
Prognostic and Immunotherapeutic Roles of KRAS in Pan-Cancer

 ,
, 
Abstract
:1. Introduction
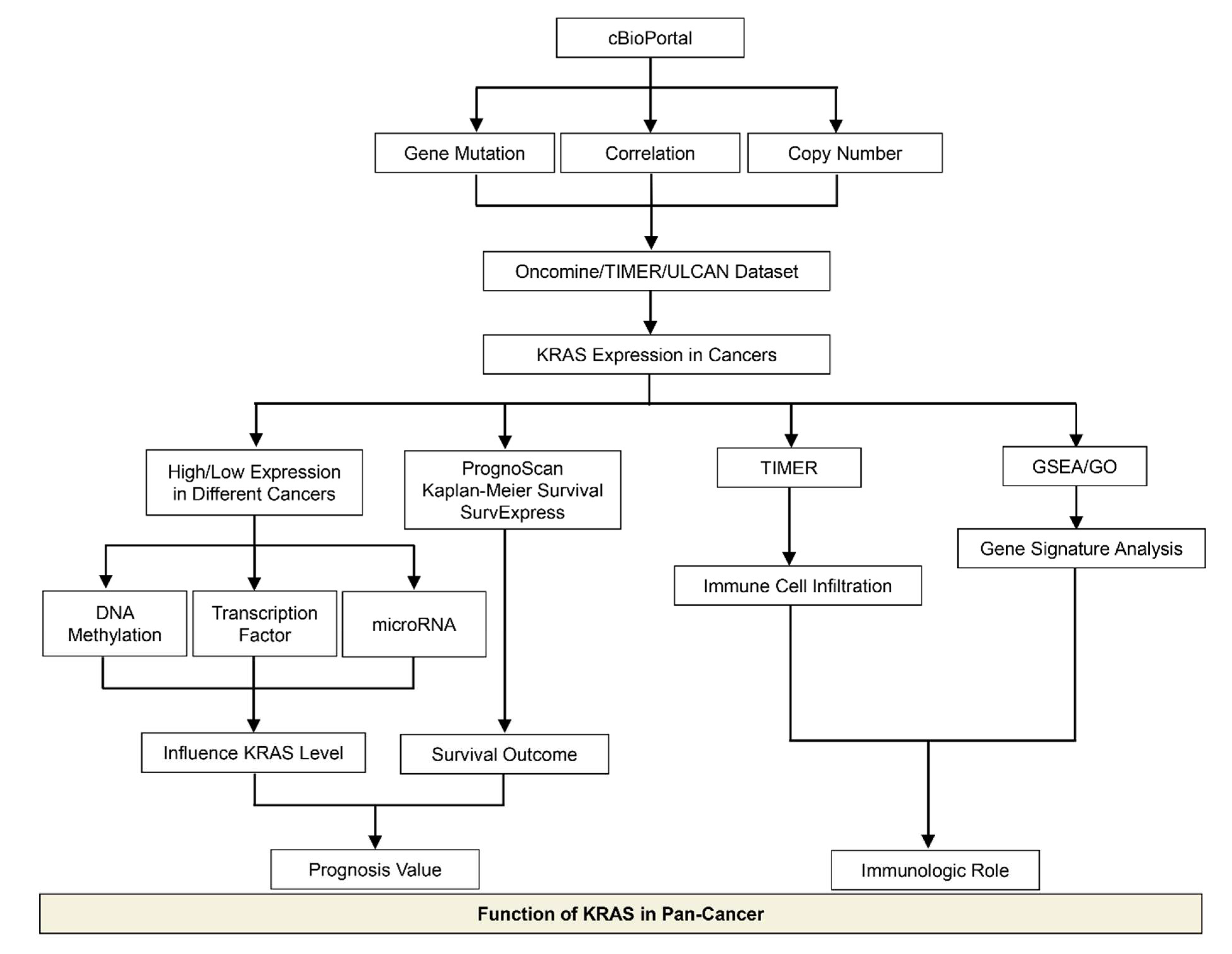
2. Materials and Methods
2.1. cBioPortal
2.2. Oncomine Database
2.3. TIMER
2.4. Survival Analysis in PrognoScan and Kaplan–Meier Plotter
2.5. Gene Signature Analysis
2.6. SurvExpress
2.7. UALCAN
2.8. SurvivalMeth
2.9. ChIP-Atlas
2.10. Protein–Protein Interaction (PPI)
2.11. TargetScan
2.12. Statistical Analysis
3. Results
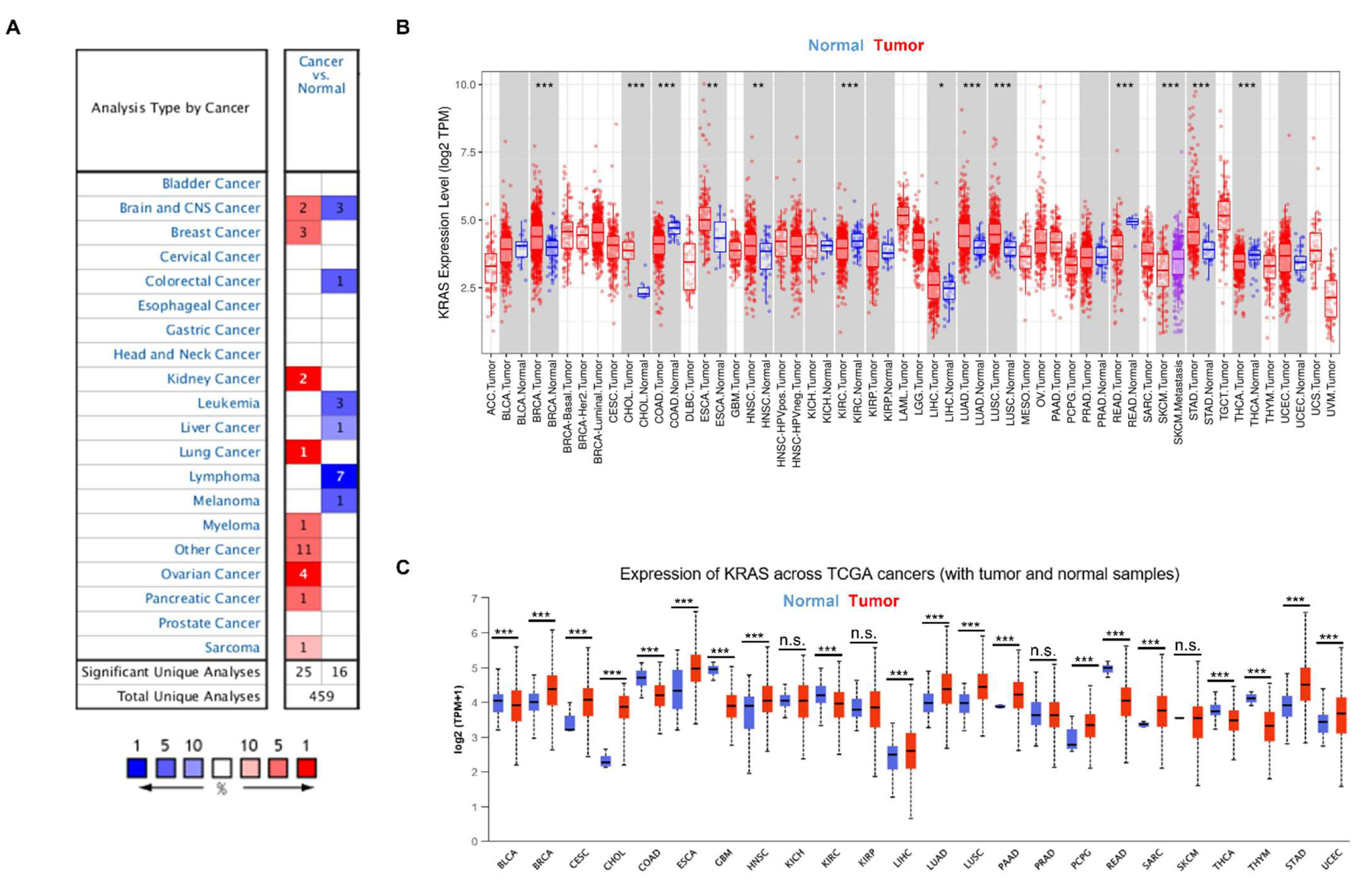
3.1. KRAS Mutation in Diverse Cancer Types
3.2. KRAS Expression in Diverse Cancers
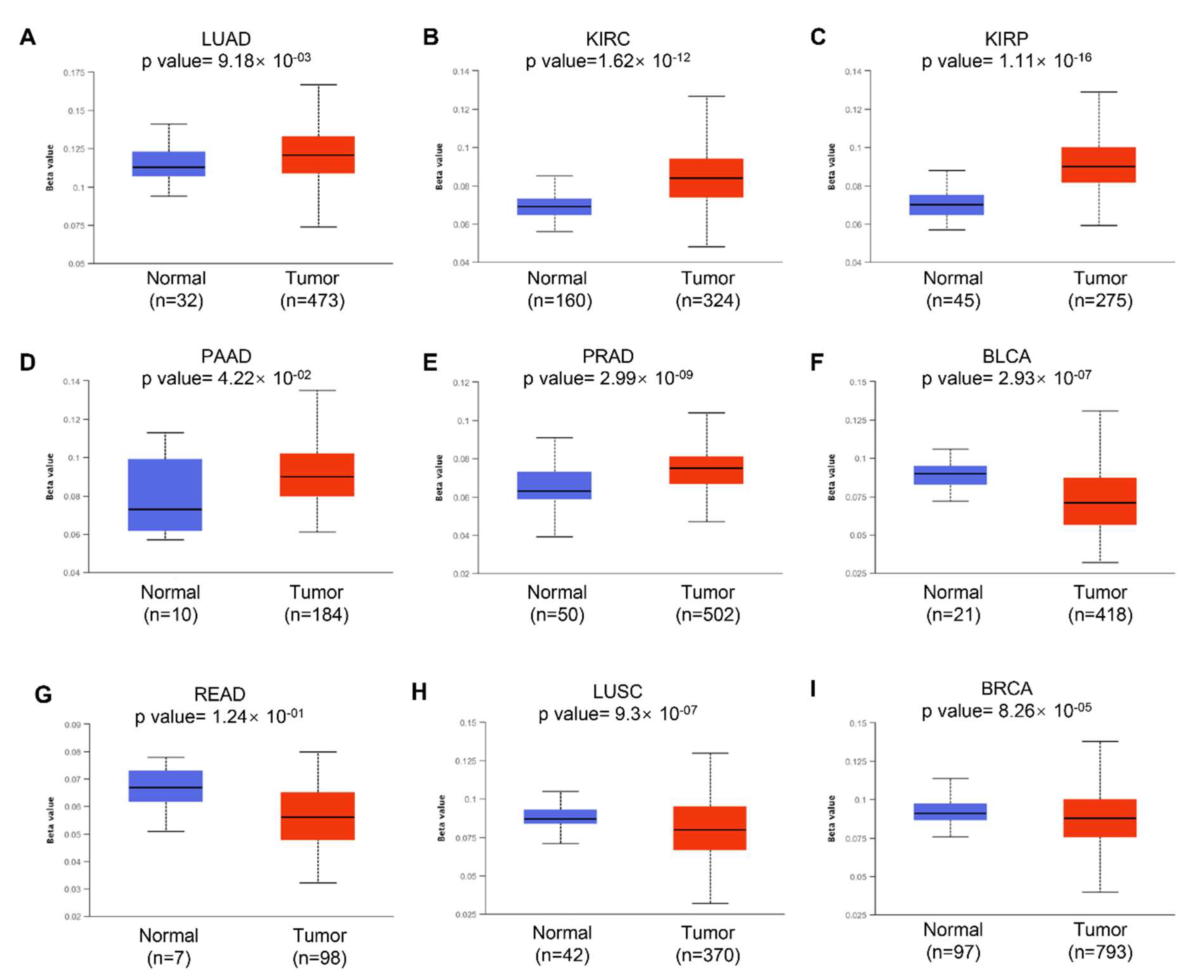
3.3. DNA Methylation of KRAS
3.4. Regulators of KRAS
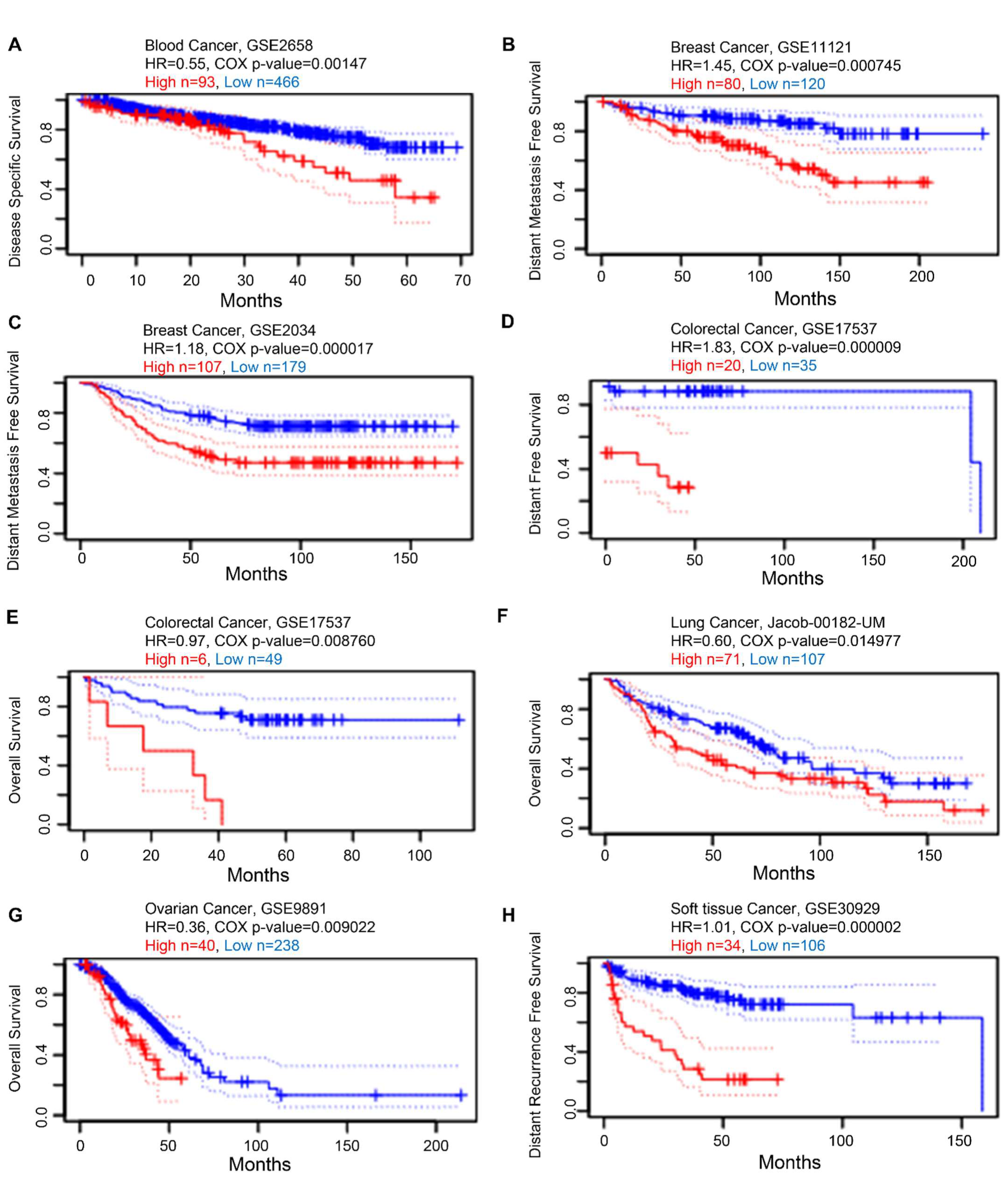
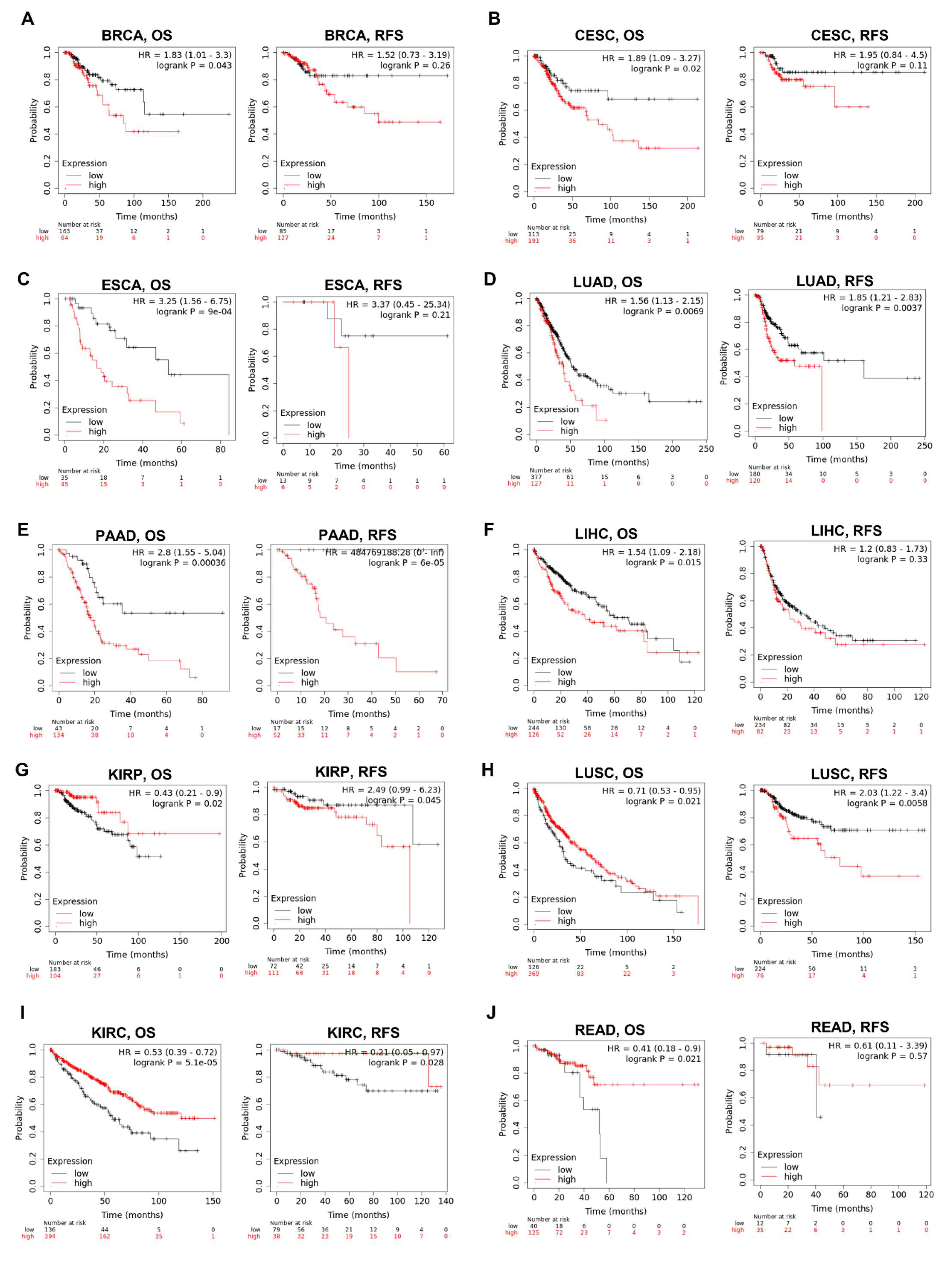
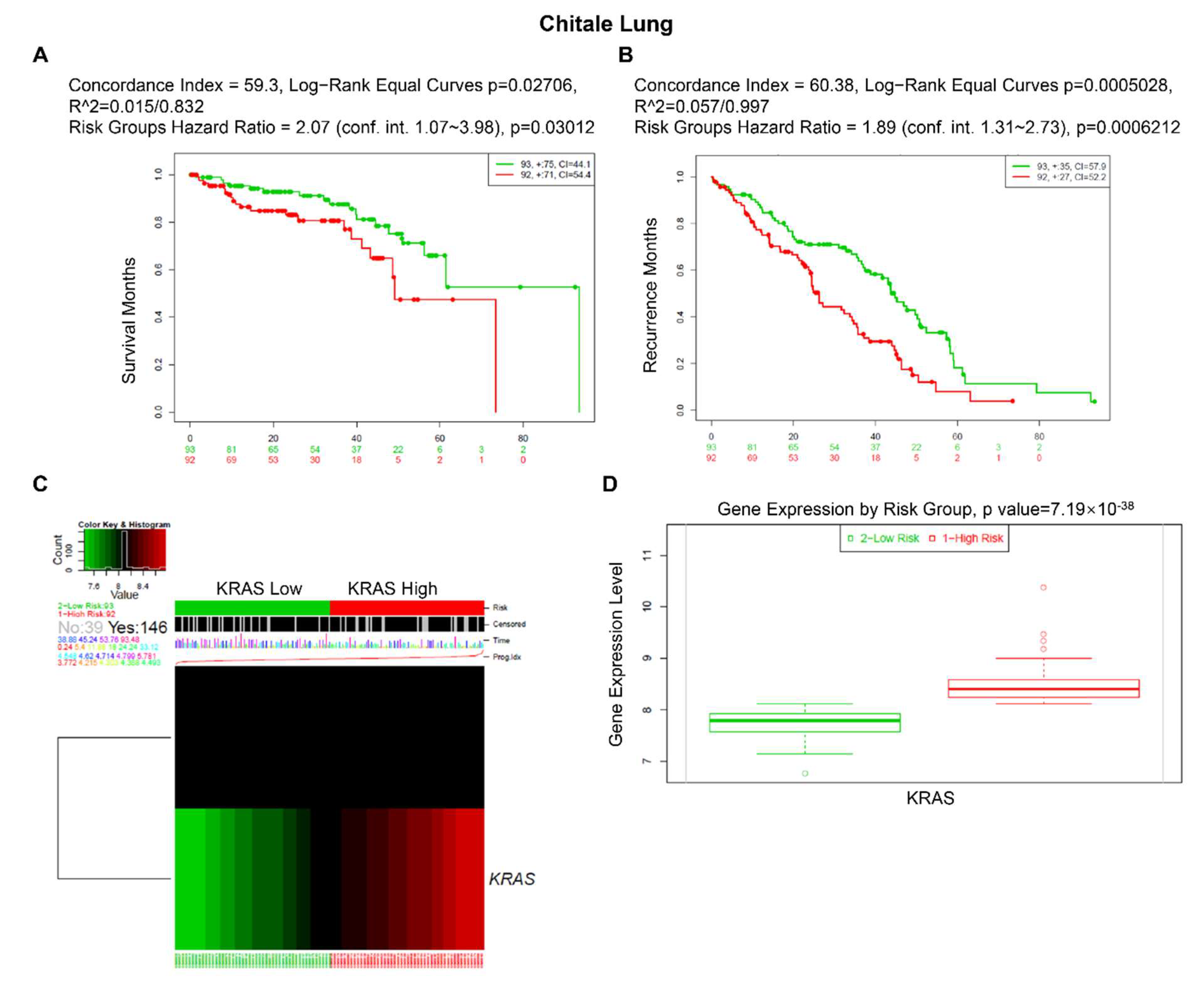
3.5. Prognostic Value of KRAS in Various Cancers
3.6. Immune Cell Infiltration of KRAS in the Pan-Cancer Analysis
3.7. Gene Signature Analysis in LUAD, LUSC, BRCA, and PAAD TCGA Datasets
4. Discussion
5. Conclusions
- Pan-cancer analysis of KRAS indicated that 33 cancers had different expressions of these genes between normal and tumor samples.
- KRAS could serve as a key prognostic factor in different cancer types.
- KRAS could affect tumor development through tumor immune cell infiltration.
- Our study illustrates the characterization of KRAS expression in various cancer types and highlights its potential value as a predictive biomarker, which sheds light on the further investigation of the prognostic and therapeutic potential of KRAS inflammation.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Pon, J.R.; Marra, M.A. Driver and passenger mutations in cancer. Annu. Rev. Pathol. 2015, 10, 25–50. [Google Scholar] [CrossRef]
