
Apoptotic and DNA Damage Effect of 1,2,3,4,6-Penta-O-galloyl-beta-D-glucose in Cisplatin-Resistant Non-Small Lung Cancer Cells via Phosphorylation of H2AX, CHK2 and p53


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. PGG Preparation
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. Cell Cycle Analysis
2.5. Annexin-V-FITC Apoptosis Assay
2.6. TUNEL Assay
2.7. DNA Fragmentation Assay
2.8. Western Blotting and Nuclear Extract Preparation
2.9. siRNA Transfection and p53 Luciferase Assay
2.10. Immunofluorescence Assay
2.11. Comet Assay
2.12. Combination Index
2.13. Tumor Xenograft Model and Immunohistochemistry
2.14. Statistical Analyses
3. Results
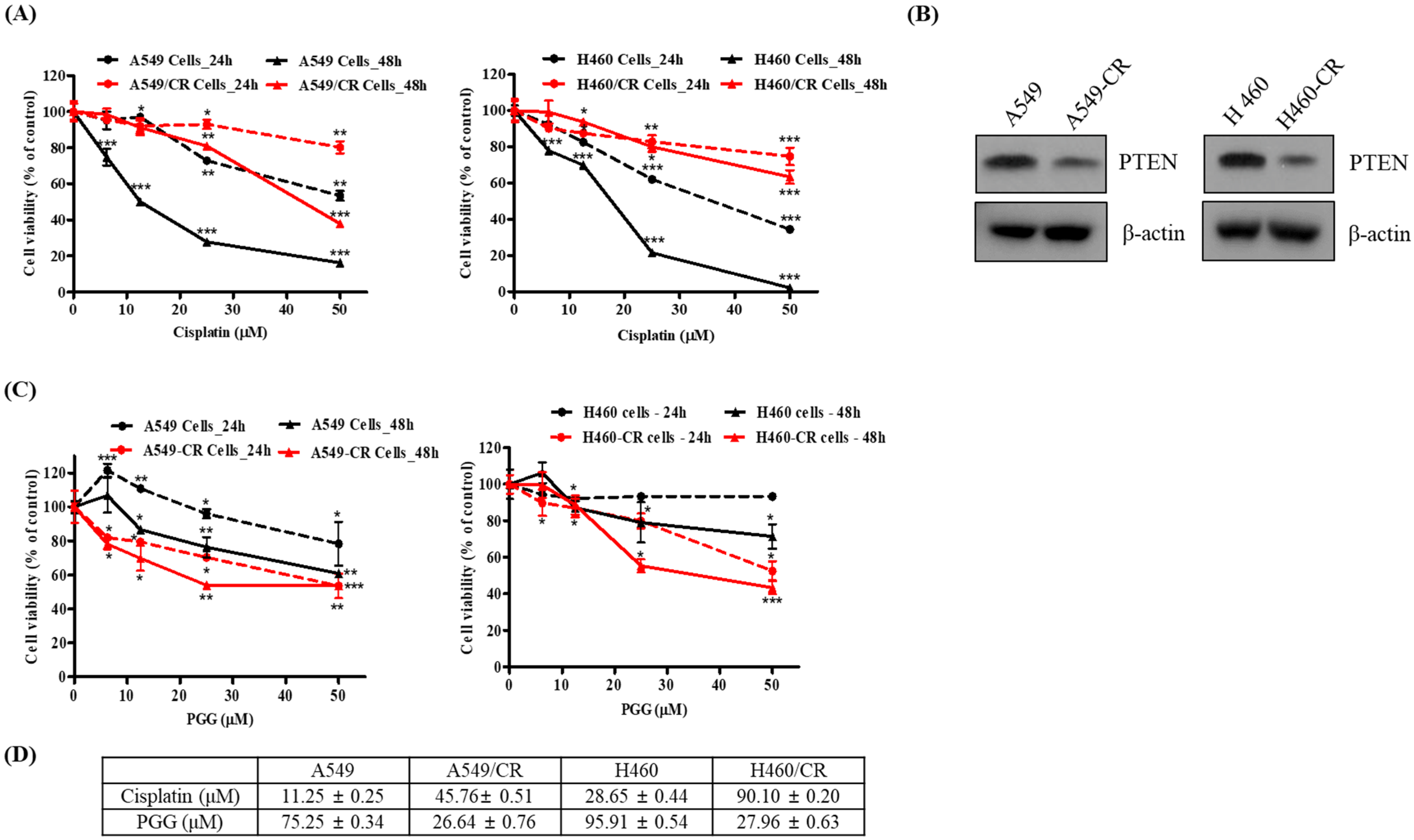
3.1. PGG Reduces the Viability of Human Cisplatin-Resistant Lung Cancer Cells Compared with Their Parental Cells
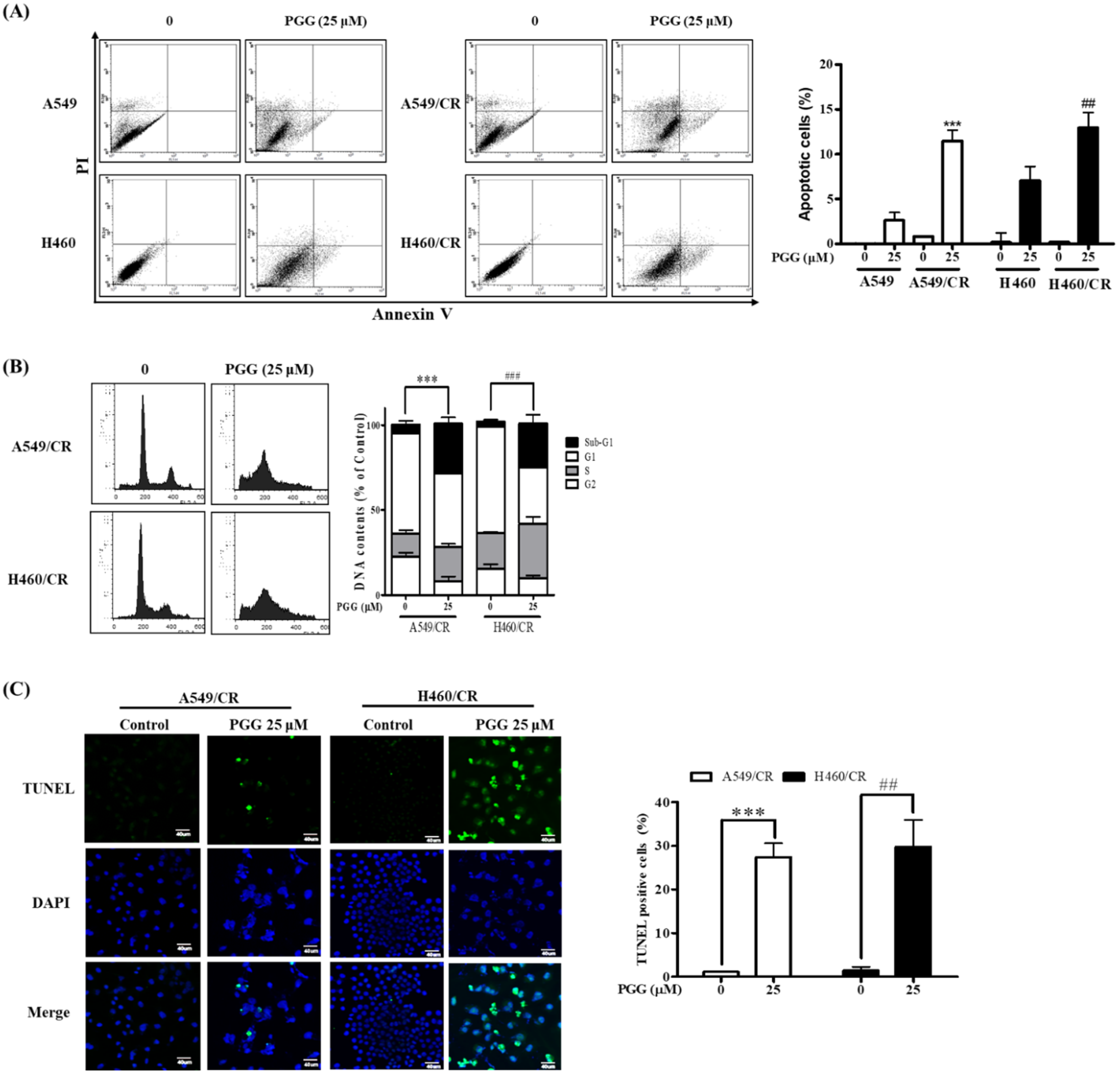
3.2. PGG Induces Apoptosis in A549/CR and H460/CR Cells
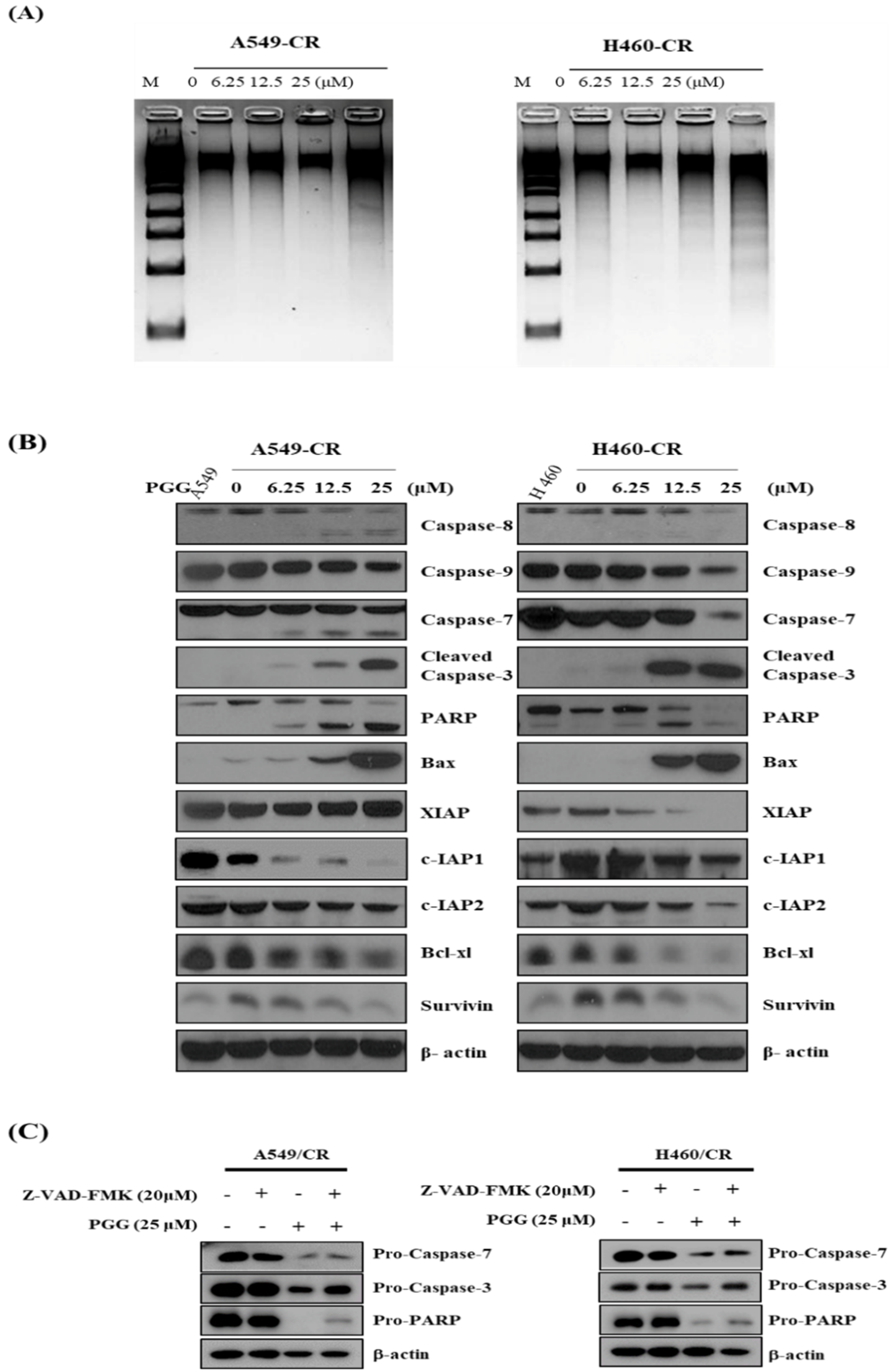
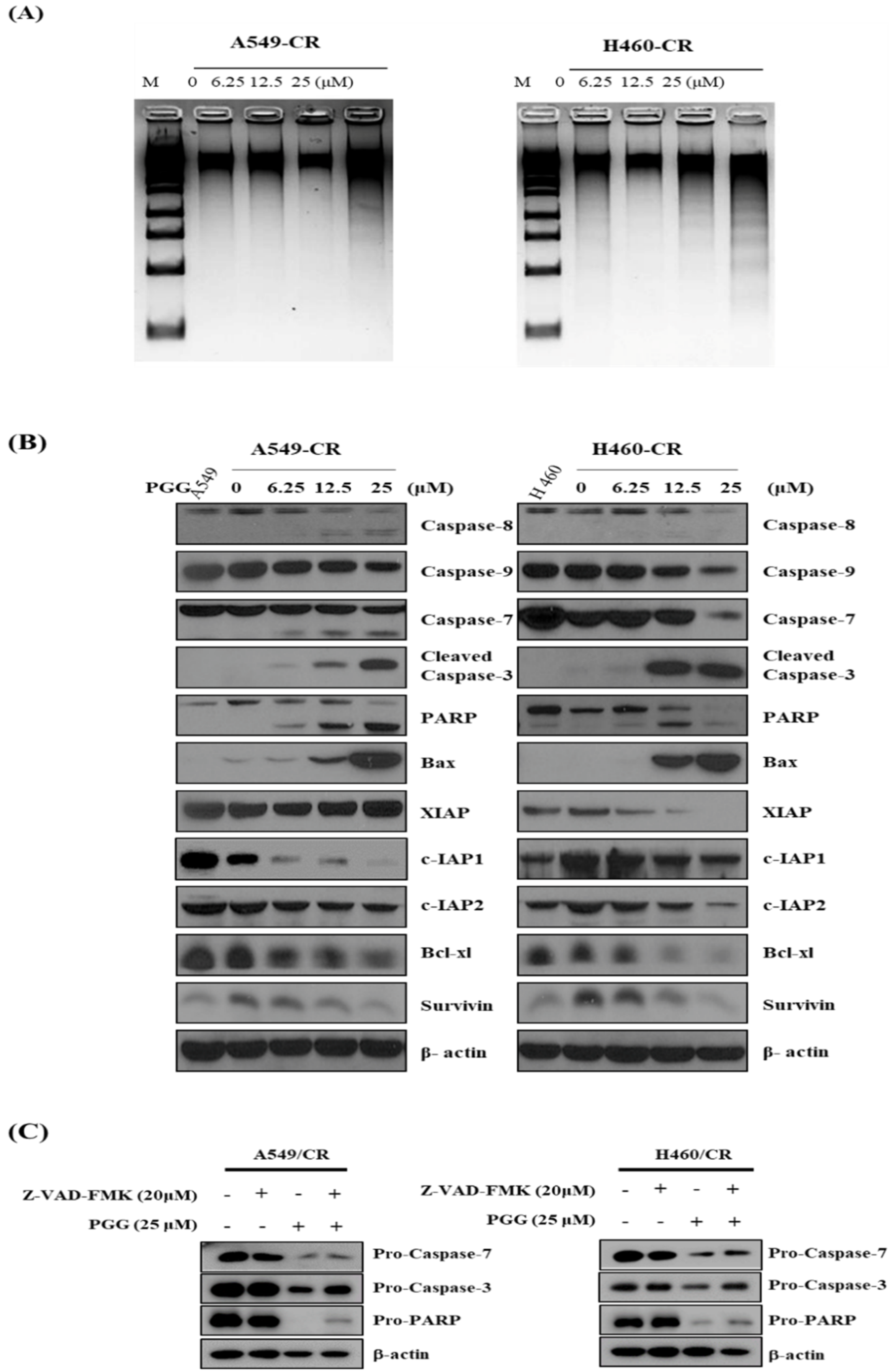
3.3. PGG Induces DNA Fragmentation and Affects Apoptosis Related Proteins in A549/CR and H640/CR Cells
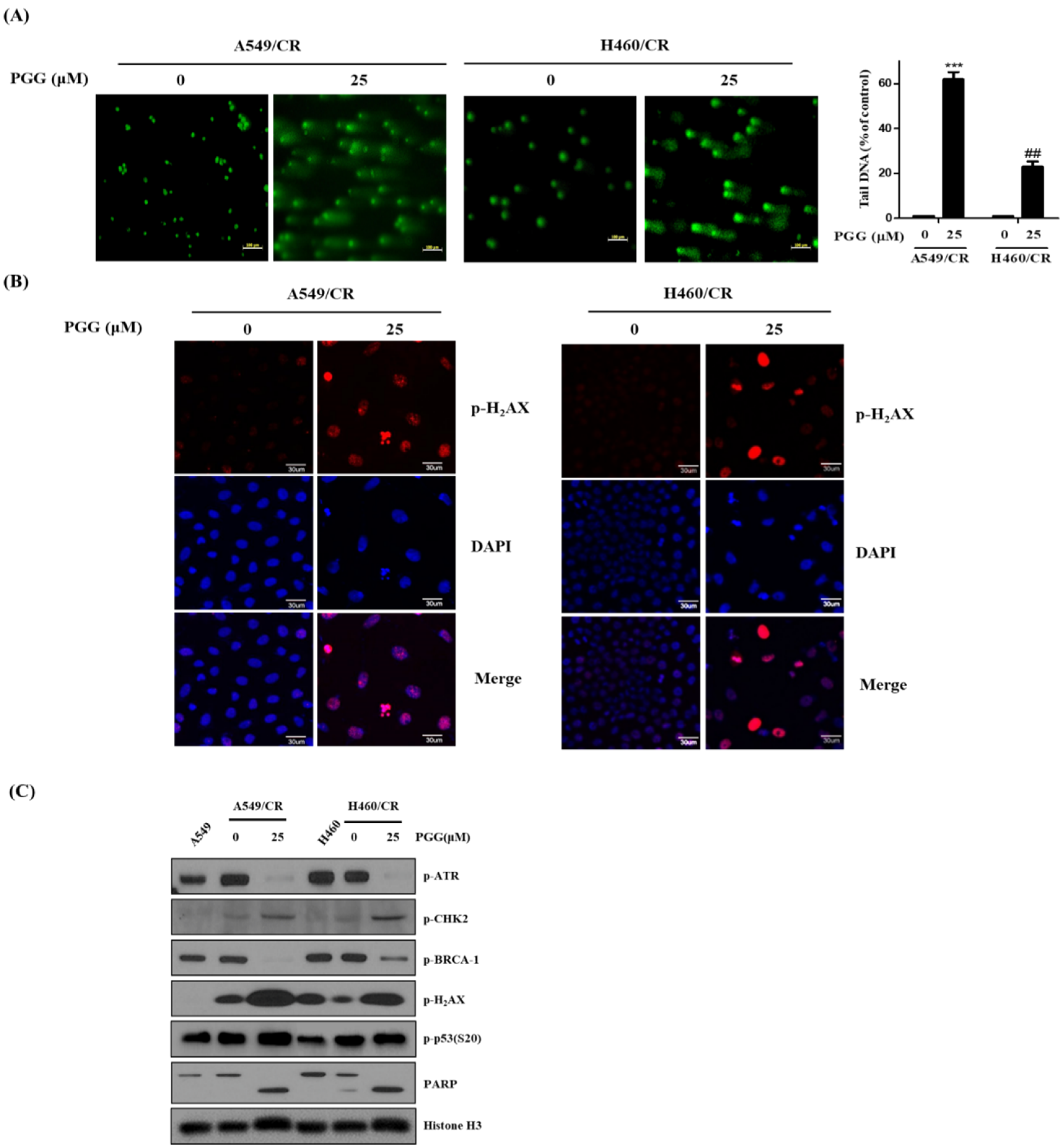
3.4. PGG Induces DNA Damage Proteins in A549/CR and H460/CR Cells
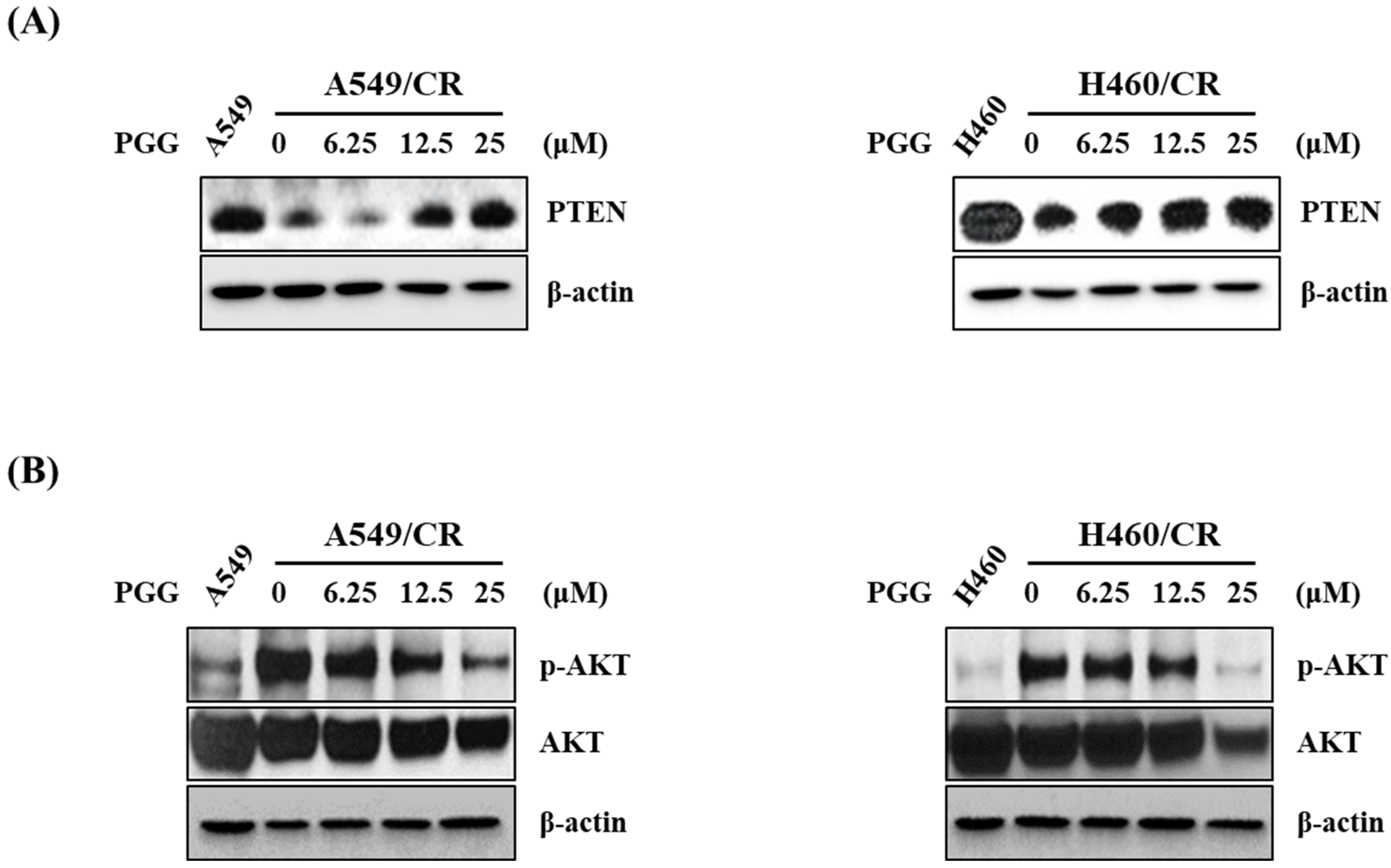
3.5. PGG Induced PTEN and Reduced p-AKT in A549/CR and H460/CR Cells
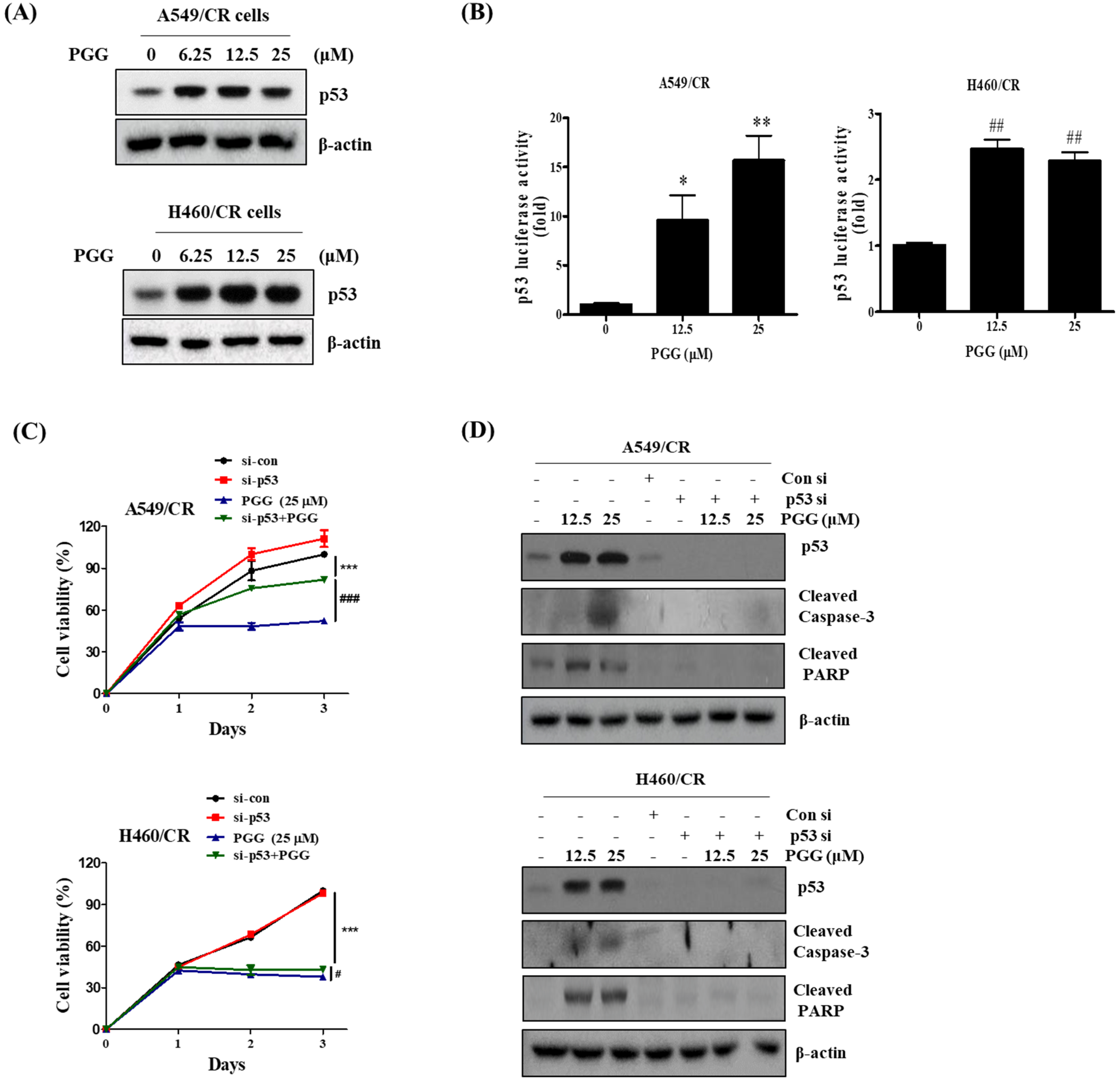
3.6. The Pivotal Role of p53 in PGG Induced Apoptosis in A549/CR and H460/CR Cells
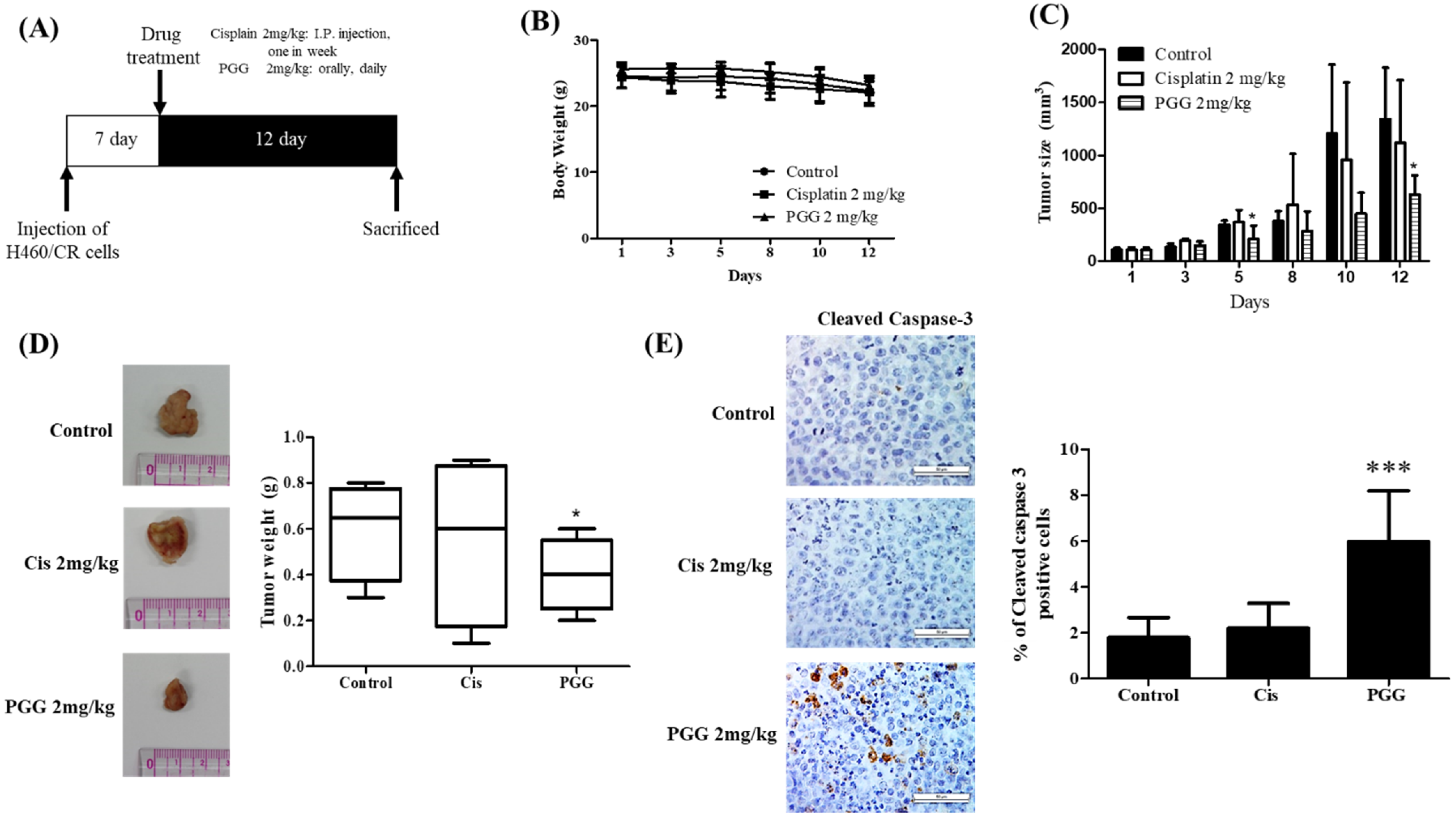
3.7. PGG Reduces the Growth of H460/CR Cells Implanted on the Right Flank of Balb/c Nude Mice with Increased Expression of Caspase 3 Compared to Cisplatin Treated Control Group
