
Organismal and Cellular Stress Responses upon Disruption of Mitochondrial Lonp1 Protease

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. C. elegans Strains and Culture Conditions
2.2. Human Cell Cultures
2.3. RNA Interference
2.4. Phenotypic Analysis of Worms
2.5. Lifespan Analysis
2.6. Microscopic Analysis
2.7. ROS Measurement
2.8. MitoTracker Staining
2.9. RNA Extraction and Quantitative Reverse Transcription PCR (qRT-PCR)
2.10. Stress Sensitivity Assays
2.11. Protein Extraction and Western Blotting
2.12. MTT Assays
2.13. Cell Cycle Analysis
2.14. Scratch-Wound Assays
2.15. Statistics
3. Results
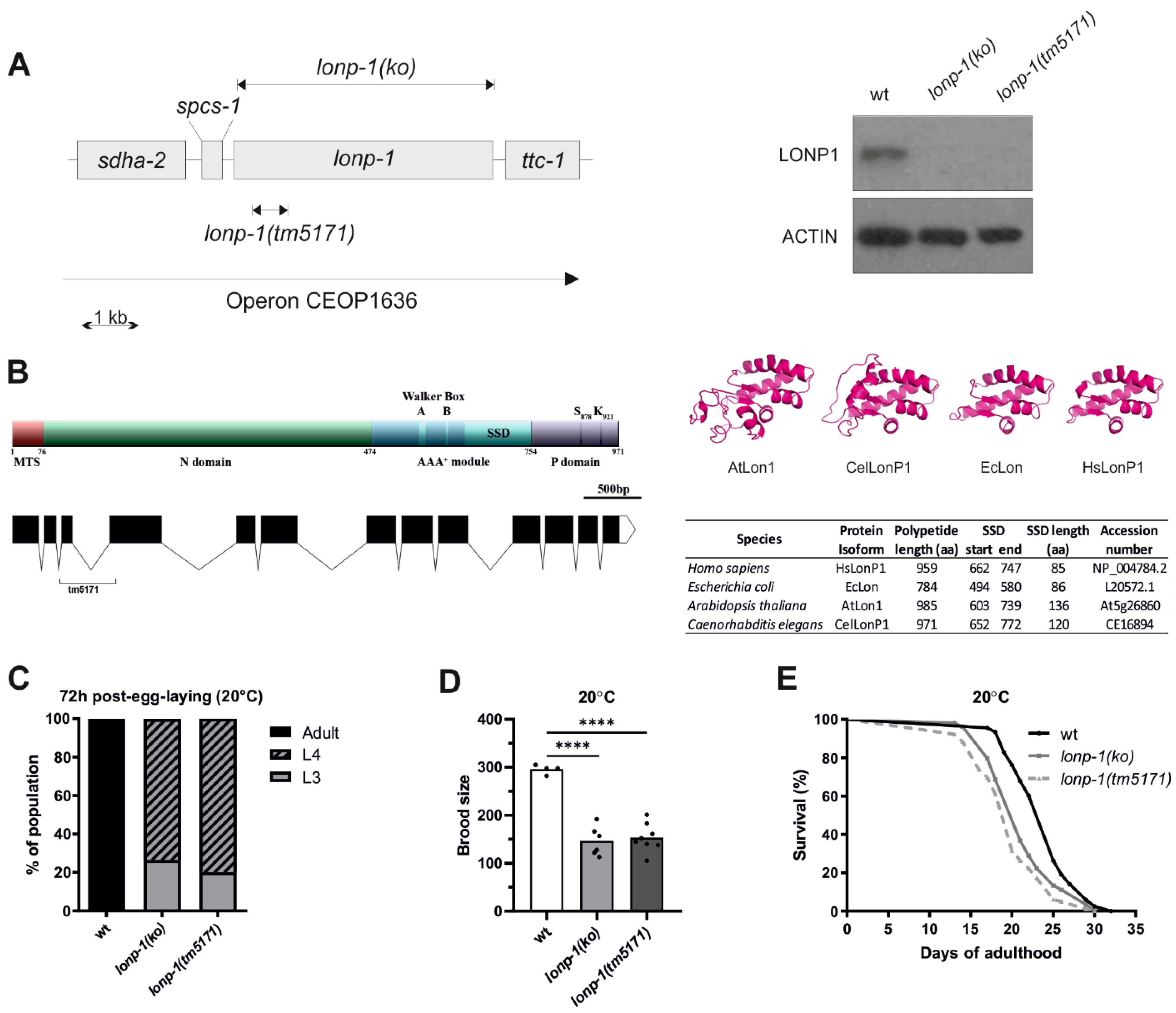
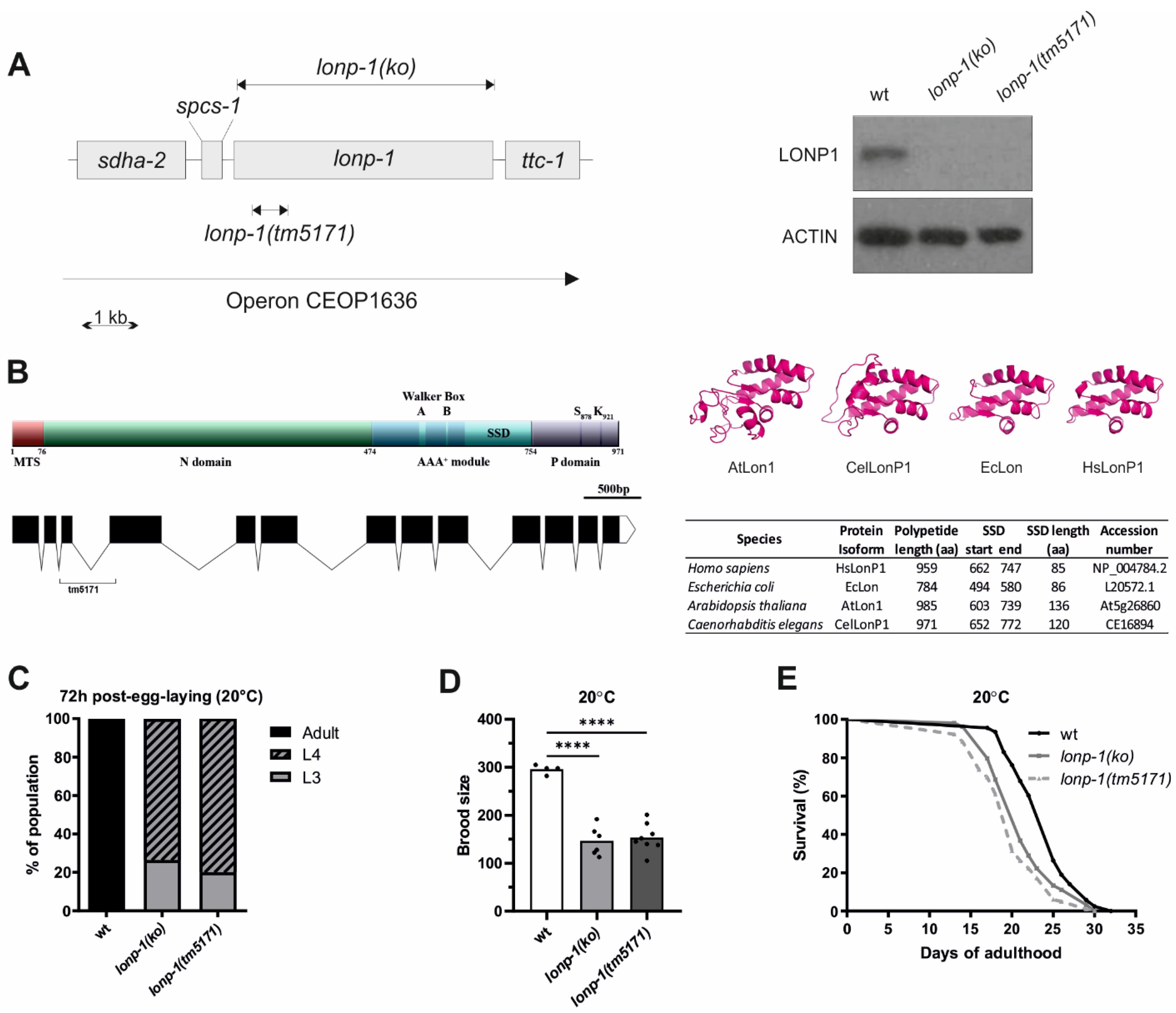
3.1. Mitochondrial Lon Protease Supports Normal Development, Fecundity, and Lifespan of C. elegans
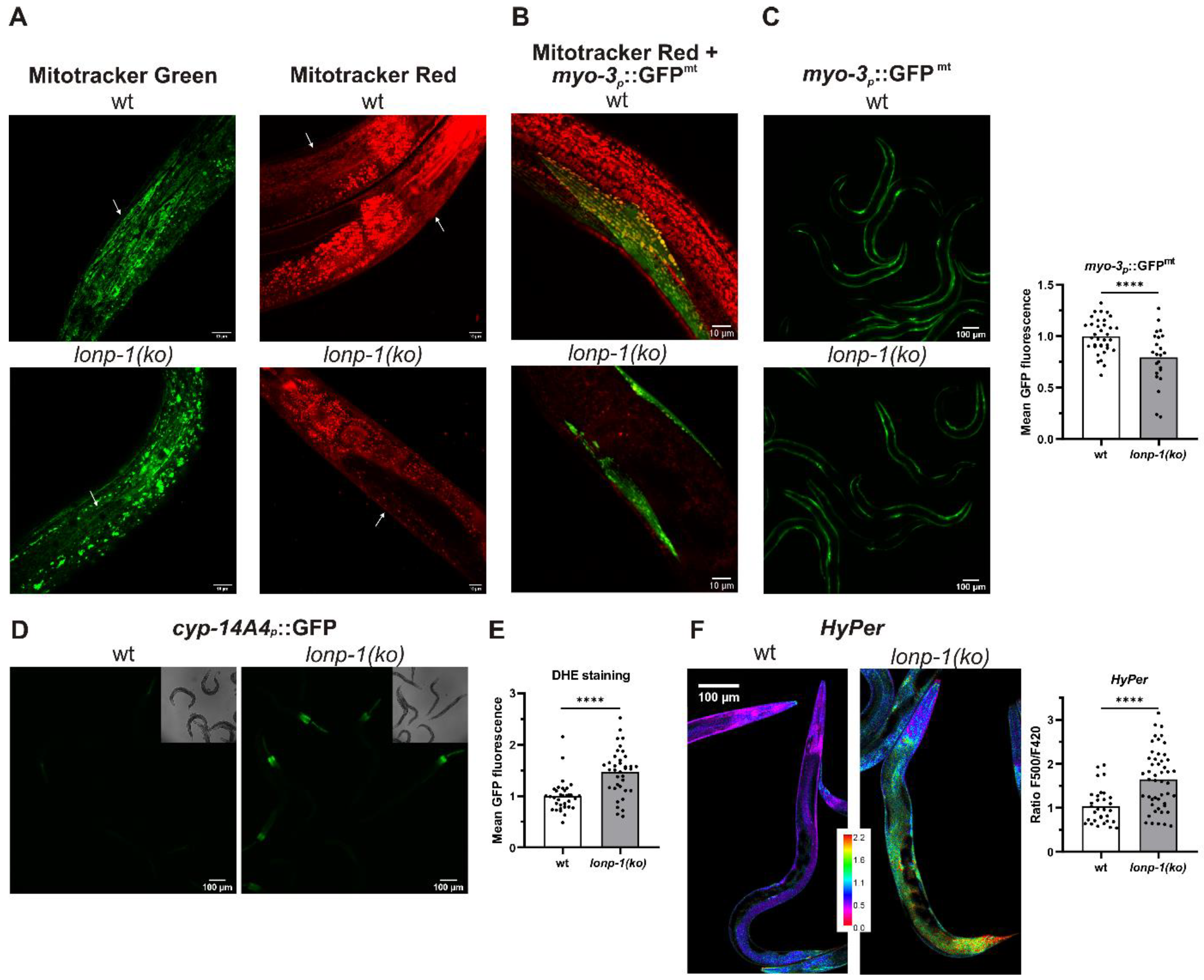
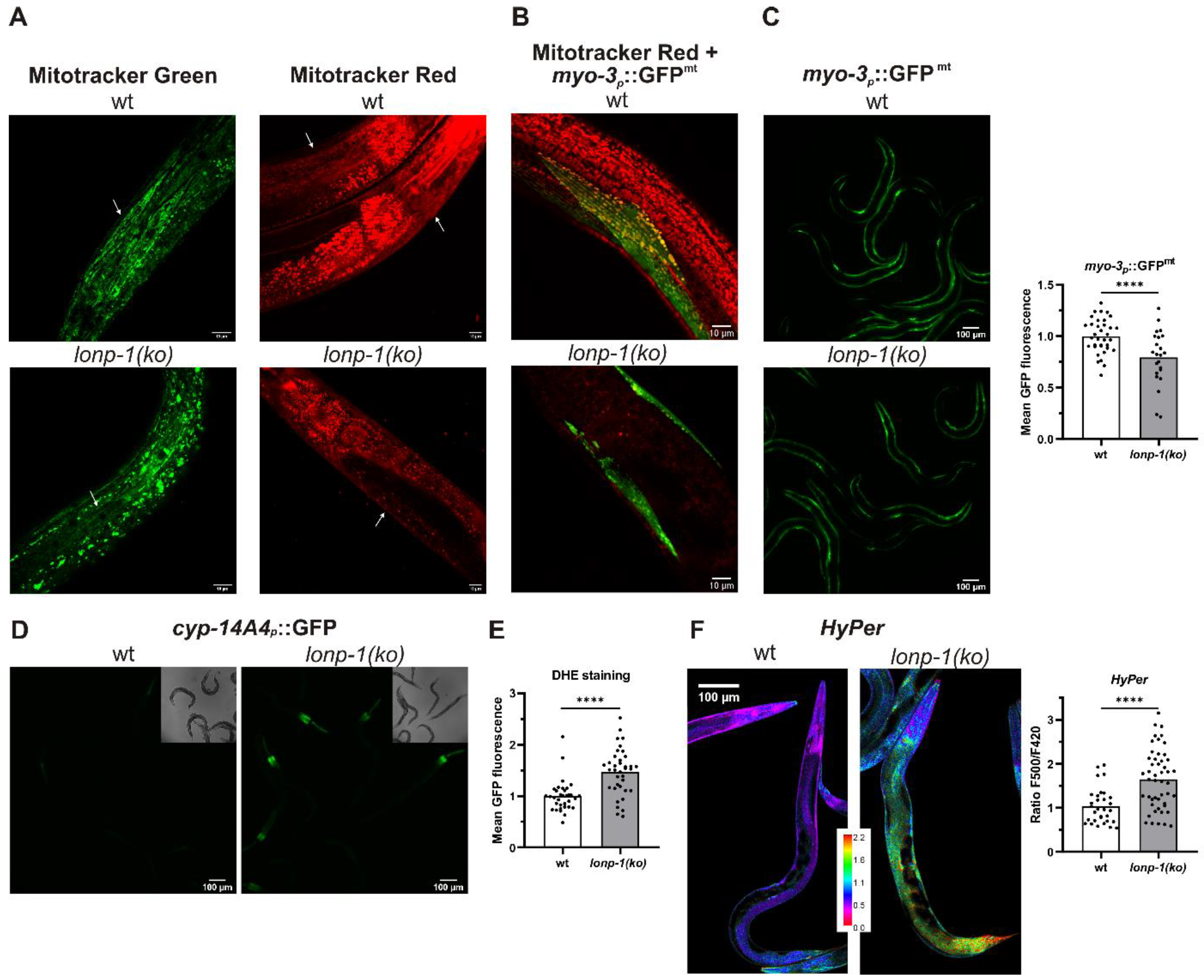
3.2. Mitochondrial and ROS Homeostasis Are Distorted in Lonp-1 Mutants
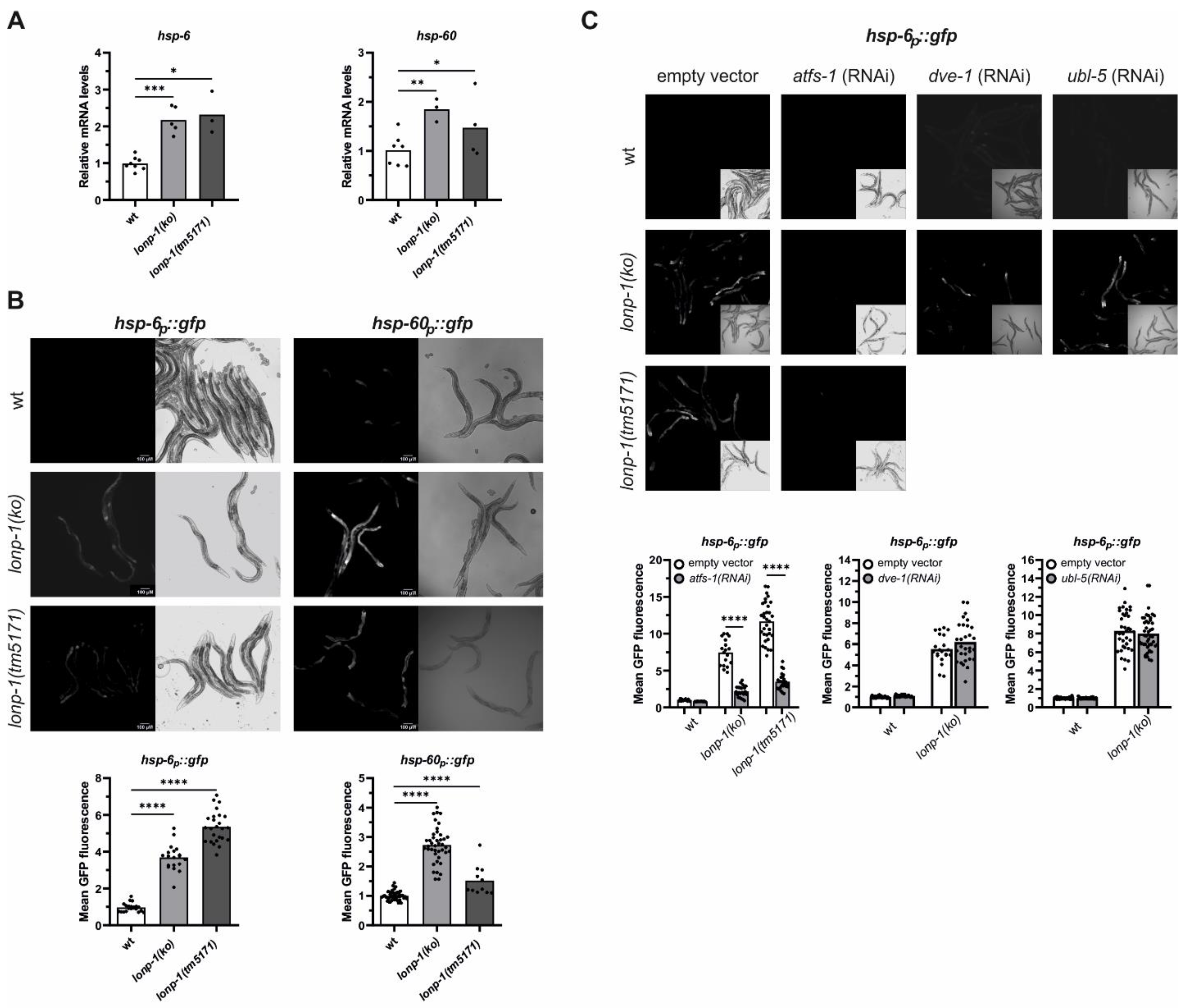
3.3. Activation of ATFS-1-Mediated Retrograde Response in Lonp-1 Mutants
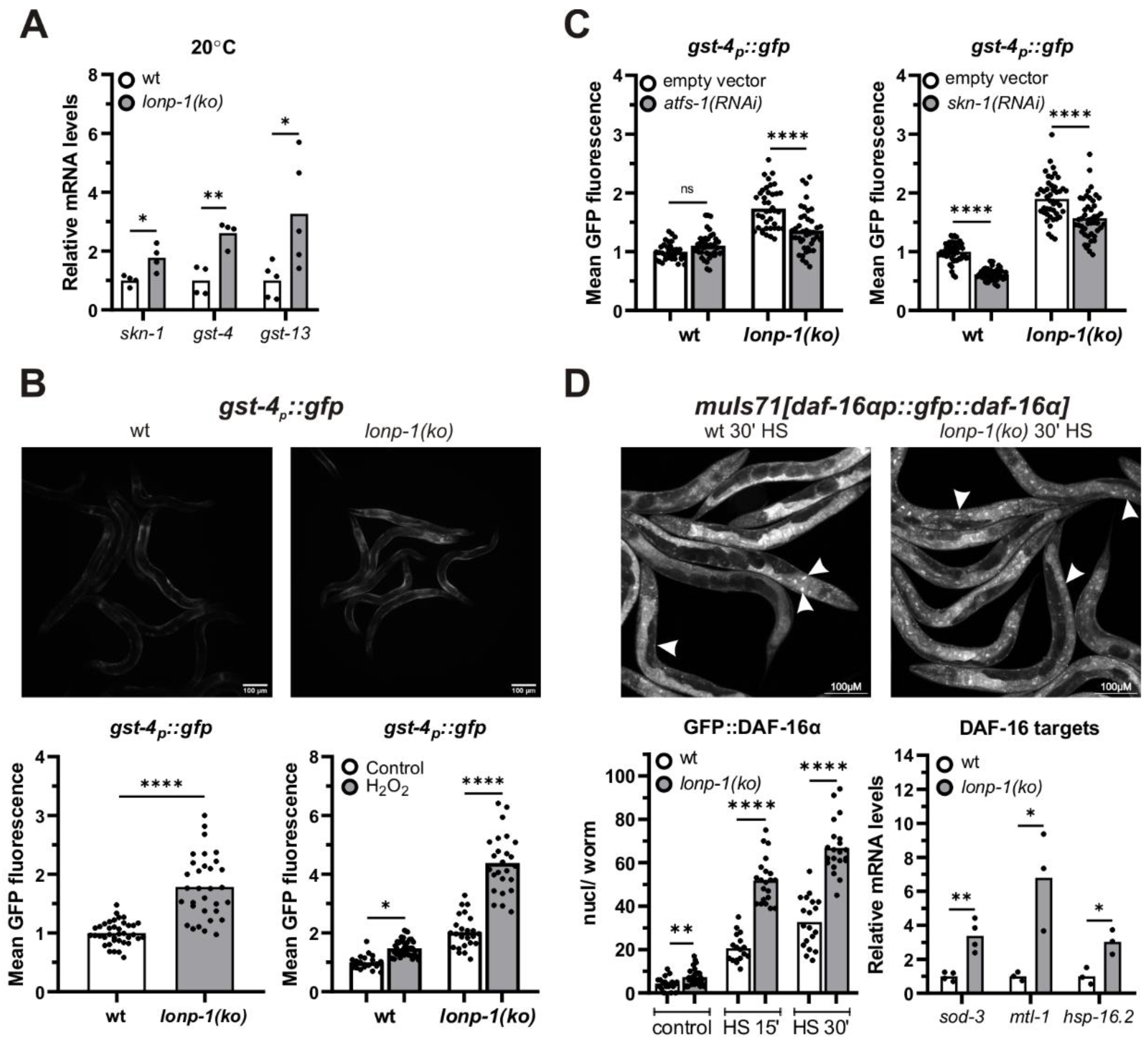
3.4. LONP-1 Deficiency Induces Cytosolic Oxidative Stress Responses
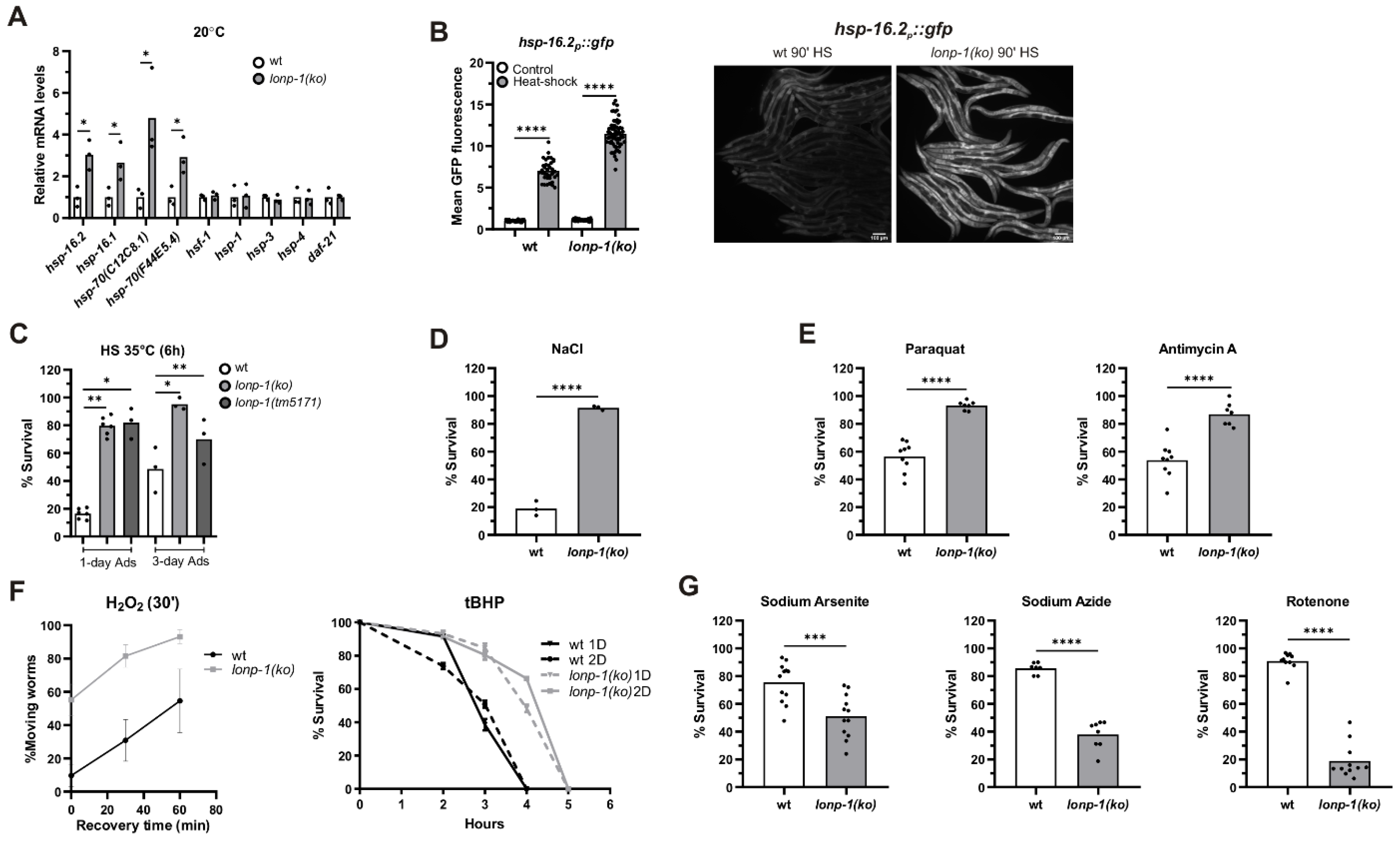
3.5. Induction of the Heat Shock Response (HSR) in Lonp-1 Mutants
3.6. Specific Responses of Lonp-1 Mutants to Exogenous Stresses
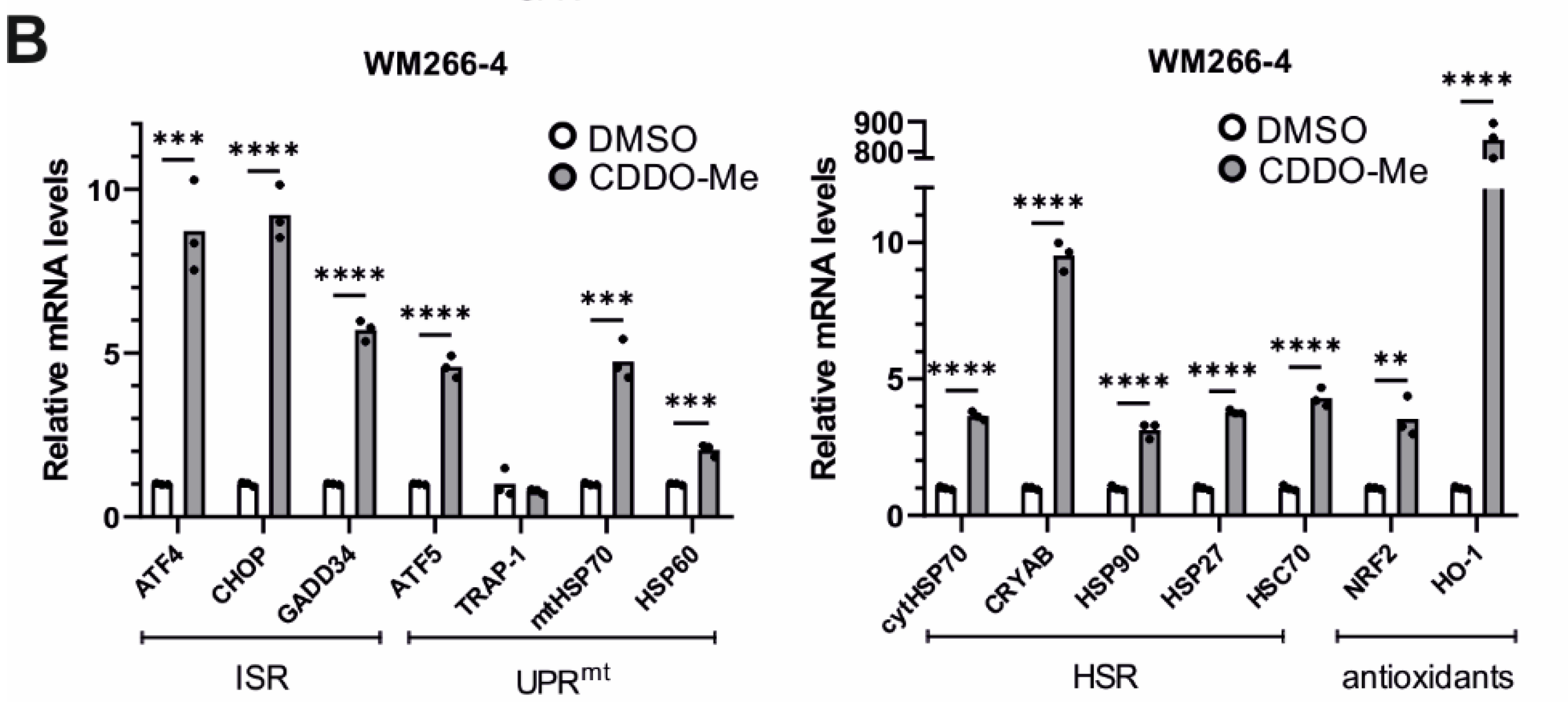
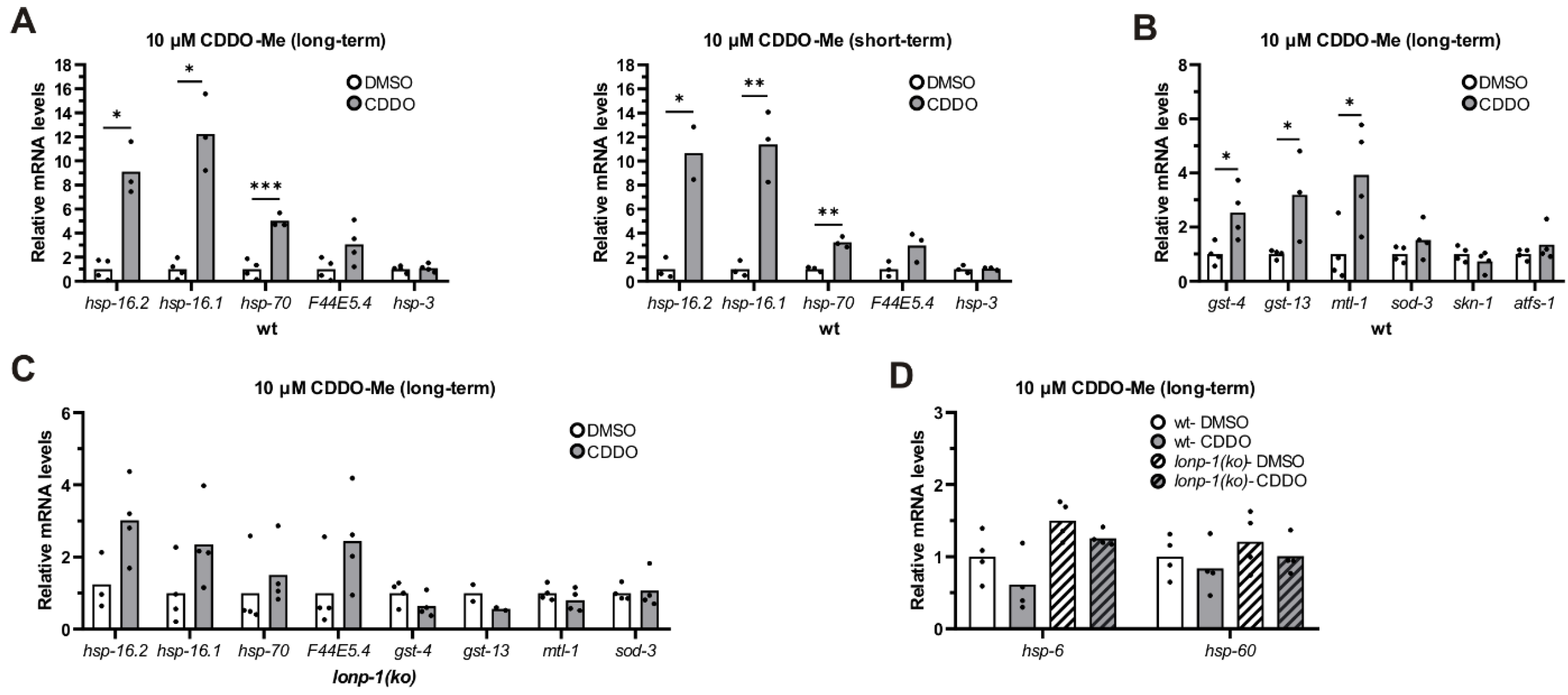
3.7. Treatment of Worms with CDDO-Me Induced Lonp-1-Specific Responses
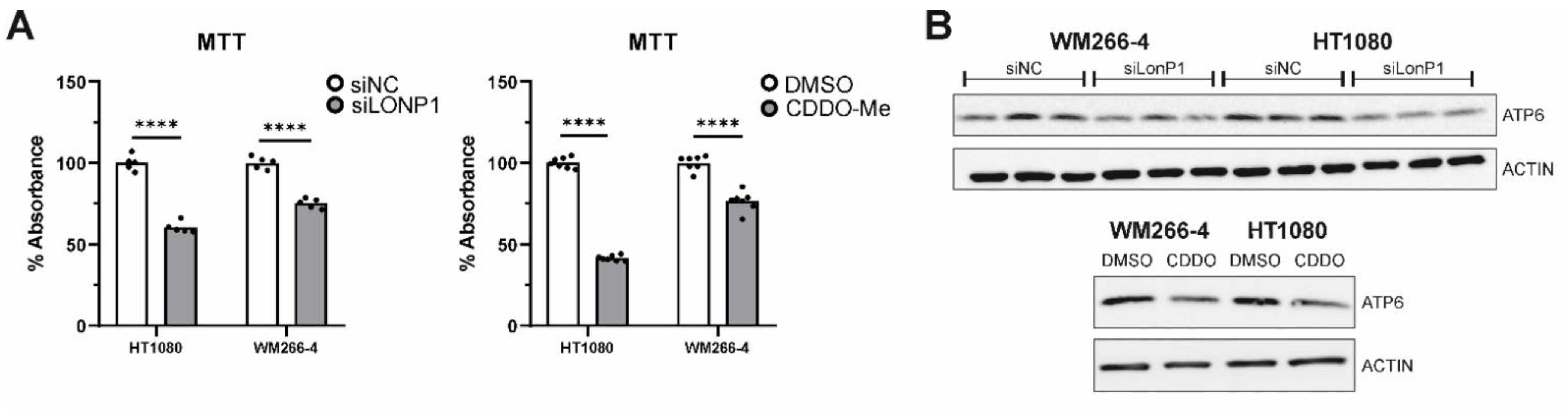
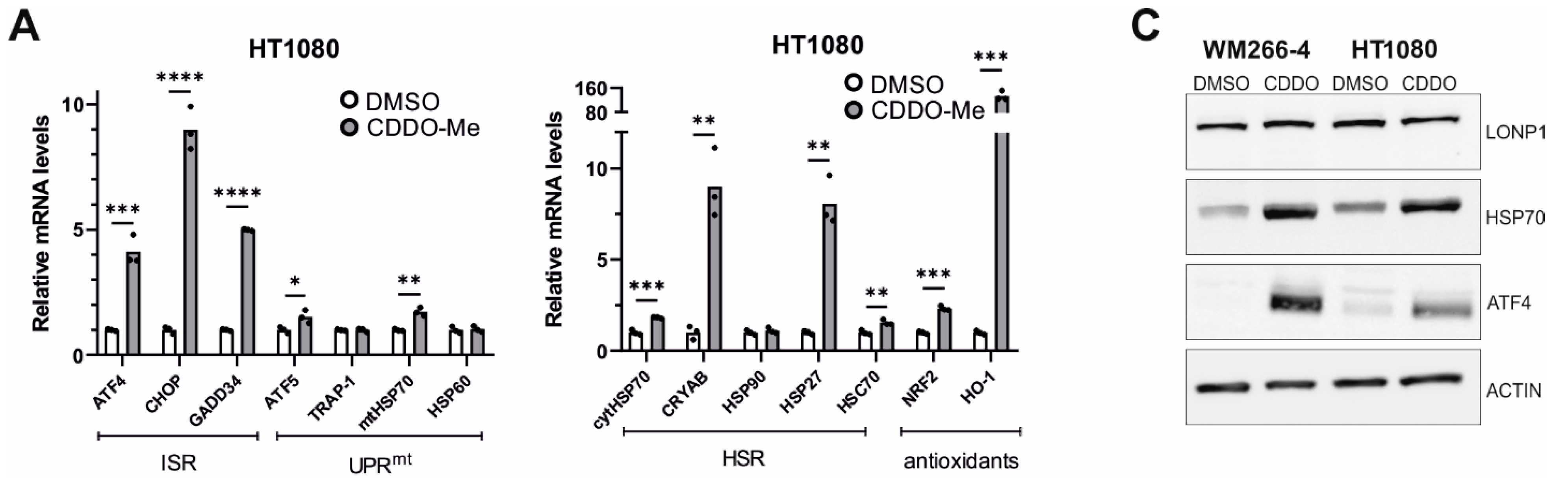
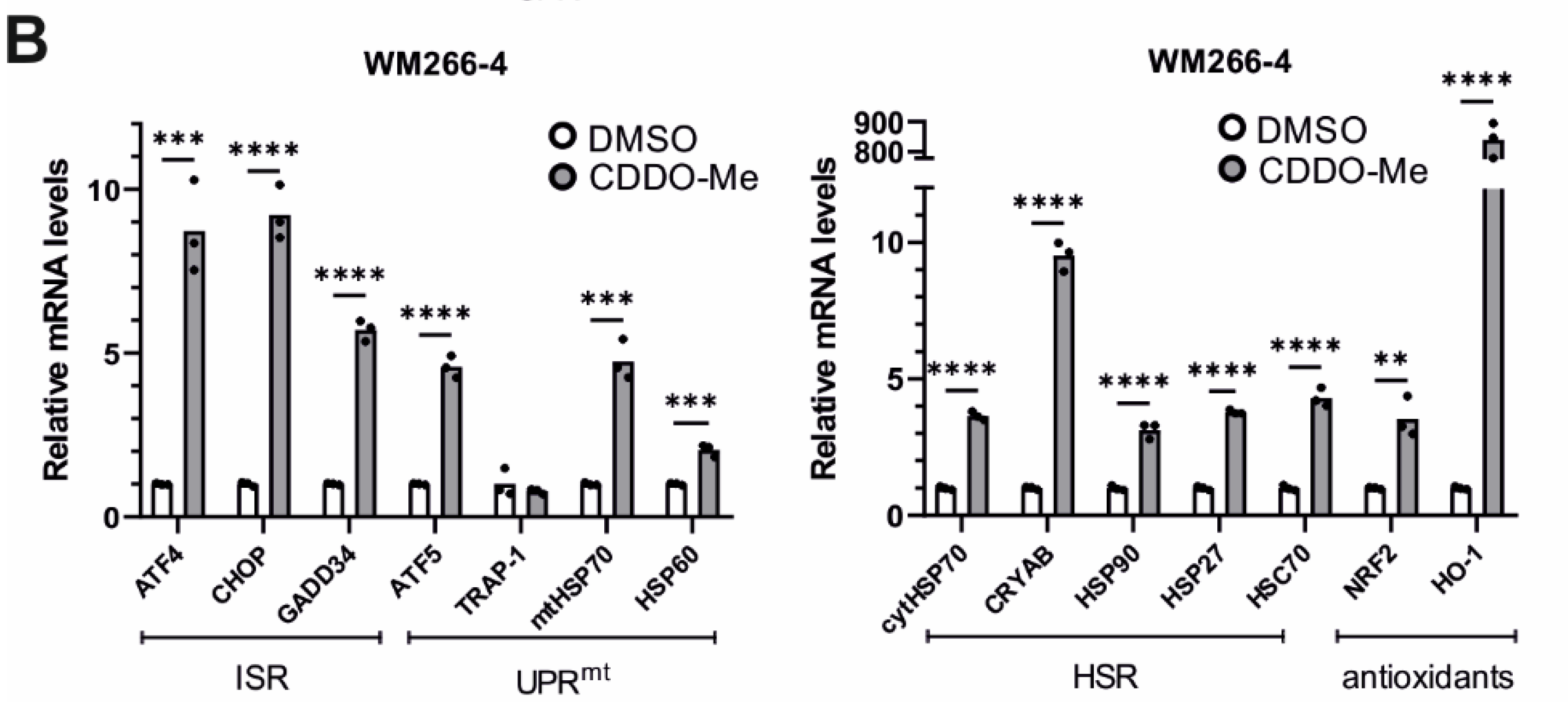
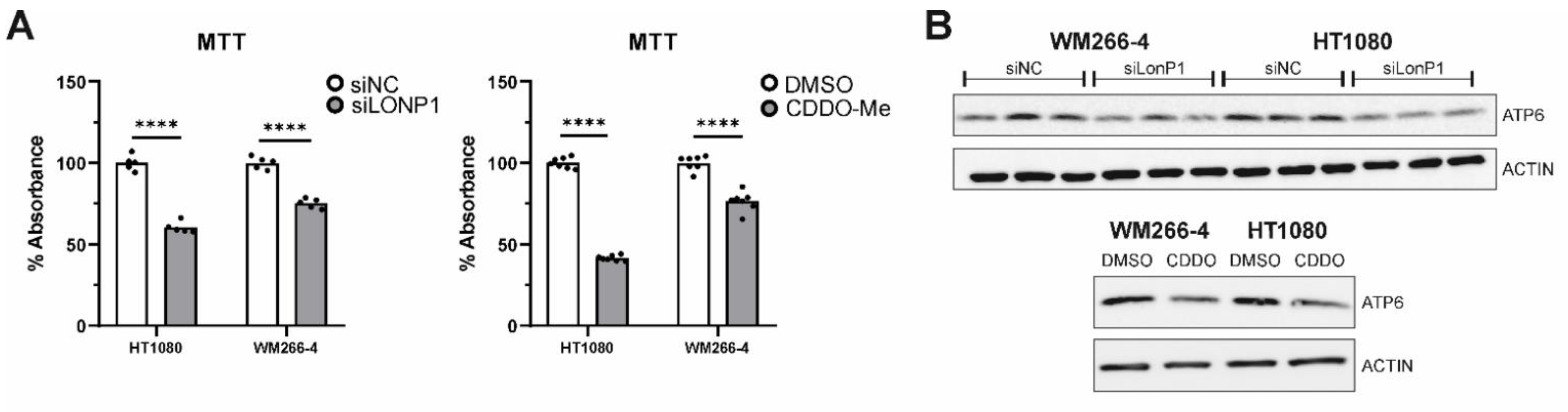
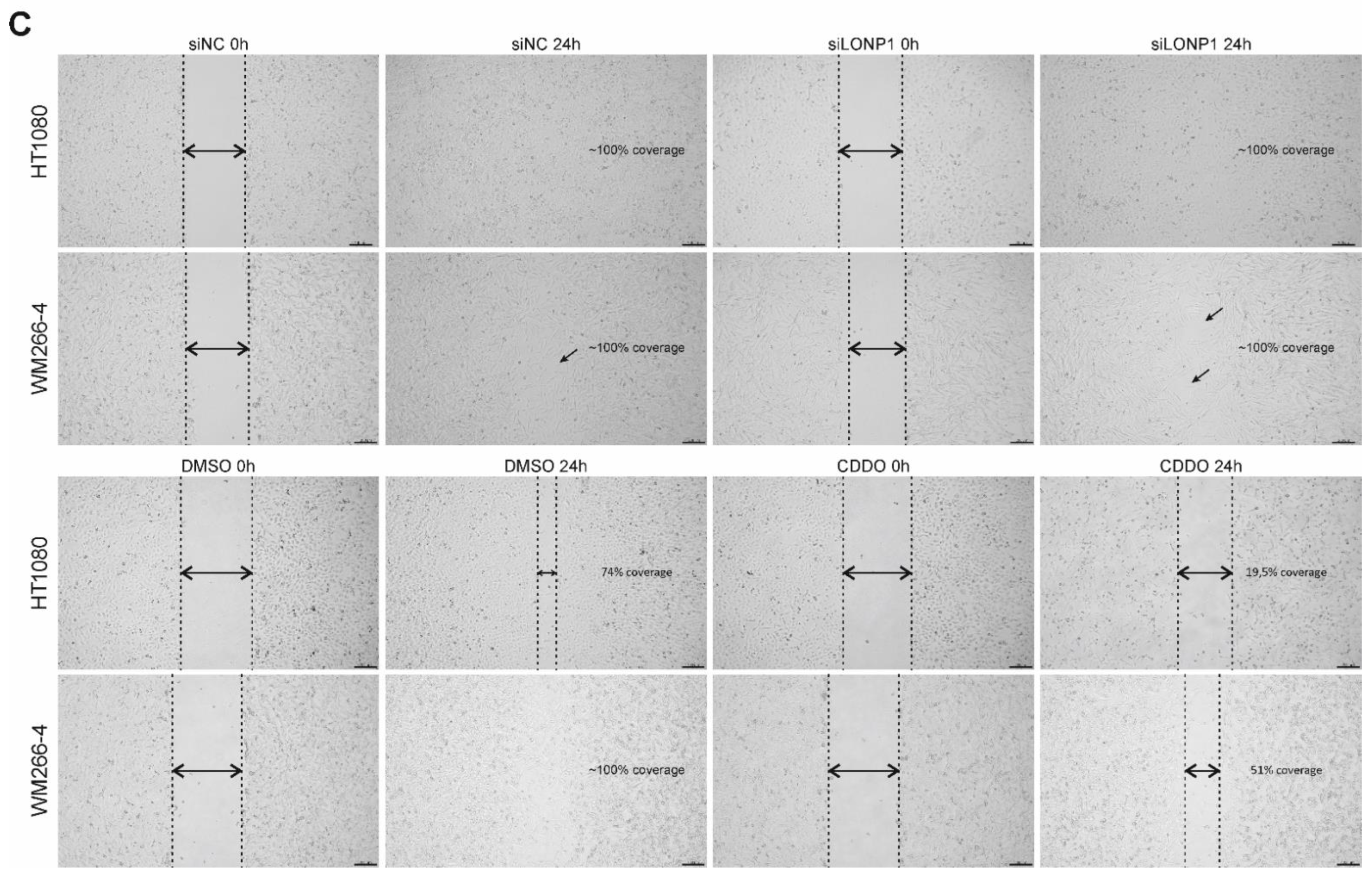
3.8. Effects of LONP1-Deficiency and CDDO-Me Treatment in Human Cancer Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rigas, S.; Daras, G.; Tsitsekian, D.; Hatzopoulos, P. The multifaceted role of Lon proteolysis in seedling establishment and maintenance of plant organelle function: Living from protein destruction. Physiol. Plant. 2012, 145, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Tsitsekian, D.; Daras, G.; Alatzas, A.; Templalexis, D.; Hatzopoulos, P.; Rigas, S. Comprehensive analysis of Lon proteases in plants highlights independent gene duplication events. J. Exp. Bot. 2019, 70, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Lee, J.; Singh, K.; Lee, I.; Suzuki, C.K. Multitasking in the mitochondrion by the ATP-dependent Lon protease. Biochim. Biophys. Acta 2012, 1823, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Chen, W.; Zhang, B.; Tian, F.; Zhou, Z.; Liao, X.; Li, C.; Zhang, Y.; Han, Y.; Wang, Y.; et al. Lon in maintaining mitochondrial and endoplasmic reticulum homeostasis. Arch. Toxicol. 2018, 92, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, C.K.; Suda, K.; Wang, N.; Schatz, G. Requirement for the yeast gene LON in intramitochondrial proteolysis and maintenance of respiration. Science 1994, 264, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, L.; Pearce, D.A.; Sherman, F. PIM1 encodes a mitochondrial ATP-dependent protease that is required for mitochondrial function in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1994, 269, 238–242. [Google Scholar] [CrossRef]