- Martinez-Jimenez, F.; Muinos, F.; Sentis, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Larsson, L.G. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin. Cancer Biol. 2011, 21, 367–376. [Google Scholar] [CrossRef]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Pantsar, T. The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 2020, 18, 189–198. [Google Scholar] [CrossRef] [PubMed]
- McCormick, F. KRAS as a Therapeutic Target. Clin. Cancer Res. 2015, 21, 1797–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosten, M.; Barbacid, M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, H.; Kitada, K.; Nakai, K.; Sarai, A. PrognoScan: A new database for meta-analysis of the prognostic value of genes. BMC Med. Genom. 2009, 2, 18. [Google Scholar] [CrossRef] [Green Version]
- Lanczky, A.; Gyorffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martinez-Ledesma, E.; Martinez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Pena, J.G.; Trevino, V. SurvExpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Men, C.; Chai, H.; Song, X.; Li, Y.; Du, H.; Ren, Q. Identification of DNA methylation associated gene signatures in endometrial cancer via integrated analysis of DNA methylation and gene expression systematically. J. Gynecol. Oncol. 2017, 28, e83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinawi, T.; Hill, V.K.; Krex, D.; Schackert, G.; Gentle, D.; Morris, M.R.; Wei, W.; Cruickshank, G.; Maher, E.R.; Latif, F. DNA methylation profiles of long- and short-term glioblastoma survivors. Epigenetics 2013, 8, 149–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Zhao, N.; Zhang, X.; Xiao, J.; Li, J.; Lv, D.; Zhou, W.; Li, Y.; Xu, J.; Li, X. SurvivalMeth: A web server to investigate the effect of DNA methylation-related functional elements on prognosis. Brief. Bioinform. 2021, 22, bbaa162. [Google Scholar] [CrossRef] [PubMed]