4. Discussion
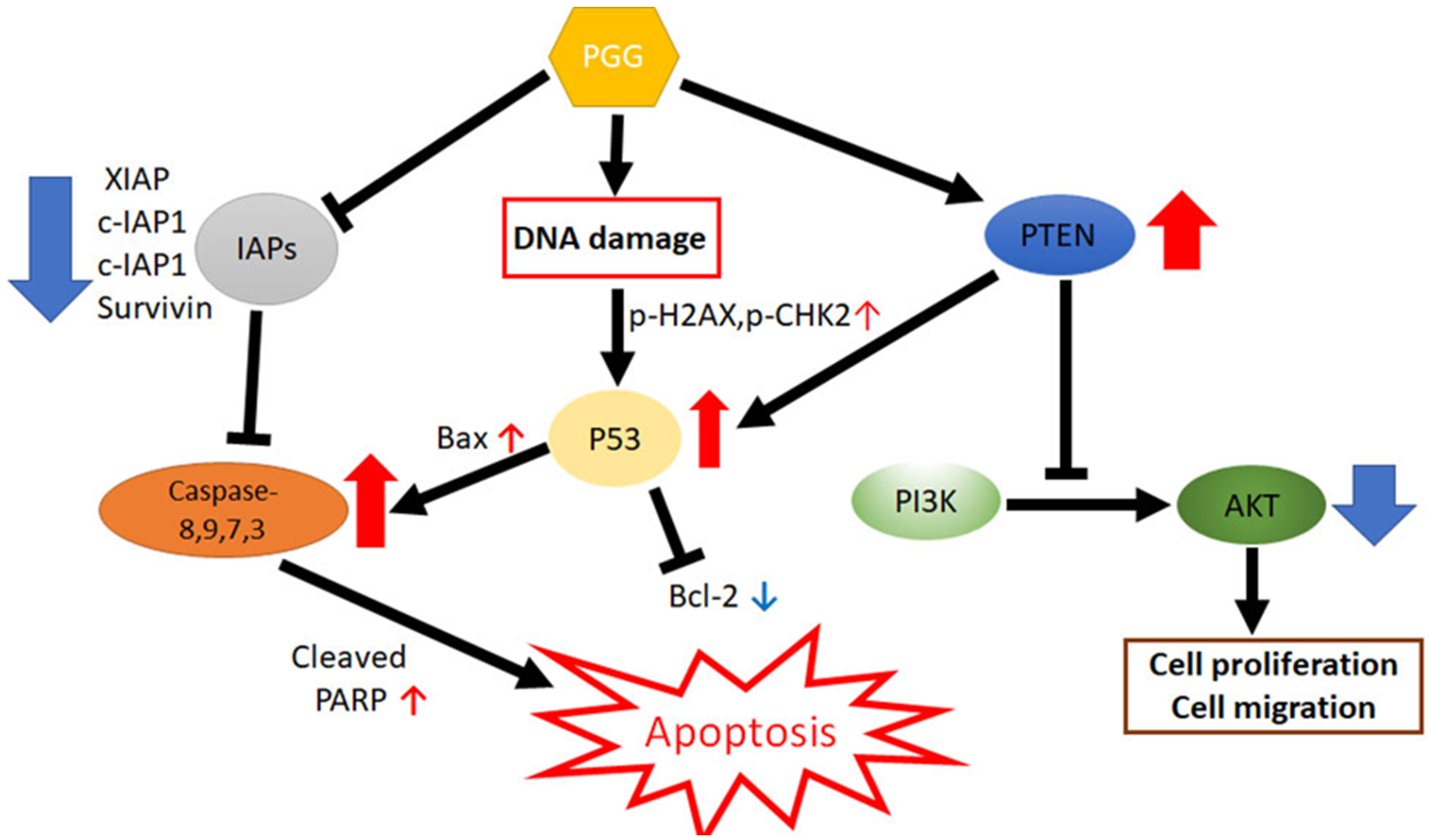
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.M.; Hynan, L.S.; Rossaro, L.; Fontana, R.J.; Stravitz, R.T.; Larson, A.M.; Davern, T.J., 2nd; Murray, N.G.; McCashland, T.; Reisch, J.S.; et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009, 137, 856–864.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florea, A.M.; Busselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, K.; Liu, W.; Hao, Q. PTEN overexpression improves cisplatin-resistance of human ovarian cancer cells through upregulating KRT10 expression. Biochem. Biophys. Res. Commun. 2014, 444, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Krishan, A.; Fitz, C.M.; Andritsch, I. Drug retention, efflux, and resistance in tumor cells. Cytometry 1997, 29, 279–285. [Google Scholar] [CrossRef]
- Ohmichi, M.; Hayakawa, J.; Tasaka, K.; Kurachi, H.; Murata, Y. Mechanisms of platinum drug resistance. Trends Pharmacol. Sci. 2005, 26, 113–116. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- Korwek, Z.; Alster, O. The role of the DNA damage response in apoptosis and cell senescence. Postepy Biochem. 2014, 60, 248–262. [Google Scholar]
- Schmitt, E.; Paquet, C.; Beauchemin, M.; Bertrand, R. DNA-damage response network at the crossroads of cell-cycle checkpoints, cellular senescence and apoptosis. J. Zhejiang Univ. Sci. B 2007, 8, 377–397. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Li, K.; Zhu, W.; Nie, R.; Wang, R.; Li, C. Penta-O-galloyl-beta-d-glucose, a hydrolysable tannin from Radix Paeoniae Alba, inhibits adipogenesis and TNF-alpha-mediated inflammation in 3T3-L1 cells. Chem. Biol. Interact. 2019, 302, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Nithitanakool, S.; Pithayanukul, P.; Bavovada, R. Antioxidant and hepatoprotective activities of thai mango seed kernel extract. Planta Med. 2009, 75, 1118–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, J.E.; Lee, E.O.; Kim, M.S.; Kang, K.S.; Kim, C.H.; Cha, B.C.; Surh, Y.J.; Kim, S.H. Penta-O-galloyl-beta-D-glucose suppresses tumor growth via inhibition of angiogenesis and stimulation of apoptosis: Roles of cyclooxygenase-2 and mitogen-activated protein kinase pathways. Carcinogenesis 2005, 26, 1436–1445. [Google Scholar] [CrossRef]
- Lee, H.J.; Seo, N.J.; Jeong, S.J.; Park, Y.; Jung, D.B.; Koh, W.; Lee, H.J.; Lee, E.O.; Ahn, K.S.; Ahn, K.S.; et al. Oral administration of penta-O-galloyl-beta-D-glucose suppresses triple-negative breast cancer xenograft growth and metastasis in strong association with JAK1-STAT3 inhibition. Carcinogenesis 2011, 32, 804–811. [Google Scholar] [CrossRef]
- Liu, X.; Malki, A.; Cao, Y.; Li, Y.; Qian, Y.; Wang, X.; Chen, X. Glucose- and Triglyceride-lowering Dietary Penta-O-galloyl-alpha-D-Glucose Reduces Expression of PPARgamma and C/EBPalpha, Induces p21-Mediated G1 Phase Cell Cycle Arrest, and Inhibits Adipogenesis in 3T3-L1 Preadipocytes. Exp. Clin. Endocrinol. Diabetes 2015, 123, 308–316. [Google Scholar] [CrossRef]
