [Google Scholar] [CrossRef]
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver Dis. 2020, 40, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Iracheta-Vellve, A.; Petrasek, J.; Gyogyosi, B.; Bala, S.; Csak, T.; Kodys, K.; Szabo, G. Interleukin-1 Inhibition Facilitates Recovery from Liver Injury and Promotes Regeneration of Hepatocytes in Alcoholic Hepatitis in Mice. Liver Int. 2017, 37, 968. [Google Scholar] [CrossRef]
- Boaru, S.G.; Borkham-Kamphorst, E.; Tihaa, L.; Haas, U.; Weiskirchen, R. Expression Analysis of Inflammasomes in Experimental Models of Inflammatory and Fibrotic Liver Disease. J. Inflamm. 2012, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Le Daré, B.; Ferron, P.J.; Gicquel, T. The Purinergic P2 × 7 Receptor-NLRP3 Inflammasome Pathway: A New Target in Alcoholic Liver Disease? Int. J. Mol. Sci. 2021, 22, 2139. [Google Scholar] [CrossRef]
- Ren, W.; Rubini, P.; Tang, Y.; Engel, T.; Illes, P. Inherent P2 × 7 Receptors Regulate Macrophage Functions during Inflammatory Diseases. Int J. Mol. Sci. 2021, 23, 232. [Google Scholar] [CrossRef]
- Khanam, A.; Saleeb, P.G.; Kottilil, S. Pathophysiology and Treatment Options for Hepatic Fibrosis: Can It Be Completely Cured? Cells 2021, 10, 1097. [Google Scholar] [CrossRef]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 Receptor Antagonist Ameliorates Inflammasome-Dependent Alcoholic Steatohepatitis in Mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, M.J.; Kim, T.H.; You, J.S.; Blaya, D.; Sancho-Bru, P.; Kim, S.G. Alcohol Dysregulates MiR-148a in Hepatocytes through FoxO1, Facilitating Pyroptosis via TXNIP Overexpression. Gut 2019, 68, 708–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanova, E.; Wu, R.; Wang, W.; Yan, R.; Chen, Y.; French, S.W.; Llorente, C.; Pan, S.Q.; Yang, Q.; Li, Y.; et al. Pyroptosis by Caspase11/4-Gasdermin-D Pathway in Alcoholic Hepatitis in Mice and Patients. Hepatology 2018, 67, 1737–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromenty, B.; Grimbert, S.; Mansouri, A.; Beaugrand, M.; Erlinger, S.; Rötig, A.; Pessayre, D. Hepatic Mitochondrial DNA Deletion in Alcoholics: Association with Microvesicular Steatosis. Gastroenterology 1995, 108, 193–200. [Google Scholar] [CrossRef]
- Bansal, S.; Anandatheerthavarada, H.K.; Prabu, G.K.; Milne, G.L.; Martin, M.V.; Guengerich, F.P.; Avadhani, N.G. Human Cytochrome P450 2E1 Mutations That Alter Mitochondrial Targeting Efficiency and Susceptibility to Ethanol-Induced Toxicity in Cellular Models. J. Biol. Chem. 2013, 288, 12627–12644. [Google Scholar] [CrossRef] [Green Version]