- Bota, D.A.; Ngo, J.K.; Davies, K.J. Downregulation of the human Lon protease impairs mitochondrial structure and function and causes cell death. Free Radic. Biol. Med. 2005, 38, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Bayot, A.; Gareil, M.; Rogowska-Wrzesinska, A.; Roepstorff, P.; Friguet, B.; Bulteau, A.L. Identification of novel oxidized protein substrates and physiological partners of the mitochondrial ATP-dependent Lon-like protease Pim1. J. Biol. Chem. 2010, 285, 11445–11457. [Google Scholar] [CrossRef] [Green Version]
- Bayot, A.; Gareil, M.; Chavatte, L.; Hamon, M.P.; L’Hermitte-Stead, C.; Beaumatin, F.; Priault, M.; Rustin, P.; Lombès, A.; Friguet, B.; et al. Effect of Lon protease knockdown on mitochondrial function in HeLa cells. Biochimie 2014, 100, 38–47. [Google Scholar] [CrossRef]
- Voos, W.; Pollecker, K. The mitochondrial Lon protease: Novel functions off the beaten track? Biomolecules 2020, 10, 253. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, Y.; Takahashi, K.; Yue, S.; Fujiyoshi, Y.; Yoshioka, H.; Aihara, M.; Setoyama, D.; Uchiumi, T.; Fukuchi, S.; Kang, D. Mitochondrial Lon protease is a gatekeeper for proteins newly imported into the matrix. Commun. Biol. 2021, 4, 974. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Lee, J.; Nie, X.; Li, M.; Morozov, Y.I.; Venkatesh, S.; Bogenhagen, D.F.; Temiakov, D.; Suzuki, C.K. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol. Cell 2013, 49, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, C.M.; Fiorese, C.J.; Lin, Y.F. Evaluating and responding to mitochondrial dysfunction: The mitochondrial unfolded-protein response and beyond. Trends Cell Biol. 2013, 23, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiros, P.M.; Espanol, Y.; Acin-Perez, R.; Rodriguez, F.; Barcena, C.; Watanabe, K.; Calvo, E.; Loureiro, M.; Fernandez-Garcia, M.S.; Fueyo, A.; et al. ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep. 2014, 8, 542–556. [Google Scholar] [CrossRef] [Green Version]
- Pomatto, L.C.D.; Carney, C.; Shen, B.; Wong, S.; Halaszynski, K.; Salomon, M.P.; Davies, K.J.A.; Tower, J. The mitochondrial Lon protease is required for age-specific and sex-specific adaptation to oxidative stress. Curr. Biol. 2017, 27, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.G.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.; et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 2015, 96, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Quirós, P.M.; Langer, T.; López-Otín, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef]