- Yang, J.; Wang, F.; Chen, X.; Qiu, S.; Cui, L.; Hu, L. beta-Pentagalloyl-Glucose Sabotages Pancreatic Cancer Cells and Ameliorates Cachexia in Tumor-Bearing Mice. Am. J. Chin. Med. 2019, 47, 675–689. [Google Scholar] [CrossRef]
- Kawk, S.H.; Kang, Y.R.; Kim, Y.H. 1,2,3,4,6-Penta-O-galloyl-beta-d-glucose suppresses colon cancer through induction of tumor suppressor. Bioorg. Med. Chem. Lett. 2018, 28, 2117–2123. [Google Scholar] [CrossRef]
- Chai, Y.; Lee, H.J.; Shaik, A.A.; Nkhata, K.; Xing, C.; Zhang, J.; Jeong, S.J.; Kim, S.H.; Lu, J. Penta-O-galloyl-beta-D-glucose induces G1 arrest and DNA replicative S-phase arrest independently of cyclin-dependent kinase inhibitor 1A, cyclin-dependent kinase inhibitor 1B and P53 in human breast cancer cells and is orally active against triple negative xenograft growth. Breast Cancer Res. 2010, 12, R67. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Yin, S.; Jiang, C.; Luo, X.; Guo, X.; Zhao, C.; Fan, L.; Meng, Y.; Lu, J.; Song, X.; et al. Involvement of autophagy induction in penta-1,2,3,4,6-O-galloyl-beta-D-glucose-induced senescence-like growth arrest in human cancer cells. Autophagy 2014, 10, 296–310. [Google Scholar] [CrossRef] [Green Version]
- Park, K.Y.; Lee, H.J.; Jeong, S.J.; Lee, H.J.; Kim, H.S.; Kim, S.H.; Lim, S.; Kim, H.C.; Lu, J.; Kim, S.H. 1,2,3,4,6-Penta-O-galloly-beta-D-glucose suppresses hypoxia-induced accumulation of hypoxia-inducible factor-1alpha and signaling in LNCaP prostate cancer cells. Biol. Pharm. Bull. 2010, 33, 1835–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, T.R.; Jeong, S.J.; Lee, H.J.; Lee, H.J.; Sohn, E.J.; Jung, J.H.; Kim, J.H.; Jung, D.B.; Lu, J.; Kim, S.H. Reactive