- Oki, S.; Ohta, T.; Shioi, G.; Hatanaka, H.; Ogasawara, O.; Okuda, Y.; Kawaji, H.; Nakaki, R.; Sese, J.; Meno, C. ChIP-Atlas: A data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018, 19, e46255. [Google Scholar] [CrossRef]
- Zhou, K.R.; Liu, S.; Sun, W.J.; Zheng, L.L.; Zhou, H.; Yang, J.H.; Qu, L.H. ChIPBase v2.0: Decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 2017, 45, D43–D50. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Magee, P.; Fassan, M.; Sahoo, S.; Leong, H.S.; Lee, D.; Sellers, R.; Brulle-Soumare, L.; Cairo, S.; Monteverde, T.; et al. A KRAS-responsive long non-coding RNA controls microRNA processing. Nat. Commun. 2021, 12, 2038. [Google Scholar] [CrossRef] [PubMed]
- Nagdas, S.; Kashatus, J.A.; Nascimento, A.; Hussain, S.S.; Trainor, R.E.; Pollock, S.R.; Adair, S.J.; Michaels, A.D.; Sesaki, H.; Stelow, E.B.; et al. Drp1 Promotes KRas-Driven Metabolic Changes to Drive Pancreatic Tumor Growth. Cell Rep. 2019, 28, 1845–1859.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Liu, G.; Zhou, F.; Su, B.; Li, Y. DNA methylation profiles in cancer diagnosis and therapeutics. Clin. Exp. Med. 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Azizi, N.; Toma, J.; Martin, M.; Khalid, M.F.; Mousavi, F.; Win, P.W.; Borrello, M.T.; Steele, N.; Shi, J.; di Magliano, M.P.; et al. Loss of activating transcription factor 3 prevents KRAS-mediated pancreatic cancer. Oncogene 2021, 40, 3118–3135. [Google Scholar] [CrossRef]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.Y.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 suppression upregulates beta-catenin and c-Myc to abrogate KRas-dependent tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Zhu, H.; Gan, X.; Jiang, X.; Diao, S.; Wu, H.; Hu, J. ALKBH5 inhibited autophagy of epithelial ovarian cancer through miR-7 and BCL-2. J. Exp. Clin. Cancer Res. 2019, 38, 163. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Mao, C.; Ouyang, L.; Liu, Y.; Lai, W.; Liu, N.; Shi, Y.; Chen, L.; Xiao, D.; Yu, F.; et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019, 26, 2329–2343. [Google Scholar] [CrossRef]