- Gibellini, L.; Losi, L.; De Biasi, S.; Nasi, M.; Lo Tartaro, D.; Pecorini, S.; Patergnani, S.; Pinton, P.; De Gaetano, A.; Carnevale, G.; et al. LonP1 differently modulates mitochondrial function and bioenergetics of primary versus metastatic colon cancer cells. Front. Oncol. 2018, 8, 254. [Google Scholar] [CrossRef]
- Ngo, J.K.; Pomatto, L.C.; Davies, K.J. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013, 1, 258–264. [Google Scholar] [CrossRef] [Green Version]
- Bota, D.A.; Van Remmen, H.; Davies, K.J. Modulation of Lon protease activity and aconitase turnover during aging and oxidative stress. FEBS Lett. 2002, 532, 103–106. [Google Scholar] [CrossRef] [Green Version]
- Bakala, H.; Delaval, E.; Hamelin, M.; Bismuth, J.; Borot-Laloi, C.; Corman, B.; Friguet, B. Changes in rat liver mitochondria with aging. Lon protease-like reactivity and N(epsilon)-carboxymethyllysine accumulation in the matrix. Eur. J. Biochem. 2003, 270, 2295–2302. [Google Scholar] [CrossRef] [PubMed]
- Erjavec, N.; Bayot, A.; Gareil, M.; Camougrand, N.; Nystrom, T.; Friguet, B.; Bulteau, A.L. Deletion of the mitochondrial Pim1/Lon protease in yeast results in accelerated aging and impairment of the proteasome. Free Radic. Biol. Med. 2013, 56, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Luce, K.; Osiewacz, H.D. Increasing organismal healthspan by enhancing mitochondrial protein quality control. Nat. Cell Biol. 2009, 11, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, M.A.; Haynes, C.M.; Pellegrino, M.W. The mitochondrial unfolded protein response: Signaling from the powerhouse. J. Biol. Chem. 2017, 292, 13500–13506. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, T.; Benedetti, C.; Urano, F.; Clark, S.G.; Harding, H.P.; Ron, D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 2004, 117, 4055–4066. [Google Scholar] [CrossRef] [Green Version]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Senchuk, M.M.; Dues, D.J.; Johnson, B.K.; Cooper, J.F.; Lew, L.; Machiela, E.; Schaar, C.E.; DeJonge, H.; Blackwell, T.K.; et al. Mitochondrial unfolded protein response transcription factor ATFS-1 promotes longevity in a long-lived mitochondrial mutant through activation of stress response pathways. BMC Biol. 2018, 16, 147. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Youle, R.J.; Finkel, T. The mitochondrial basis of aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.F.; Vander Wende, H.; Simko, M.; Klum, S.; Barfield, S.; Choi, H.; Pineda, V.V.; Kaeberlein, M. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun. 2014, 5, 3483. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nargund, A.M.; Fiorese, C.J.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol. Cell 2015, 58, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.M.; Nargund, A.M.; Sun, T.; Haynes, C.M. Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 2012, 8, e1002760. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef] [Green Version]