- Robin, M.A.; Anandatheerthavarada, H.K.; Fang, J.K.; Cudic, M.; Otros, L.; Avadhani, N.G. Mitochondrial Targeted Cytochrome P450 2E1 (P450 MT5) Contains an Intact N Terminus and Requires Mitochondrial Specific Electron Transfer Proteins for Activity. J. Biol. Chem. 2001, 276, 24680–24689. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Cederbaum, A.I. Ethanol Cytotoxicity to a Transfected HepG2 Cell Line Expressing Human Cytochrome P4502E1. J. Biol. Chem. 1996, 271, 23914–23919. [Google Scholar] [CrossRef] [Green Version]
- Angireddy, R.; Chowdhury, A.R.; Zielonka, J.; Ruthel, G.; Kalyanaraman, B.; Avadhani, N.G. Alcohol-Induced CYP2E1, Mitochondrial Dynamics and Retrograde Signaling in Human Hepatic 3D Organoids. Free Radic. Biol. Med. 2020, 159, 1–14. [Google Scholar] [CrossRef]
- Knockaert, L.; Fromenty, B.; Robin, M.A. Mechanisms of Mitochondrial Targeting of Cytochrome P450 2E1: Physiopathological Role in Liver Injury and Obesity. FEBS J. 2011, 278, 4252–4260. [Google Scholar] [CrossRef]
- Abdelmegeed, M.A.; Ha, S.-K.; Choi, Y.; Akbar, M.; Song, B.-J. Role of CYP2E1 in Mitochondrial Dysfunction and Hepatic Tissue Injury in Alcoholic and Non-Alcoholic Diseases. Curr. Mol. Pharm. 2017, 10, 207. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; He, X.; Zhao, J.; Huang, W. Hepatoprotection by Ginsenoside Rg1 in Alcoholic Liver Disease. Int. Immunopharmacol. 2021, 92, 107327. [Google Scholar] [CrossRef]
- Lu, Y.; Wu, D.; Wang, X.; Ward, S.C.; Cederbaum, A.I. Chronic Alcohol-Induced Liver Injury and Oxidant Stress Are Decreased in Cytochrome P4502E1 Knockout Mice and Restored in Humanized Cytochrome P4502E1 Knock-in Mice. Free Radic. Biol. Med. 2010, 49, 1406–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; He, B.; Zhang, S.; Huang, W.; Li, X. Ginsenoside Rg1 Alleviates Acute Liver Injury through the Induction of Autophagy and Suppressing NF-ΚB/NLRP3 Inflammasome Signaling Pathway. Int. J. Med. Sci. 2021, 18, 1382. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, D.; Yang, L.; Gan, L.; Cederbaum, A.I. Cyto Chrome P450 2E1 Potentiates Ethanol Induction of Hypoxia and HIF-1α In Vivo. Free Radic. Biol. Med. 2013, 63, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial Dysfunction Resulting from Loss of Cytochrome c Impairs Cellular Oxygen Sensing and Hypoxic HIF-α Activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial Complex III Is Required for Hypoxia-Induced ROS Production and Cellular Oxygen Sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Xuan, W.; Li, J.; Yao, H.; Huang, C.; Li, J. AMPK Protects against Alcohol-Induced Liver Injury through UQCRC2 to up-Regulate Mitophagy. Autophagy 2021, 17, 3622. [Google Scholar] [CrossRef]