- Diakos, C.I.; Charles, K.A.; McMillan, D.C.; Clarke, S.J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014, 15, e493–e503. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Huang, X.; Liu, H.; Wei, C.; Ru, H.; Qin, H.; Lai, H.; Meng, Y.; Wu, G.; Xie, W.; et al. Immune landscape and prognostic immune-related genes in KRAS-mutant colorectal cancer patients. J. Transl. Med. 2021, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Sim, M.J.W.; Lu, J.; Spencer, M.; Hopkins, F.; Tran, E.; Rosenberg, S.A.; Long, E.O.; Sun, P.D. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc. Natl. Acad. Sci. USA 2020, 117, 12826–12835. [Google Scholar] [CrossRef]
- McAllister, F.; Bailey, J.M.; Alsina, J.; Nirschl, C.J.; Sharma, R.; Fan, H.; Rattigan, Y.; Roeser, J.C.; Lankapalli, R.H.; Zhang, H.; et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 2014, 25, 621–637. [Google Scholar] [CrossRef] [Green Version]
- Hamarsheh, S.; Gross, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: Finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef]
- Timar, J.; Kashofer, K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metastasis Rev. 2020, 39, 1029–1038. [Google Scholar] [CrossRef]
- Kerk, S.A.; Papagiannakopoulos, T.; Shah, Y.M.; Lyssiotis, C.A. Metabolic networks in mutant KRAS-driven tumours: Tissue specificities and the microenvironment. Nat. Rev. Cancer 2021, 21, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Ginder, G.D.; Williams, D.C., Jr. Readers of DNA methylation, the MBD family as potential therapeutic targets. Pharmacol. Ther. 2018, 184, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Huang, M.; Xing, L.; Yang, R.; Wang, X.; Jiang, R.; Zhang, L.; Chen, J. The circRNA circSEPT9 mediated by E2F1 and EIF4A3 facilitates the carcinogenesis and development of triple-negative breast cancer. Mol. Cancer 2020, 19, 73. [Google Scholar] [CrossRef] [Green Version]