oxygen species-mediated activation of JNK and down-regulation of DAXX are critically involved in penta-O-galloyl-beta-d-glucose-induced apoptosis in chronic myeloid leukemia K562 cells. Biochem. Biophys. Res. Commun. 2012, 424, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Lee, C. Reversal of Cisplatin resistance by epigallocatechin gallate is mediated by downregulation of axl and tyro 3 expression in human lung cancer cells. Korean J. Physiol. Pharmacol. 2014, 18, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Jeong, S.J.; Kwon, T.R.; Yun, S.M.; Jung, J.H.; Kim, M.; Lee, H.J.; Lee, M.H.; Ko, S.G.; Chen, C.Y.; et al. Cryptotanshinone enhances TNF-alpha-induced apoptosis in chronic myeloid leukemia KBM-5 cells. Apoptosis 2011, 16, 696–707. [Google Scholar] [CrossRef]
- Lee, H.; Lee, H.J.; Jung, J.H.; Shin, E.A.; Kim, S.H. Melatonin disturbs SUMOylation-mediated crosstalk between c-Myc and nestin via MT1 activation and promotes the sensitivity of paclitaxel in brain cancer stem cells. J. Pineal Res. 2018, 65, e12496. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Shin, E.A.; Kim, J.H.; Sim, D.Y.; Lee, H.; Park, J.E.; Lee, H.J.; Kim, S.H. NEDD9 Inhibition by miR-25-5p Activation Is Critically Involved in Co-Treatment of Melatonin- and Pterostilbene-Induced Apoptosis in Colorectal Cancer Cells. Cancers 2019, 11, 1684. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.H.; Jung, D.B.; Kim, H.; Lee, H.; Kang, S.E.; Srivastava, S.K.; Yun, M.; Kim, S.H. Zinc finger protein 746 promotes colorectal cancer progression via c-Myc stability mediated by glycogen synthase kinase 3beta and F-box and WD repeat domain-containing 7. Oncogene 2018, 37, 3715–3728. [Google Scholar] [CrossRef]