- Inagaki, T.; Koike, M.; Ikuta, K.; Kobayashi, S.; Suzuki, M.; Kato, K.; Kato, K. Ultrastructural Identification and Clinical Significance of Light Microscopic Giant Mitochondria in Alcoholic Liver Injuries. Gastroenterol. Jpn. 1989, 24, 46–53. [Google Scholar] [CrossRef]
- Palma, E.; Riva, A.; Moreno, C.; Odena, G.; Mudan, S.; Manyakin, N.; Miquel, R.; Degré, D.; Trepo, E.; Sancho-Bru, P.; et al. Perturbations in Mitochondrial Dynamics Are Closely Involved in the Progression of Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2020, 44, 856–865. [Google Scholar] [CrossRef]
- Palma, E.; Ma, X.; Riva, A.; Iansante, V.; Dhawan, A.; Wang, S.; Ni, H.M.; Sesaki, H.; Williams, R.; Ding, W.X.; et al. Dynamin-1-Like Protein Inhibition Drives Megamitochondria Formation as an Adaptive Response in Alcohol-Induced Hepatotoxicity. Am. J. Pathol. 2019, 189, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Colell, A.; García-Ruiz, C.; Miranda, M.; Ardite, E.; Marí, M.; Morales, A.; Corrales, F.; Kaplowitz, N.; Fernández-Checa, J.C. Selective Glutathione Depletion of Mitochondria by Ethanol Sensitizes Hepatocytes to Tumor Necrosis Factor. Gastroenterology 1998, 115, 1541–1551. [Google Scholar] [CrossRef]
- Li, Q.; Zhong, W.; Qiu, Y.; Kang, X.; Sun, X.; Tan, X.; Zhao, Y.; Sun, X.; Jia, W.; Zhou, Z. Preservation of Hepatocyte Nuclear Factor-4α Contributes to the Beneficial Effect of Dietary Medium Chain Triglyceride on Alcohol-Induced Hepatic Lipid Dyshomeostasis in Rats. Alcohol. Clin. Exp. Res. 2013, 37, 587–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ruiz, C.; Kaplowitz, N.; Fernandez-Checa, J.C. Role of Mitochondria in Alcoholic Liver Disease. Curr. Pathobiol. Rep. 2013, 1, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, S.; Baulies, A.; Insausti-Urkia, N.; Alarcón-Vila, C.; Fucho, R.; Solsona-Vilarrasa, E.; Núñez, S.; Robles, D.; Ribas, V.; Wakefield, L.; et al. Endoplasmic Reticulum Stress-Induced Upregulation of STARD1 Promotes Acetaminophen-Induced Acute Liver Failure. Gastroenterology 2019, 157, 552–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, A.; Matias, N.; Fucho, R.; Ribas, V.; Von Montfort, C.; Nuño, N.; Baulies, A.; Martinez, L.; Tarrats, N.; Mari, M.; et al. ASMase Is Required for Chronic Alcohol Induced Hepatic Endoplasmic Reticulum Stress and Mitochondrial Cholesterol Loading. J. Hepatol. 2013, 59, 805–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, S.; Solsona-Vilarrasa, E.; Nuñez, S.; Matías, N.; Insausti-Urkia, N.; Castro, F.; Casasempere, M.; Fabriás, G.; Casas, J.; Enrich, C.; et al. Acid Ceramidase Improves Mitochondrial Function and Oxidative Stress in Niemann-Pick Type C Disease by Repressing STARD1 Expression and Mitochondrial Cholesterol Accumulation. Redox Biol. 2021, 45, 102052. [Google Scholar] [CrossRef]