- Caramel, J.; Ligier, M.; Puisieux, A. Pleiotropic Roles for ZEB1 in Cancer. Cancer Res. 2018, 78, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Chun, S.Y.; Johnson, C.; Washburn, J.G.; Cruz-Correa, M.R.; Dang, D.T.; Dang, L.H. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1alpha and HIF-2alpha target genes. Mol. Cancer 2010, 9, 293. [Google Scholar] [CrossRef] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Marraco, S.A.F.; Calderon-Copete, S.; Ferreira, D.P.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Zhang, Q.; Lou, Y.; Wang, J.; Zhao, X.; Wang, L.; Zhang, X.; Li, S.; Zhao, Y.; Chen, Q.; et al. USP22 Deubiquitinates CD274 to Suppress Anticancer Immunity. Cancer Immunol. Res. 2019, 7, 1580–1590. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Nennig, S.E.; Schank, J.R. The Role of NFkB in Drug Addiction: Beyond Inflammation. Alcohol Alcohol. 2017, 52, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.A.; Rajan, A.; Keen, C.; Szabo, E.; Khozin, S.; Thomas, A.; Brzezniak, C.; Guha, U.; Doyle, L.A.; Steinberg, S.M.; et al. Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced nonsmall-cell lung cancer. Ann. Oncol. 2016, 27, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Affected Cases | CNV Gain | CNV Loss | Mutations |
|---|---|---|---|---|
| TP53 | 4796/12,538 (38.25%) | 144/10,473 (1.37%) | 495/10,473 (4.73%) | 1423 |
| PIK3CA | 1621/12,538 (12.93%) | 1367/10,473 (13.05%) | 201/10,473 (1.92%) | 450 |
| FAT4 | 1415/12,538 (11.29%) | 213/10,473 (2.03%) | 378/10,473 (3.61%) | 2225 |
