
The Role of Transglutaminase 2 in the Radioresistance of Melanoma Cells

 , , ,
, , ,  , and
, and
Abstract
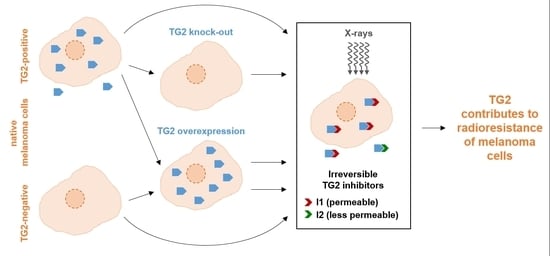
:
1. Introduction
2. Materials and Methods
2.1. Generation and Cultivation of Melanoma Sub-Cell Lines
2.2. Treatment of Melanoma Sub-Cell Lines
2.3. Proliferation and Clonal Expansion
2.4. Spheroidal Expansion
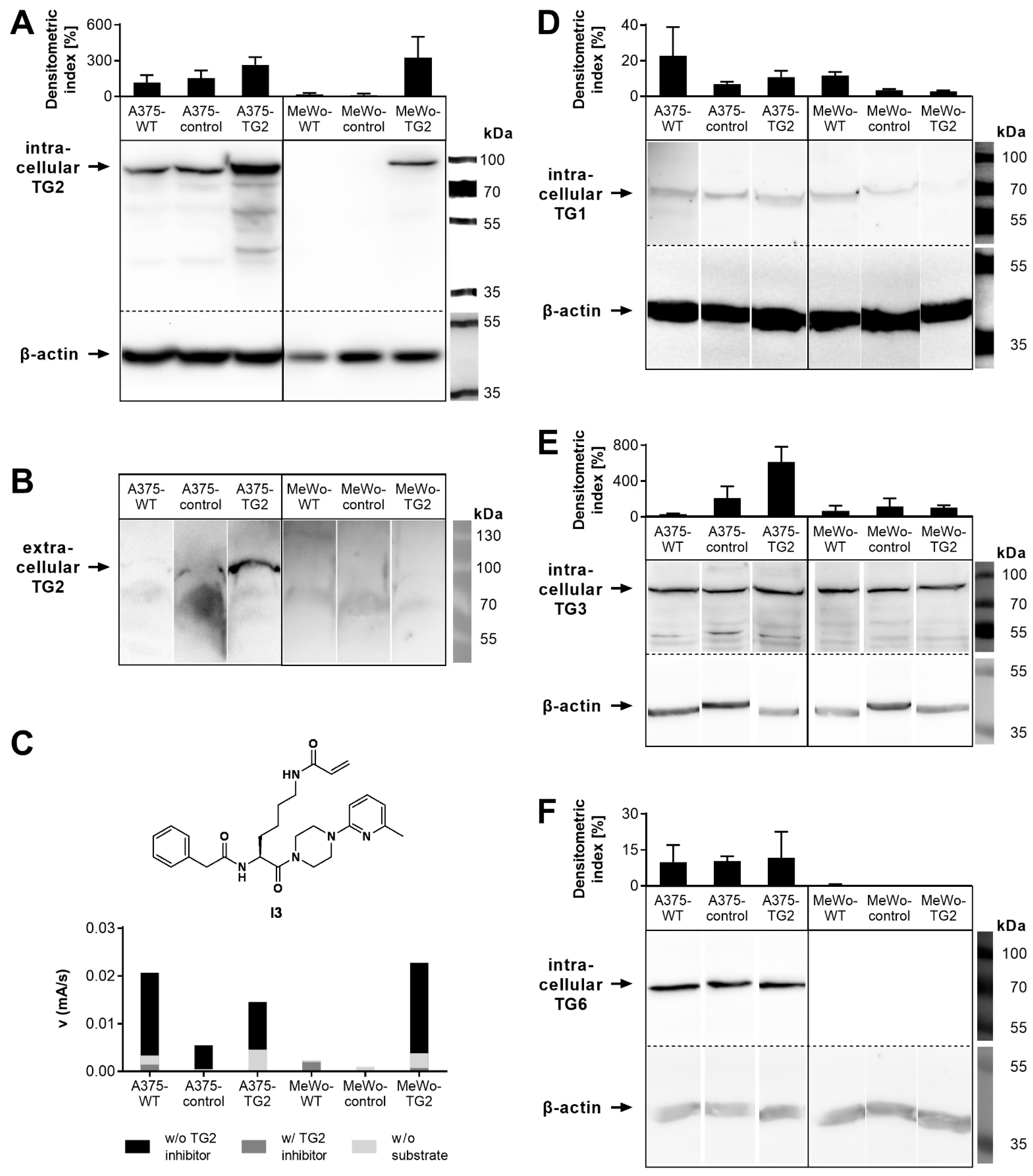
2.5. Western Blot Analysis
2.6. In Vitro TG2 Activity
2.7. Statistical Analysis
3. Results
3.1. TG Biosynthesis, Secretion and Activity of Melanoma Cells
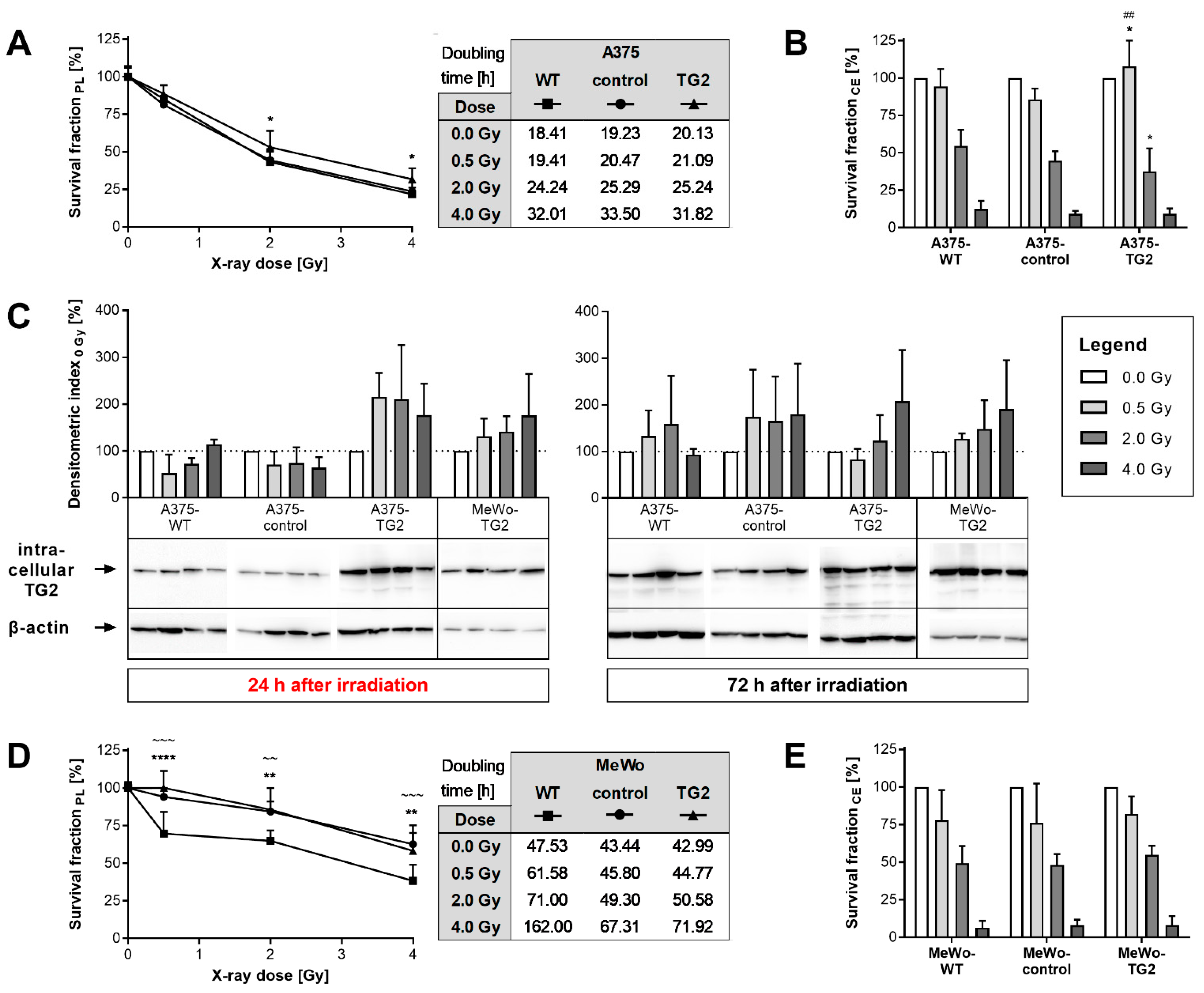
3.2. Proliferation, Clonal Expansion and TG2 Biosynthesis of Melanoma Cells after Irradiation
3.3. Effects of TG2 Knock-Out on Cellular Reaction towards Irradiation
3.4. Effects of TG2 Inhibitors on Cellular Reactions towards Irradiation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| ATP | Adenosine triphosphate |
| DNA | Desoxyribonucleic acid |
| CE | Clonal expansion |
| DMC | N,N-dimethylated casein |
| ECM | Extracellular matrix |
| FAK | Focal adhesion kinase |
| GDP | Guanosine diphosphate |
| GTP | Guanosine triphosphate |
| I1 | Nα-Phenylacetyl-Nε-acryloyl-l-lysine-4-(6-nitropyridin-2-yl) piperazide |
| I2 | Nα-Phenylacetyl-Nε-acryloyl-l-lysine-4-(pyridin-4-yl) piperazide |
| I3 | Nα-Phenylacetyl-Nε-acryloyl-l-lysine-4-(6-methylpyridin-2-yl) piperazide |
| IL | Interleukin |
| KO | Knock-out |
| mRNA | Messenger ribonucleic acid |
| MT | Mesenchymal transdifferentiation |
| NF | Nuclear factor |
| PDGF | Platelet-derived growth factor |
| PL | Proliferation |
| SA | Spheroidal area |
| SD | Standard deviation |