- Conde de la Rosa, L.; Garcia-Ruiz, C.; Vallejo, C.; Baulies, A.; Nuñez, S.; Monte, M.J.; Marin, J.J.G.; Baila-Rueda, L.; Cenarro, A.; Civeira, F.; et al. STARD1 Promotes NASH-Driven HCC by Sustaining the Generation of Bile Acids through the Alternative Mitochondrial Pathway. J. Hepatol. 2021, 74, 1429–1441. [Google Scholar] [CrossRef]
- Gao, B.; Argemi, J.; Bataller, R.; Schnabl, B. Serum Acylcarnitines Associated with High Short-Term Mortality in Patients with Alcoholic Hepatitis. Biomolecules 2021, 11, 281. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Holmuhamedov, E.L.; Czerny, C.; Zhong, Z.; Maldonado, E.N. Regulation of Mitochondrial Function by Voltage Dependent Anion Channels in Ethanol Metabolism and the Warburg Effect. Biochim. Biophys. Acta 2012, 1818, 1536–1544. [Google Scholar] [CrossRef] [Green Version]
- Byrne, C.D.; Targher, G. NAFLD: A Multisystem Disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [Green Version]
- Gariani, K.; Jornayvaz, F.R. Pathophysiology of NASH in Endocrine Diseases. Endocr. Connect. 2021, 10, R52–R65. [Google Scholar] [CrossRef] [PubMed]
- Pappachan, J.M.; Babu, S.; Krishnan, B.; Ravindran, N.C. Non-Alcoholic Fatty Liver Disease: A Clinical Update. J. Clin. Transl. Hepatol. 2017, 5, 384–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease—Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, P.; Sanyal, A.J. Nonalcoholic Fatty Liver Disease: Definitions, Risk Factors, and Workup. Clin. Liver Dis. 2012, 1, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Colak, Y.; Tuncer, I.; Senates, E.; Ozturk, O.; Doganay, L.; Yilmaz, Y. Nonalcoholic Fatty Liver Disease: A Nutritional Approach. Metab. Syndr. Relat. Disord. 2012, 10, 161–166. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [Green Version]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte Dysfunctions Linking Obesity to Insulin Resistance and Type 2 Diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.J.; Harrison, S.A.; Brunt, E.M.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a Premature Change in Terminology. Hepatology 2021, 73, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Xian, Y.X.; Weng, J.P.; Xu, F. MAFLD vs. NAFLD: Shared Features and Potential Changes in Epidemiology, Pathophysiology, Diagnosis, and Pharmacotherapy. Chin. Med. J. 2020, 134, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Simões, I.C.M.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in Non-Alcoholic Fatty Liver Disease. Int. J. Biochem. Cell Biol. 2018, 95, 93–99. [Google Scholar] [CrossRef]