| KRAS | 1333/12,538 (10.63%) | 518/10,473 (4.95%) | 182/10,473 (1.74%) | 182 |
| KMT2D | 1310/12,538 (10.45%) | 283/10,473 (2.70%) | 300/10,473 (2.86%) | 1673 |
| PTEN | 1205/12,538 (9.61%) | 203/10,473 (1.94%) | 949/10,473 (9.06%) | 893 |
| KMT2C | 1137/12,538 (9.07%) | 500/10,473 (4.77%) | 718/10,473 (6.86%) | 1535 |
| ARID1A | 1083/12,538 (8.64%) | 212/10,473 (2.02%) | 1128/10,473 (10.77%) | 1006 |
| FAT1 | 1058/12,538 (8.44%) | 304/10,473 (2.90%) | 836/10,473 (7.98%) | 1478 |
| APC | 1049/12,538 (8.37%) | 268/10,473 (2.56%) | 518/10,473 (4.95%) | 1128 |
| BRAF | 924/12,538 (7.37%) | 539/10,473 (5.15%) | 501/10,473 (4.78%) | 235 |
| NF1 | 913/12,538 (7.28%) | 418/10,473 (3.99%) | 450/10,473 (4.30%) | 1110 |
| ATRX | 881/12,538 (7.03%) | 184/10,473 (1.76%) | 167/10,473 (1.59%) | 1143 |
| RNF213 | 881/12,538 (7.03%) | 1047/10,473 (10.00%) | 206/10,473 (1.97%) | 1299 |
| ZFHX3 | 833/12,538 (6.64%) | 232/10,473 (2.22%) | 430/10,473 (4.11%) | 1114 |
| GRIN2A | 799/12,538 (6.37%) | 355/10,473 (3.39%) | 159/10,473 (1.52%) | 906 |
| TRRAP | 763/12,538 (6.09%) | 475/10,473 (4.54%) | 177/10,473 (1.69%) | 1020 |
| ATM | 732/12,538 (5.84%) | 247/10,473 (2.36%) | 731/10,473 (6.98%) | 891 |
| ERBB4 | 709/12,538 (5.65%) | 338/10,473 (3.23%) | 795/10,473 (7.59%) | 836 |
| AKAP9 | 709/12,538 (5.65%) | 473/10,473 (4.52%) | 160/10,473 (1.53%) | 957 |
| Cancer | Cancer Number | Normal Number | Cancer Expression | Normal Expression | Fold Change | p-Value | FDR |
|---|---|---|---|---|---|---|---|
| BLCA | 411 | 19 | 7.9 | 6.67 | 1.18 | 0.63 | 0.83 |
| BRCA | 1104 | 113 | 9.25 | 5.86 | 1.58 | 2.00 × 10−18 | 1.60 × 10−17 |
| CHOL | 36 | 9 | 6.39 | 2.5 | 2.55 | 4.20 × 10−9 | 4.20 × 10−8 |
| COAD | 471 | 41 | 8.49 | 11.55 | 0.73 | 1.00 × 10−7 | 6.70 × 10−7 |
| ESCA | 162 | 11 | 21.36 | 10.35 | 2.06 | 0.23 | 0.52 |
| HNSC | 502 | 44 | 7.93 | 6.61 | 1.2 | 0.049 | 0.11 |