| SHP | Src homology 2 domain-containing tyrosine Phosphatase |
| SP | Specificity protein |
| TAA | Transamidase activity |
| TG | Transglutaminase |
| TGF | Transforming growth factor |
| TNF | Tumor necrosis factor |
References
- Mehta, K.; Kumar, A.; Kim, H.I. Transglutaminase 2: A multi-tasking protein in the complex circuitry of inflammation and cancer. Biochem. Pharmacol. 2010, 80, 1921–1929. [Google Scholar] [CrossRef]
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417. [Google Scholar] [CrossRef] [Green Version]
- Smethurst, P.A.; Griffin, M. Measurement of tissue transglutaminase activity in a permeabilized cell system: Its regulation by Ca2+ and nucleotides. Biochem. J. 1996, 313 Pt 3, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Verderio, E.A.; Johnson, T.; Griffin, M. Tissue transglutaminase in normal and abnormal wound healing: Review article. Amino Acids 2004, 26, 387–404. [Google Scholar] [CrossRef]
- Pavlyukov, M.S.; Antipova, N.V.; Balashova, M.V.; Shakhparonov, M.I. Detection of Transglutaminase 2 conformational changes in living cell. Biochem. Biophys. Res. Commun. 2012, 421, 773–779. [Google Scholar] [CrossRef]
- Achyuthan, K.E.; Greenberg, C.S. Identification of a guanosine triphosphate-binding site on guinea pig liver transglutaminase. Role of GTP and calcium ions in modulating activity. J. Biol. Chem. 1987, 262, 1901–1906. [Google Scholar] [CrossRef]
- Kiraly, R.; Csosz, E.; Kurtan, T.; Antus, S.; Szigeti, K.; Simon-Vecsei, Z.; Korponay-Szabo, I.R.; Keresztessy, Z.; Fesus, L. Functional significance of five noncanonical Ca2+-binding sites of human transglutaminase 2 characterized by site-directed mutagenesis. FEBS J. 2009, 276, 7083–7096. [Google Scholar] [CrossRef] [Green Version]