- Zhang, N.P.; Liu, X.J.; Xie, L.; Shen, X.Z.; Wu, J. Impaired Mitophagy Triggers NLRP3 Inflammasome Activation during the Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis. Lab. Investig. 2019, 99, 749–763. [Google Scholar] [CrossRef]
- Farruggio, S.; Cocomazzi, G.; Marotta, P.; Romito, R.; Surico, D.; Calamita, G.; Bellan, M.; Pirisi, M.; Grossini, E. Genistein and 17β-Estradiol Protect Hepatocytes from Fatty Degeneration by Mechanisms Involving Mitochondria, Inflammasome and Kinases Activation. Cell Physiol. Biochem. 2020, 54, 401–416. [Google Scholar] [CrossRef]
- Lee, J.; Homma, T.; Fujii, J. Mice in the Early Stage of Liver Steatosis Caused by a High Fat Diet Are Resistant to Thioacetamide-Induced Hepatotoxicity and Oxidative Stress. Toxicol. Lett. 2017, 277, 92–103. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondria, Cholesterol and Cancer Cell Metabolism. Clin. Transl. Med. 2016, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Léveillé, M.; Estall, J.L. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites 2019, 9, 233. [Google Scholar] [CrossRef] [Green Version]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty Acid and Endotoxin Activate Inflammasomes in Mouse Hepatocytes That Release Danger Signals to Stimulate Immune Cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Colak, Y.; Hasan, B.; Erkalma, B.; Tandon, K.; Zervos, X.; Menzo, E.L.; Erim, T. Pathogenetic Mechanisms of Nonalcoholic Fatty Liver Disease and Inhibition of the Inflammasome as a New Therapeutic Target. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101710. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Akter, A.; Hossain, S.; Sarker, T.; Safa, S.A.; Mustafa, Q.G.; Muhammad, S.A.; Munir, F. Role of NLRP3 Inflammasome in Liver Disease. J. Dig. Dis. 2020, 21, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 2011, 17, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 Inflammasome Blockade Reduces Liver Inflammation and Fibrosis in Experimental NASH in Mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; de La Rosa, L.C.; Robles, D.; Núñez, S.; Segalés, P.; Insausti-Urkia, N.; Solsona-Vilarrasa, E.; Fernández-Checa, J.C.; García-Ruiz, C. Dietary and Genetic Cholesterol Loading Rather Than Steatosis Promotes Liver Tumorigenesis and NASH-Driven HCC. Cancers 2021, 13, 4091. [Google Scholar] [CrossRef]