| KICH | 65 | 24 | 7.71 | 6.74 | 1.14 | 0.26 | 0.43 |
| KIRC | 535 | 72 | 6.45 | 7.33 | 0.88 | 0.00021 | 0.00051 |
| KIRP | 289 | 32 | 6.58 | 5.73 | 1.15 | 0.42 | 0.63 |
| LIHC | 374 | 50 | 3.31 | 2.5 | 1.32 | 0.029 | 0.065 |
| LUAD | 526 | 59 | 11.56 | 6.59 | 1.75 | 3.10 × 10−9 | 1.90 × 10−8 |
| LUSC | 501 | 49 | 11.27 | 6.43 | 1.75 | 2.10 × 10−11 | 1.10 × 10−10 |
| PAAD | 178 | 4 | 8.17 | 6.62 | 1.23 | 0.49 | 0.94 |
| PRAD | 499 | 52 | 5.45 | 5.25 | 1.04 | 0.85 | 0.91 |
| STAD | 375 | 32 | 16.21 | 6.6 | 2.46 | 0.0003 | 0.0011 |
| THCA | 510 | 58 | 4.42 | 4.74 | 0.93 | 0.023 | 0.052 |
| UCEC | 548 | 35 | 6.59 | 4.73 | 1.4 | 0.034 | 0.09 |
| Tumor | Probe ID | Average of Tumor Samples | Average of Normal Samples | p-Value |
|---|---|---|---|---|
| LUAD | cg20836156 | 0.619649 | 0.666993 | 1.89740 × 10−9 |
| PAAD | cg12990174 | 0.768317 | 0.843160 | 0.00126474 |
| cg20836156 | 0.633778 | 0.707345 | 0.004233613 | |
| READ | cg17197538 | 0.051156 | 0.040148 | 9.32 × 10−7 |
| COAD | cg01269191 | 0.056962 | 0.047081 | 9.95 × 10−5 |
| Clinicopathological Features | BRCA OS | LUAD OS | ||||
|---|---|---|---|---|---|---|
| N | Hazard Ratio | p | N | Hazard Ratio | p | |
| Stage | ||||||
| Stage 1 | 180 | 0.45 (0.16–1.2) | 0.1 | 270 | 1.78 (1.04–3.03) | 0.033 |
| Stage 2 | 619 | 1.76 (1.08–2.86) | 0.02 | 119 | 1.92 (1.09–3.35) | 0.021 |
| Stage 3 | 247 | 1.83 (1.01–3.3) | 0.043 | 81 | 1.51 (0.82–2.79) | 0.18 |
| Stage 4 | 20 | 0 (0–inf) | 7.8 × 10−7 | 26 | 2.93 (0.66–13.14) | 0.14 |
| Mutation Burden | ||||||
| High | 493 | 1.9 (1.18–3.07) | 0.0071 | 255 | 1.72 (1.12–2.64) | 0.012 |
| Low | 485 | 1.54 (0.93–2.54) | 0.093 | 244 | 1.54 (0.97–2.44) | 0.067 |
| Gender | ||||||
| Male | --- | --- | --- | 234 | 2.26 (1.48–3.46) | 0.00011 |
| Female | 1077 | 1.48 (1.07–2.04) | 0.016 | 270 | 1.23 (0.79–1.92) | 0.36 |