- Gao, K.; Li, Y.; Hu, S.; Liu, Y. SUMO peptidase ULP-4 regulates mitochondrial UPR-mediated innate immunity and lifespan extension. Elife 2019, 8. [Google Scholar] [CrossRef]
- Kim, S.; Sieburth, D. FSHR-1/GPCR regulates the mitochondrial unfolded protein response in Caenorhabditis elegans. Genetics 2020, 214, 409–418. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. Embo J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [Green Version]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.S.; Haynes, C.M. Folding the mitochondrial UPR into the integrated stress response. Trends Cell Biol. 2020, 30, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Yamazaki, H.; Tanji, K.; Engler, M.J.; Matsumiya, T.; Itoh, K. Role of the ISR-ATF4 pathway and its cross talk with Nrf2 in mitochondrial quality control. J. Clin. Biochem. Nutr. 2019, 64, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef] [PubMed]
- Dokshin, G.A.; Ghanta, K.S.; Piscopo, K.M.; Mello, C.C. Robust Genome Editing with Short Single-Stranded and Long, Partially Single-Stranded DNA Donors in Caenorhabditis elegans. Genetics 2018, 210, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Farboud, B.; Severson, A.F.; Meyer, B.J. Strategies for efficient genome editing using CRISPR-Cas9. Genetics 2019, 211, 431–457. [Google Scholar] [CrossRef] [Green Version]
- Borbolis, F.; Rallis, J.; Kanatouris, G.; Kokla, N.; Karamalegkos, A.; Vasileiou, C.; Vakaloglou, K.M.; Diallinas, G.; Stravopodis, D.J.; Zervas, C.G.; et al. mRNA decapping is an evolutionarily conserved modulator of neuroendocrine signaling that controls development and ageing. Elife 2020, 9. [Google Scholar] [CrossRef]
- Syntichaki, P.; Troulinaki, K.; Tavernarakis, N. eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature 2007, 445, 922–926. [Google Scholar] [CrossRef]
- Back, P.; De Vos, W.H.; Depuydt, G.G.; Matthijssens, F.; Vanfleteren, J.R.; Braeckman, B.P. Exploring real-time in vivo redox biology of developing and aging Caenorhabditis elegans. Free. Radic. Biol. Med. 2012, 52, 850–859. [Google Scholar] [CrossRef]
- Aspernig, H.; Heimbucher, T.; Qi, W.; Gangurde, D.; Curic, S.; Yan, Y.; Donner von Gromoff, E.; Baumeister, R.; Thien, A. Mitochondrial Perturbations Couple mTORC2 to Autophagy in C. elegans. Cell Rep. 2019, 29, 1399–1409.e5. [Google Scholar] [CrossRef] [PubMed]
- Ewald, C.Y.; Hourihan, J.M.; Blackwell, T.K. Oxidative stress assays (arsenite and tBHP) in Caenorhabditis elegans. Bio. Protoc. 2017, 7, e2365. [Google Scholar] [CrossRef] [PubMed]