- Lu, X.X.; Cao, L.Y.; Chen, X.; Xiao, J.; Zou, Y.; Chen, Q. PTEN Inhibits Cell Proliferation, Promotes Cell Apoptosis, and Induces Cell Cycle Arrest via Downregulating the PI3K/AKT/hTERT Pathway in Lung Adenocarcinoma A549 Cells. BioMed Res. Int. 2016, 2016, 2476842. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yuan, D.; Li, Y.; Qi, Q.; Guo, B.; Yang, S.; Zhou, J.; Xu, L.; Chen, T.; Yang, C.; et al. Involvement of Phosphatase and Tensin Homolog in Cyclin-Dependent Kinase 4/6 Inhibitor-Induced Blockade of Glioblastoma. Front. Pharmacol. 2019, 10, 1316. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Zhu, F.; Cho, Y.Y.; Tang, F.; Zykova, T.; Ma, W.Y.; Bode, A.M.; Dong, Z. Cell apoptosis: Requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell 2006, 23, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Yu, D. PI(3)king apart PTEN’s role in cancer. Clin. Cancer Res. 2010, 16, 4325–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, S.H.; Hengartner, M.O. Programmed cell death: Alive and well in the new millennium. Trends Cell Biol. 2001, 11, 526–534. [Google Scholar] [CrossRef]
- Zhou, J.; Li, P.; Xue, X.; He, S.; Kuang, Y.; Zhao, H.; Chen, S.; Zhi, Q.; Guo, X. Salinomycin induces apoptosis in cisplatin-resistant colorectal cancer cells by accumulation of reactive oxygen species. Toxicol. Lett. 2013, 222, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Eze, N.; Lee, J.W.; Yang, D.H.; Zhu, F.; Neumeister, V.; Sandoval-Schaefer, T.; Mehra, R.; Ridge, J.A.; Forastiere, A.; Chung, C.H.; et al. PTEN loss is associated with resistance to cetuximab in patients with head and neck squamous cell carcinoma. Oral Oncol. 2019, 91, 69–78. [Google Scholar] [CrossRef]
- Kumar, K.; Mishra, J.P.N.; Singh, R.P. Usnic acid induces apoptosis in human gastric cancer cells through ROS generation and DNA damage and causes up-regulation of DNA-PKcs and gamma-H2A.X phosphorylation. Chem. Biol. Interact. 2020, 315, 108898. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef]
- Feng, W.; Dean, D.C.; Hornicek, F.J.; Wang, J.; Jia, Y.; Duan, Z.; Shi, H. ATR and p-ATR are emerging prognostic biomarkers and DNA damage response targets in ovarian cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920982853. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Wengner, A.M.; Siemeister, G.; Lucking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bomer, U.; Moosmayer, D.; Eberspacher, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, P.Y.; Peng, S.F.; Lee, C.Y.; Lu, C.C.; Tsai, S.C.; Shieh, T.M.; Wu, T.S.; Tu, M.G.; Chen, M.Y.; Yang, J.S. Curcumin-loaded nanoparticles induce apoptotic cell death through regulation of the function of MDR1 and reactive oxygen species in cisplatin-resistant CAR human oral cancer cells. Int. J. Oncol. 2013, 43, 1141–1150. [Google Scholar] [CrossRef] [Green Version]