- Caballero, F.; Fernández, A.; de Lacy, A.M.; Fernández-Checa, J.C.; Caballería, J.; García-Ruiz, C. Enhanced Free Cholesterol, SREBP-2 and StAR Expression in Human NASH. J. Hepatol. 2009, 50, 789–796. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Mari, M.; Colell, A.; Morales, A.; Caballero, F.; Montero, J.; Terrones, O.; Basañez, G.; Fernández-Checa, J.C. Mitochondrial Cholesterol in Health and Disease. Histol. Histopathol. 2009, 24, 117–132. [Google Scholar] [CrossRef]
- Hendrikx, T.; Bieghs, V.; Walenbergh, S.M.A.; Van Gorp, P.J.; Verheyen, F.; Jeurissen, M.L.J.; Steinbusch, M.M.F.; Vaes, N.; Binder, C.J.; Koek, G.H.; et al. Macrophage Specific Caspase-1/11 Deficiency Protects against Cholesterol Crystallization and Hepatic Inflammation in Hyperlipidemic Mice. PLoS ONE 2013, 8, e78792. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N.; Haigh, W.G.; Thorning, D.; Savard, C. Hepatic Cholesterol Crystals and Crown-like Structures Distinguish NASH from Simple Steatosis. J. Lipid Res. 2013, 54, 1326–1334. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.W.; Yen, C.C.; Kuo, L.L.; Lo, C.W.; Huang, C.S.; Chen, C.C.; Lii, C.K. Benzyl Isothiocyanate Ameliorates High-Fat/Cholesterol/Cholic Acid Diet-Induced Nonalcoholic Steatohepatitis through Inhibiting Cholesterol Crystal-Activated NLRP3 Inflammasome in Kupffer Cells. Toxicol. Appl. Pharmacol. 2020, 393, 114941. [Google Scholar] [CrossRef]
- Bashiri, A.; Nesan, D.; Tavallaee, G.; Sue-Chue-Lam, I.; Chien, K.; Maguire, G.F.; Naples, M.; Zhang, J.; Magomedova, L.; Adeli, K.; et al. Cellular Cholesterol Accumulation Modulates High Fat High Sucrose (HFHS) Diet-Induced ER Stress and Hepatic Inflammasome Activation in the Development of Non-Alcoholic Steatohepatitis. Biochim. Biophys. Acta 2016, 1861, 594–605. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Van Rooyen, D.M.; Savard, C.; Haigh, W.G.; Yeh, M.M.; Teoh, N.C.; Farrell, G.C. Cholesterol-Lowering Drugs Cause Dissolution of Cholesterol Crystals and Disperse Kupffer Cell Crown-like Structures during Resolution of NASH. J. Lipid Res. 2015, 56, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactiv.ve Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Hajduch, E.; Lachkar, F.; Ferré, P.; Foufelle, F. Roles of Ceramides in Non-Alcoholic Fatty Liver Disease. J. Clin. Med. 2021, 10, 792. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Mato, J.M.; Vance, D.; Kaplowitz, N.; Fernández-Checa, J.C. Acid Sphingomyelinase-Ceramide System in Steatohepatitis: A Novel Target Regulating Multiple Pathways. J. Hepatol. 2015, 62, 219–233. [Google Scholar] [CrossRef]