- Pareek, G.; Pallanck, L.J. Inactivation of Lon protease reveals a link between mitochondrial unfolded protein stress and mitochondrial translation inhibition. Cell Death Dis. 2018, 9, 1168. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Suzuki, C.K. Functional mechanics of the ATP-dependent Lon protease-lessons from endogenous protein and synthetic peptide substrates. Biochim. Biophys. Acta 2008, 1784, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrousse, A.M.; Zappaterra, M.D.; Rube, D.A.; van der Bliek, A.M.C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol. Cell 1999, 4, 815–826. [Google Scholar] [CrossRef]
- Byrne, J.J.; Soh, M.S.; Chandhok, G.; Vijayaraghavan, T.; Teoh, J.S.; Crawford, S.; Cobham, A.E.; Yapa, N.M.B.; Mirth, C.K.; Neumann, B. Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans. Cell Mol. Life Sci. 2019, 76, 1967–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Jin, P.; Yu, L.; Wang, Y.; Han, L.; Shi, T.; Li, X. Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS ONE 2014, 9, e92810. [Google Scholar] [CrossRef]
- Mao, K.; Ji, F.; Breen, P.; Sewell, A.; Han, M.; Sadreyev, R.; Ruvkun, G. Mitochondrial dysfunction in C. elegans activates mitochondrial relocalization and nuclear hormone receptor-dependent detoxification genes. Cell Metab. 2019, 29, 1182–1191.e4. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.W.; Lee, W.H.; Kim, P.H. Oxidative mechanisms of IL-4-induced IL-6 expression in vascular endothelium. Cytokine 2010, 49, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Schaar, C.E.; Dues, D.J.; Spielbauer, K.K.; Machiela, E.; Cooper, J.F.; Senchuk, M.; Hekimi, S.; Van Raamsdonk, J.M. Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 2015, 11, e1004972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runkel, E.D.; Liu, S.; Baumeister, R.; Schulze, E. Surveillance-activated defenses block the ROS-induced mitochondrial unfolded protein response. PLoS Genet. 2013, 9, e1003346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousakis, A.; Vlassis, A.; Vlanti, A.; Patera, S.; Thireos, G.; Syntichaki, P. The general control nonderepressible-2 kinase mediates stress response and longevity induced by target of rapamycin inactivation in Caenorhabditis elegans. Aging Cell 2013, 12, 742–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.K.; See, R.; Batchelder, C.; Kophengnavong, T.; Gronniger, J.T.; Shi, Y.; Blackwell, T.K. A conserved transcription motif suggesting functional parallels between Caenorhabditis elegans SKN-1 and Cap’n’Collar-related basic leucine zipper proteins. J. Biol. Chem. 2000, 275, 22166–22171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, R.P.; Porter Abate, J.; Dilks, K.; Landis, J.; Ashraf, J.; Murphy, C.T.; Blackwell, T.K. Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell 2009, 8, 524–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.H.; Blackwell, T.K. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 2003, 17, 1882–1893. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, T.K.; Steinbaugh, M.J.; Hourihan, J.M.; Ewald, C.Y.; Isik, M. SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic. Biol. Med. 2015, 88, 290–301. [Google Scholar] [CrossRef] [Green Version]