- Parajuli, B.; Lee, H.G.; Kwon, S.H.; Cha, S.D.; Shin, S.J.; Lee, G.H.; Bae, I.; Cho, C.H. Salinomycin inhibits Akt/NF-kappaB and induces apoptosis in cisplatin resistant ovarian cancer cells. Cancer Epidemiol. 2013, 37, 512–517. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-H.; Im, E.; Lee, J.; Lee, H.-J.; Sim, D.Y.; Park, J.E.; Ahn, C.-H.; Kwon, H.H.; Shim, B.S.; Kim, B.; et al. Apoptotic and DNA Damage Effect of 1,2,3,4,6-Penta-O-galloyl-beta-D-glucose in Cisplatin-Resistant Non-Small Lung Cancer Cells via Phosphorylation of H2AX, CHK2 and p53. Cells 2022, 11, 1343. https://doi.org/10.3390/cells11081343
Kim J-H, Im E, Lee J, Lee H-J, Sim DY, Park JE, Ahn C-H, Kwon HH, Shim BS, Kim B, et al. Apoptotic and DNA Damage Effect of 1,2,3,4,6-Penta-O-galloyl-beta-D-glucose in Cisplatin-Resistant Non-Small Lung Cancer Cells via Phosphorylation of H2AX, CHK2 and p53. Cells. 2022; 11(8):1343. https://doi.org/10.3390/cells11081343
Chicago/Turabian StyleKim, Ji-Hyun, Eunji Im, Jihyun Lee, Hyo-Jung Lee, Deok Yong Sim, Ji Eon Park, Chi-Hoon Ahn, Hyeon Hee Kwon, Bum Sang Shim, Bonglee Kim, and et al. 2022. "Apoptotic and DNA Damage Effect of 1,2,3,4,6-Penta-O-galloyl-beta-D-glucose in Cisplatin-Resistant Non-Small Lung Cancer Cells via Phosphorylation of H2AX, CHK2 and p53" Cells 11, no. 8: 1343. https://doi.org/10.3390/cells11081343
APA StyleKim, J.-H., Im, E., Lee, J., Lee, H.-J., Sim, D. Y., Park, J. E., Ahn, C.-H., Kwon, H. H., Shim, B. S., Kim, B., & Kim, S.-H. (2022). Apoptotic and DNA Damage Effect of 1,2,3,4,6-Penta-O-galloyl-beta-D-glucose in Cisplatin-Resistant Non-Small Lung Cancer Cells via Phosphorylation of H2AX, CHK2 and p53. Cells, 11(8), 1343. https://doi.org/10.3390/cells11081343