- Mallampalli, R.K.; Peterson, E.J.; Carter, A.B.; Salome, R.G.; Mathur, S.N.; Koretzky, G.A. TNF-Alpha Increases Ceramide without Inducing Apoptosis in Alveolar Type II Epithelial Cells. Am. J. Physiol. 1999, 276, L481–L490. [Google Scholar] [CrossRef]
- Krogh-Madsen, R.; Plomgaard, P.; Møller, K.; Mittendorfer, B.; Pedersen, B.K. Influence of TNF-Alpha and IL-6 Infusions on Insulin Sensitivity and Expression of IL-18 in Humans. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E108–E114. [Google Scholar] [CrossRef]
- Chaurasia, B.; Talbot, C.L.; Summers, S.A. Adipocyte Ceramides—The Nexus of Inflammation and Metabolic Disease. Front. Immunol. 2020, 11, 2282. [Google Scholar] [CrossRef]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-Induced Insulin Resistance Mediated by the Proinflammatory Receptor TLR4 Requires Saturated Fatty Acid-Induced Ceramide Biosynthesis in Mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.; Torres, S.; Baulies, A.; Alarcón-Vila, C.; Elena, M.; Fabriàs, G.; Casas, J.; Caballeria, J.; Fernandez-Checa, J.C.; García-Ruiz, C. Myristic Acid Potentiates Palmitic Acid-Induced Lipotoxicity and Steatohepatitis Associated with Lipodystrophy by Sustaning de Novo Ceramide Synthesis. Oncotarget 2015, 6, 41479–41496. [Google Scholar] [CrossRef] [Green Version]
- Ussher, J.R.; Koves, T.R.; Cadete, V.J.J.; Zhang, L.; Jaswal, J.S.; Swyrd, S.J.; Lopaschuk, D.G.; Proctor, S.D.; Keung, W.; Muoio, D.M.; et al. Inhibition of de Novo Ceramide Synthesis Reverses Diet-Induced Insulin Resistance and Enhances Whole-Body Oxygen Consumption. Diabetes 2010, 59, 2453–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurek, K.; Piotrowska, D.M.; Wiesiołek-Kurek, P.; Łukaszuk, B.; Chabowski, A.; Górski, J.; Zendzian-Piotrowska, M. Inhibition of Ceramide de Novo Synthesis Reduces Liver Lipid Accumulation in Rats with Nonalcoholic Fatty Liver Disease. Liver Int. 2014, 34, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Kasumov, T.; Li, L.; Li, M.; Gulshan, K.; Kirwan, J.P.; Liu, X.; Previs, S.; Willard, B.; Smith, J.D.; McCullough, A. Ceramide as a Mediator of Non-Alcoholic Fatty Liver Disease and Associated Atherosclerosis. PLoS ONE 2015, 10, e0126910. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.H.; Yoon, J.E.; Ko, M.S.; Leem, J.; Yun, J.Y.; Hong, C.H.; Cho, Y.K.; Lee, S.E.; Jang, J.E.; Baek, J.Y.; et al. Sphingomyelin Synthase 1 Mediates Hepatocyte Pyroptosis to Trigger Non-Alcoholic Steatohepatitis. Gut 2021, 70, 1954–1964. [Google Scholar] [CrossRef]