- Kahn, N.W.; Rea, S.L.; Moyle, S.; Kell, A.; Johnson, T.E. Proteasomal dysfunction activates the transcription factor SKN-1 and produces a selective oxidative-stress response in Caenorhabditis elegans. Biochem. J. 2008, 409, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, K.; Heidler, T.; Moch, J.; Daniel, H.; Wenzel, U. Feeding a ROS-generator to Caenorhabditis elegans leads to increased expression of small heat shock protein HSP-16.2 and hormesis. Genes Nutr. 2009, 4, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Glover-Cutter, K.M.; Lin, S.; Blackwell, T.K. Integration of the unfolded protein and oxidative stress responses through SKN-1/Nrf. PLoS Genet. 2013, 9, e1003701. [Google Scholar] [CrossRef] [Green Version]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senchuk, M.M.; Dues, D.J.; Schaar, C.E.; Johnson, B.K.; Madaj, Z.B.; Bowman, M.J.; Winn, M.E.; Van Raamsdonk, J.M. Activation of DAF-16/FOXO by reactive oxygen species contributes to longevity in long-lived mitochondrial mutants in Caenorhabditis elegans. PLoS Genet. 2018, 14, e1007268. [Google Scholar] [CrossRef] [PubMed]
- Libina, N.; Berman, J.R.; Kenyon, C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 2003, 115, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.T.; McCarroll, S.A.; Bargmann, C.I.; Fraser, A.; Kamath, R.S.; Ahringer, J.; Li, H.; Kenyon, C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 2003, 424, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Tepper, R.G.; Ashraf, J.; Kaletsky, R.; Kleemann, G.; Murphy, C.T.; Bussemaker, H.J. PQM-1 complements DAF-16 as a key transcriptional regulator of DAF-2-mediated development and longevity. Cell 2013, 154, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Williams, R.; Laskovs, M.; Williams, R.I.; Mahadevan, A.; Labbadia, J. A Mitochondrial Stress-Specific Form of HSF1 Protects against Age-Related Proteostasis Collapse. Dev. Cell 2020, 54, 758–772.e5. [Google Scholar] [CrossRef]
- Ngo, J.K.; Davies, K.J. Mitochondrial Lon protease is a human stress protein. Free Radic. Biol. Med. 2009, 46, 1042–1048. [Google Scholar] [CrossRef] [Green Version]