| Race | ||||||
| White | 752 | 1.76 (1.2–2.57) | 0.0032 | 387 | 1.53 (1.08–2.15) | 0.015 |
| Asian | 61 | 7204224608 (0–inf) | 0.0054 | --- | --- | --- |
| Black of African American | 181 | 0.4 (0.17–0.94) | 0.03 | 52 | 1.96 (0.61–6.32) | 0.25 |
| Compound | Interaction Types | PMID | Interactions Score |
|---|---|---|---|
| AZD-4785 | n/a | 28615361 | 1.42 |
| Panitumumab | n/a | 20978259, 21398618, 18316791 | 1.31 |
| Cetuximab | n/a | 20978259, 28632865 | 0.84 |
| Pelareorep | Inhibitor | 24798549, 26156229 | 0.63 |
| PD-0325901 | n/a | 21325073, 20570890, 26582713 | 0.52 |
| XMT-1536 | n/a | - | 0.47 |
| Chembl217354 | n/a | 25665005 | 0.47 |
| Rilotumumab | n/a | 24919569 | 0.47 |
| Ralimetnib | n/a | 26725216 | 0.47 |
| SAR-125844 | n/a | 25504634 | 0.47 |
| Necitumumab | n/a | 26766738 | 0.47 |
| Imgatuzumab | n/a | 23209031 | 0.47 |
| Selumetinib | Inhibitor | 25870145, 27312529, 27556948 | 0.43 |
| Ridaforolimus | n/a | 26725216 | 0.32 |
| Phenformin | n/a | 26574479 | 0.32 |
| Teprotumumab | n/a | 21985784 | 0.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.; Li, C.; Liu, Y.; Gu, X.; Jiang, L.; Shi, L. Prognostic and Immunotherapeutic Roles of KRAS in Pan-Cancer. Cells 2022, 11, 1427. https://doi.org/10.3390/cells11091427
Yang K, Li C, Liu Y, Gu X, Jiang L, Shi L. Prognostic and Immunotherapeutic Roles of KRAS in Pan-Cancer. Cells. 2022; 11(9):1427. https://doi.org/10.3390/cells11091427
Chicago/Turabian StyleYang, Kaixin, Chengyun Li, Yang Liu, Xueyan Gu, Longchang Jiang, and Lei Shi. 2022. "Prognostic and Immunotherapeutic Roles of KRAS in Pan-Cancer" Cells 11, no. 9: 1427. https://doi.org/10.3390/cells11091427
APA StyleYang, K., Li, C., Liu, Y., Gu, X., Jiang, L., & Shi, L. (2022). Prognostic and Immunotherapeutic Roles of KRAS in Pan-Cancer. Cells, 11(9), 1427. https://doi.org/10.3390/cells11091427