- Hwan Hong, C.; Ko, M.S.; Kim, J.H.; Cho, H.; Lee, C.-H.; Yoon, J.E.; Yun, J.-Y.; Baek, I.-J.; Jang, J.E.; Lee, S.E.; et al. Sphingosine 1-Phosphate Receptor 4 Promotes Nonalcoholic Steatohepatitis by Activating NLRP3 Inflammasome. Cell Mol. Gastroenterol. Hepatol. 2021, 13, 925–947. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and Inflammasomes in Alcoholic Liver Disease/Acute Alcoholic Hepatitis and Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Hepatology 2016, 64, 955–965. [Google Scholar] [CrossRef]
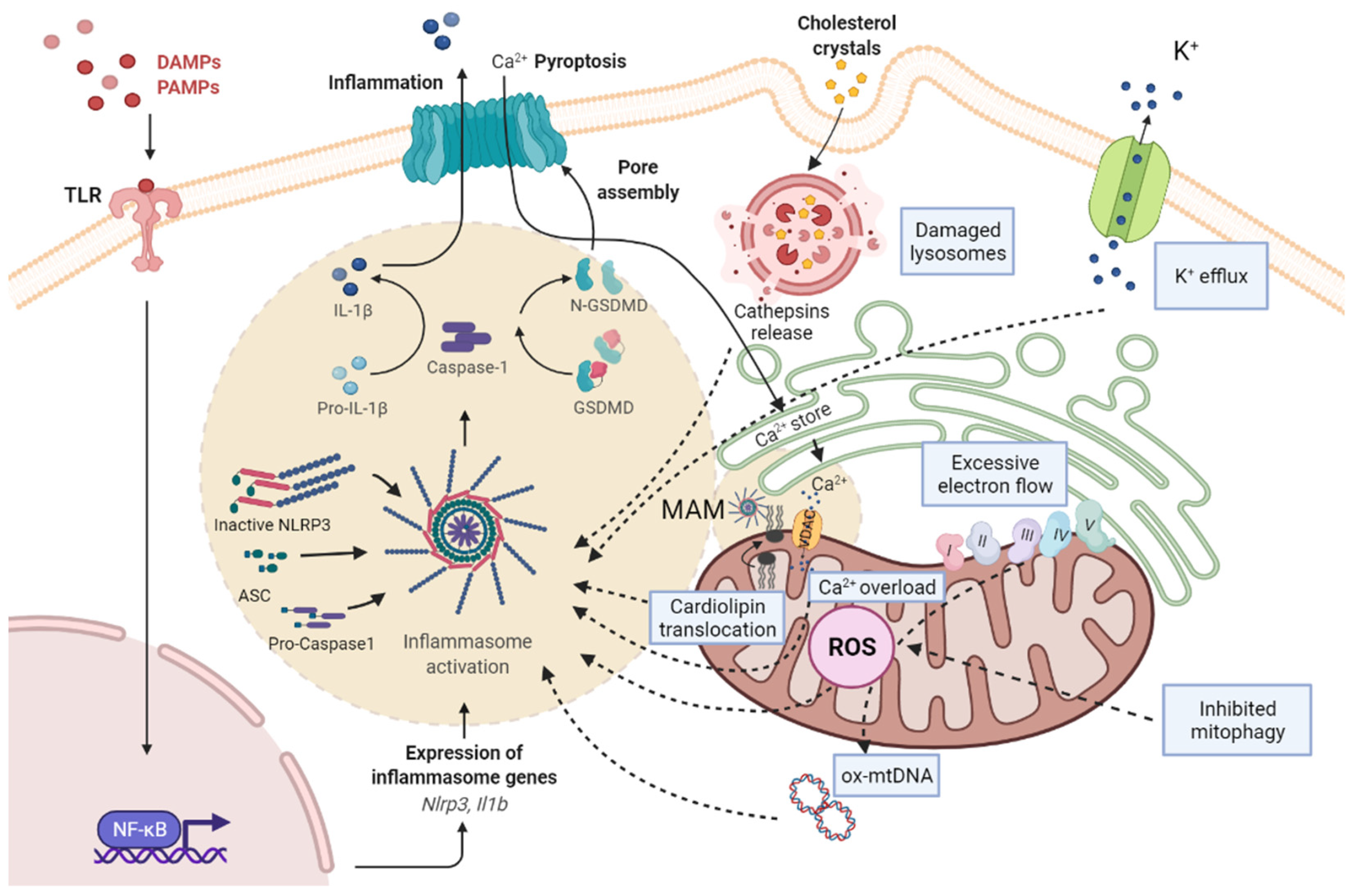
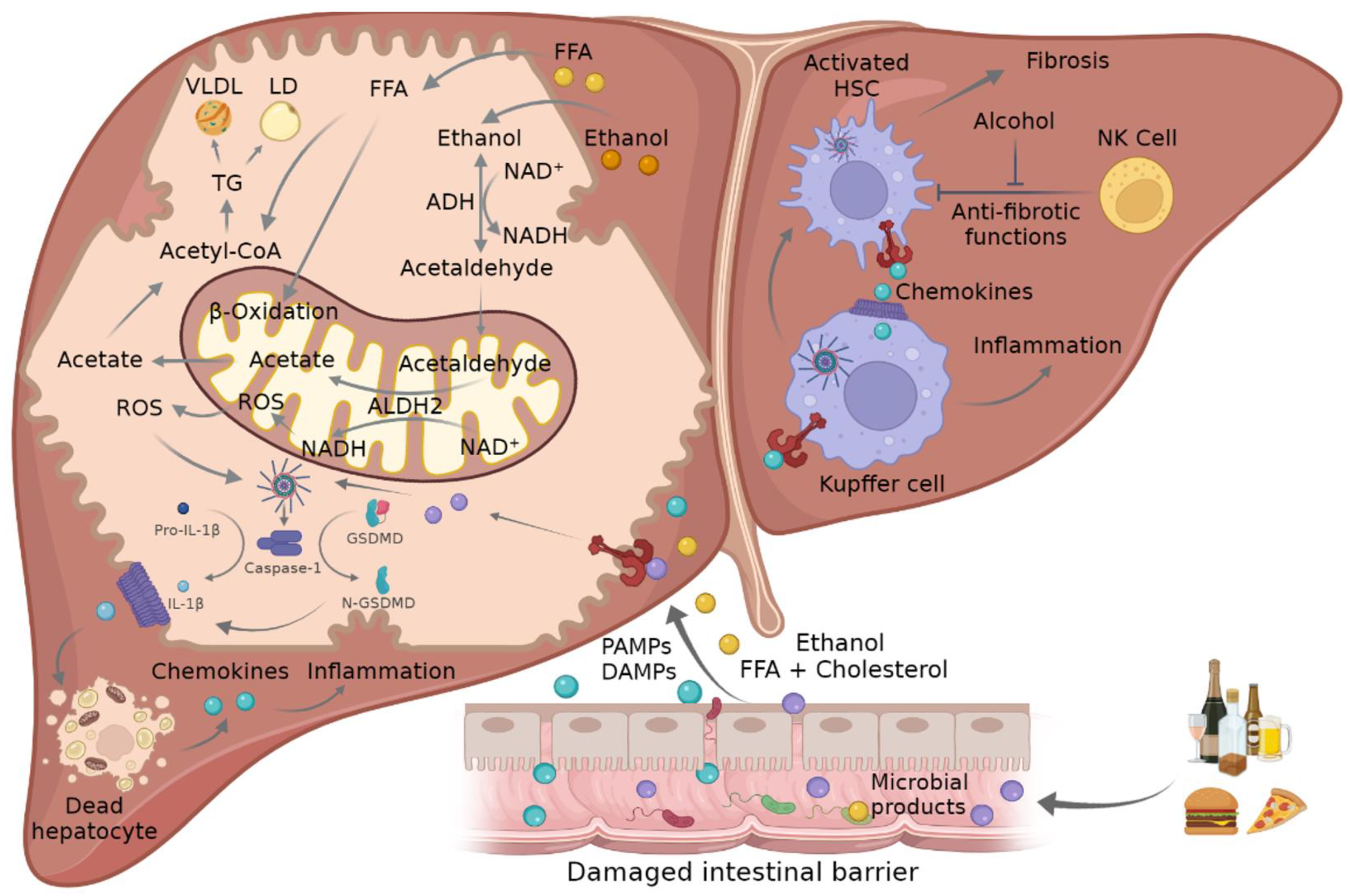

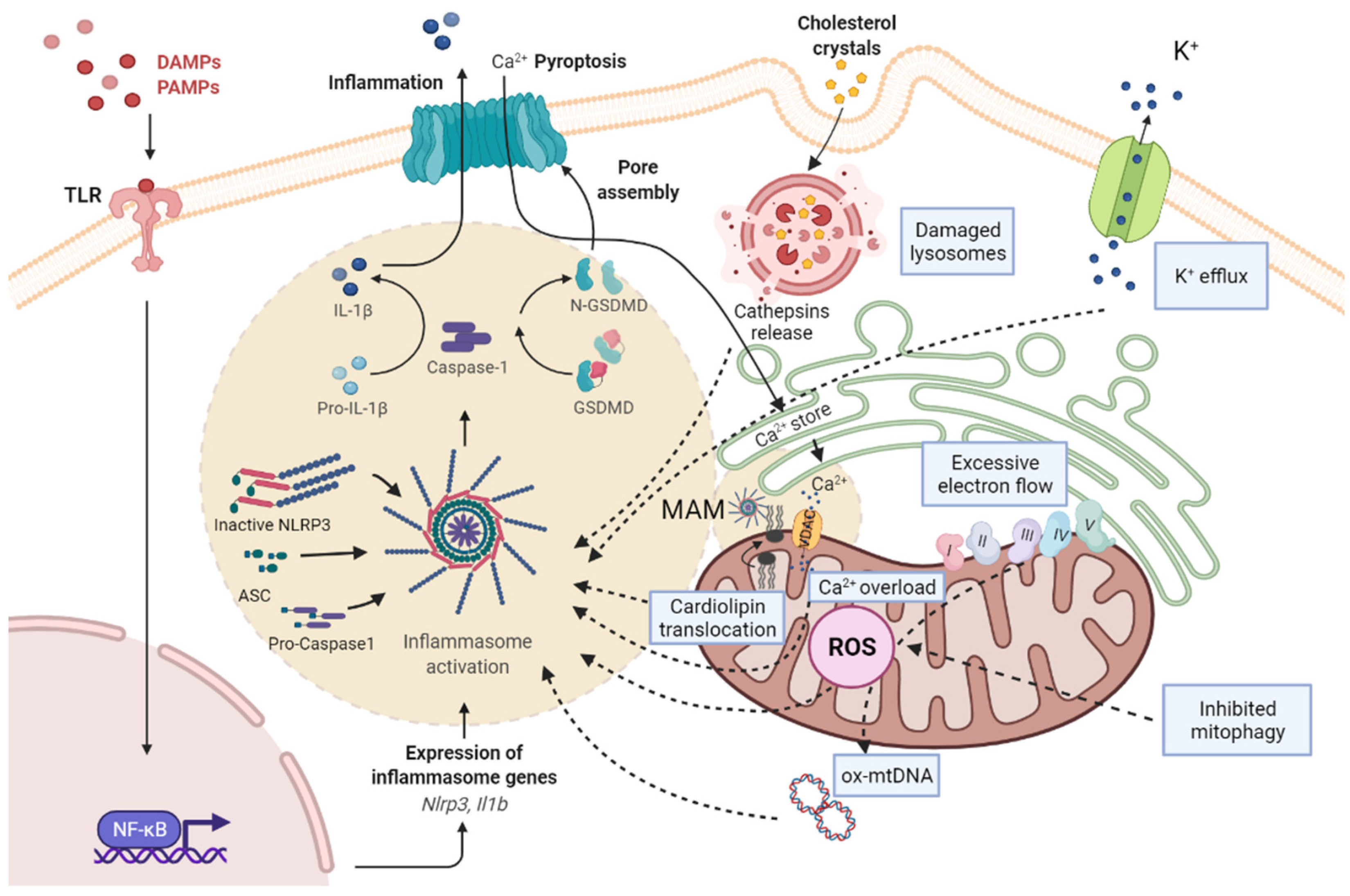{kind=link}
{kind=link}
| ASH | NASH | |
|---|---|---|
| Pathological characteristics | >5% Steatosis Hepatocyte ballooning Liver fibrosis (HSC activation, extracellular matrix production) Mallory–Denk bodies formation Lobular inflammation (neutrophils and macrophages infiltration) | |
| >20 g alcohol/day Ethanol is metabolized by ADH and ALDH Activation CYP2E1 and ROS production Disruption of hepatic lipid metabolism (4-HNE, MDA) Induction of lipogenic factors (SREBP1c, PPARα) Gut dysbiosis (ethanol) | <20 g alcohol/day Obesity Insulin resistance Associated T2DM Hepatic FFA and TG Oxidative phosphorylation, TCA and FA β-oxidation Induction of lipogenic factors (SREBP2) Gut dysbiosis (LPS, cholesterol crystals) | |
| Mitochondrial mechanisms | Mitochondrial cholesterol accumulation (STARD1) Calcium overload (VDAC) Reduction in ATP production Cardiolipin translocation Decreased mGSH Ox-mtDNA Loss of MMP Impaired mitophagy Inflammasome complex activation (MAMs) | |
| ROS derived from acetaldehyde metabolization | ROS derived from FA β-oxidation | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, S.; Segalés, P.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondria and the NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Cells 2022, 11, 1475. https://doi.org/10.3390/cells11091475
Torres S, Segalés P, García-Ruiz C, Fernández-Checa JC. Mitochondria and the NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Cells. 2022; 11(9):1475. https://doi.org/10.3390/cells11091475
Chicago/Turabian StyleTorres, Sandra, Paula Segalés, Carmen García-Ruiz, and José C. Fernández-Checa. 2022. "Mitochondria and the NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis" Cells 11, no. 9: 1475. https://doi.org/10.3390/cells11091475
APA StyleTorres, S., Segalés, P., García-Ruiz, C., & Fernández-Checa, J. C. (2022). Mitochondria and the NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Cells, 11(9), 1475. https://doi.org/10.3390/cells11091475