- Labbadia, J.; Morimoto, R.I. Repression of the heat shock response is a programmed event at the onset of reproduction. Mol. Cell 2015, 59, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Cochemé, H.M.; Murphy, M.P. Complex I is the major site of mitochondrial superoxide production by paraquat. J. Biol. Chem. 2008, 283, 1786–1798. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Li, J.; Lou, B.; Wu, R.; Wang, G.; Lu, C.; Wang, H.; Pi, J.; Xu, Y. The role of reactive oxygen species in arsenic toxicity. Biomolecules 2020, 10, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, M.; Kwon, J.Y.; Shim, J.; Lee, J. Differential hypoxia response of hsp-16 genes in the nematode. J. Mol. Biol 2004, 344, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hunt, C.P.; Luz, A.L.; Ryde, I.T.; Turner, E.A.; Ilkayeva, O.R.; Bhatt, D.P.; Hirschey, M.D.; Meyer, J.N. Multiple metabolic changes mediate the response of Caenorhabditis elegans to the complex I inhibitor rotenone. Toxicology 2021, 447, 152630. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, S.H.; Venkatesh, S.; Li, M.; Lee, J.; Lu, B.; Hilchey, S.P.; Morse, K.M.; Metcalfe, H.M.; Skalska, J.; Andreeff, M.; et al. The mitochondrial ATP-dependent Lon protease: A novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood 2012, 119, 3321–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibellini, L.; Pinti, M.; Bartolomeo, R.; De Biasi, S.; Cormio, A.; Musicco, C.; Carnevale, G.; Pecorini, S.; Nasi, M.; De Pol, A.; et al. Inhibition of Lon protease by triterpenoids alters mitochondria and is associated to cell death in human cancer cells. Oncotarget 2015, 6, 25466–25483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moehle, E.A.; Shen, K.; Dillin, A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 2019, 294, 5396–5407. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci. Rep. 2015, 5, 17397. [Google Scholar] [CrossRef] [Green Version]
- Zurita Rendón, O.; Shoubridge, E.A. LONP1 Is required for maturation of a subset of mitochondrial proteins, and its loss elicits an integrated stress response. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinti, M.; Gibellini, L.; Nasi, M.; De Biasi, S.; Bortolotti, C.A.; Iannone, A.; Cossarizza, A. Emerging role of Lon protease as a master regulator of mitochondrial functions. Biochim. Biophys. Acta 2016, 1857, 1300–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soo, S.K.; Traa, A.; Rudich, P.D.; Mistry, M.; Van Raamsdonk, J.M. Activation of mitochondrial unfolded protein response protects against multiple exogenous stressors. Life Sci. Alliance 2021, 4, e202101182. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Brielmann, R.M.; Neto, M.F.; Lin, Y.F.; Haynes, C.M.; Morimoto, R.I. Mitochondrial stress restores the heat shock response and prevents proteostasis collapse during aging. Cell Rep. 2017, 21, 1481–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjahjono, E.; McAnena, A.P.; Kirienko, N.V. The evolutionarily conserved ESRE stress response network is activated by ROS and mitochondrial damage. BMC Biol. 2020, 18, 74. [Google Scholar] [CrossRef]
- Kim, S.; Sieburth, D. Sphingosine kinase activates the mitochondrial unfolded protein response and is targeted to mitochondria by stress. Cell Rep. 2018, 24, 2932–2945.e4. [Google Scholar] [CrossRef] [Green Version]
- Dues, D.J.; Schaar, C.E.; Johnson, B.K.; Bowman, M.J.; Winn, M.E.; Senchuk, M.M.; Van Raamsdonk, J.M. Uncoupling of oxidative stress resistance and lifespan in long-lived isp-1 mitochondrial mutants in Caenorhabditis elegans. Free Radic. Biol. Med. 2017, 108, 362–373. [Google Scholar] [CrossRef]
- Hamon, M.P.; Gergondey, R.; L’Honoré, A.; Friguet, B. Mitochondrial Lon protease-depleted HeLa cells exhibit proteome modifications related to protein quality control, stress response and energy metabolism. Free Radic. Biol. Med. 2020, 148, 83–95. [Google Scholar] [CrossRef]
- Pollecker, K.; Sylvester, M.; Voos, W. Proteomic analysis demonstrates the role of the quality control protease LONP1 in mitochondrial protein aggregation. JBC 2021, 297, 101134. [Google Scholar] [CrossRef]
- Borella, R.; Fonti, L.; Gibellini, L.; De Gaetano, A.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Synthesis and anticancer activity of CDDO and CDDO-Me, two derivatives of natural triterpenoids. Molecules 2019, 24, 4097. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. CDDO-Me selectively attenuates CA1 neuronal death induced by status epilepticus via facilitating mitochondrial fission independent of LONP1. Cells 2019, 8, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bota, D.A.; Davies, K.J. Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders. Free Radic. Biol. Med. 2016, 100, 188–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taouktsi, E.; Kyriakou, E.; Smyrniotis, S.; Borbolis, F.; Bondi, L.; Avgeris, S.; Trigazis, E.; Rigas, S.; Voutsinas, G.E.; Syntichaki, P. Organismal and Cellular Stress Responses upon Disruption of Mitochondrial Lonp1 Protease. Cells 2022, 11, 1363. https://doi.org/10.3390/cells11081363
Taouktsi E, Kyriakou E, Smyrniotis S, Borbolis F, Bondi L, Avgeris S, Trigazis E, Rigas S, Voutsinas GE, Syntichaki P. Organismal and Cellular Stress Responses upon Disruption of Mitochondrial Lonp1 Protease. Cells. 2022; 11(8):1363. https://doi.org/10.3390/cells11081363
Chicago/Turabian StyleTaouktsi, Eirini, Eleni Kyriakou, Stefanos Smyrniotis, Fivos Borbolis, Labrina Bondi, Socratis Avgeris, Efstathios Trigazis, Stamatis Rigas, Gerassimos E. Voutsinas, and Popi Syntichaki. 2022. "Organismal and Cellular Stress Responses upon Disruption of Mitochondrial Lonp1 Protease" Cells 11, no. 8: 1363. https://doi.org/10.3390/cells11081363
APA StyleTaouktsi, E., Kyriakou, E., Smyrniotis, S., Borbolis, F., Bondi, L., Avgeris, S., Trigazis, E., Rigas, S., Voutsinas, G. E., & Syntichaki, P. (2022). Organismal and Cellular Stress Responses upon Disruption of Mitochondrial Lonp1 Protease. Cells, 11(8), 1363. https://doi.org/10.3390/cells11081363