- Tatsukawa, H.; Fukaya, Y.; Frampton, G.; Martinez-Fuentes, A.; Suzuki, K.; Kuo, T.F.; Nagatsuma, K.; Shimokado, K.; Okuno, M.; Wu, J.; et al. Role of transglutaminase 2 in liver injury via cross-linking and silencing of transcription factor Sp1. Gastroenterology 2009, 136, 1783–1795.e10. [Google Scholar] [CrossRef] [Green Version]
- Mehta, K.; McQueen, T.; Neamati, N.; Collins, S.; Andreeff, M. Activation of retinoid receptors RAR alpha and RXR alpha induces differentiation and apoptosis, respectively, in HL-60 cells. Cell Growth Differ. 1996, 7, 179–186. [Google Scholar]
- Suto, N.; Ikura, K.; Sasaki, R. Expression induced by interleukin-6 of tissue-type transglutaminase in human hepatoblastoma HepG2 cells. J. Biol. Chem. 1993, 268, 7469–7473. [Google Scholar] [CrossRef]
- Kuncio, G.S.; Tsyganskaya, M.; Zhu, J.; Liu, S.L.; Nagy, L.; Thomazy, V.; Davies, P.J.; Zern, M.A. TNF-alpha modulates expression of the tissue transglutaminase gene in liver cells. Am. J. Physiol. 1998, 274, G240–G245. [Google Scholar]
- Quan, G.; Choi, J.Y.; Lee, D.S.; Lee, S.C. TGF-beta1 up-regulates transglutaminase two and fibronectin in dermal fibroblasts: A possible mechanism for the stabilization of tissue inflammation. Arch. Dermatol. Res. 2005, 297, 84–90. [Google Scholar] [CrossRef]
- Ai, L.; Skehan, R.R.; Saydi, J.; Lin, T.; Brown, K.D. Ataxia-Telangiectasia, Mutated (ATM)/Nuclear Factor kappa light chain enhancer of activated B cells (NFkappaB) signaling controls basal and DNA damage-induced transglutaminase 2 expression. J. Biol. Chem. 2012, 287, 18330–18341. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, Y.S.; Choi, D.H.; Bang, M.S.; Han, T.R.; Joh, T.H.; Kim, S.Y. Transglutaminase 2 induces nuclear factor-kappaB activation via a novel pathway in BV-2 microglia. J. Biol. Chem. 2004, 279, 53725–53735. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, Y.; Ohashi, H.; Birckbichler, P.J.; Ikejima, T. Nuclear translocation of tissue type transglutaminase during sphingosine-induced cell death: A novel aspect of the enzyme with DNA hydrolytic activity. Z. Naturforsch. 1998, 53, 352–358. [Google Scholar] [CrossRef]
- Lei, X.; Cao, K.; Chen, Y.; Shen, H.; Liu, Z.; Qin, H.; Cai, J.; Gao, F.; Yang, Y. Nuclear Transglutaminase 2 interacts with topoisomerase II to promote DNA damage repair in lung cancer cells. J. Exp. Clin. Cancer Res. 2021, 40, 224. [Google Scholar] [CrossRef]
- Furini, G.; Schroeder, N.; Huang, L.; Boocock, D.; Scarpellini, A.; Coveney, C.; Tonoli, E.; Ramaswamy, R.; Ball, G.; Verderio, C.; et al. Proteomic Profiling Reveals the Transglutaminase-2 Externalization Pathway in Kidneys after Unilateral Ureteric Obstruction. J. Am. Soc. Nephrol. 2018, 29, 880–905. [Google Scholar] [CrossRef] [Green Version]
- Adamczyk, M.; Griffiths, R.; Dewitt, S.; Knauper, V.; Aeschlimann, D. P2X7 receptor activation regulates rapid unconventional export of transglutaminase-2. J. Cell Sci. 2015, 128, 4615–4628. [Google Scholar] [CrossRef] [Green Version]
- Aeschlimann, D.; Thomazy, V. Protein crosslinking in assembly and remodelling of extracellular matrices: The role of transglutaminases. Connect. Tissue Res. 2000, 41, 1–27. [Google Scholar] [CrossRef]
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003, 4, 140–156. [Google Scholar] [CrossRef]
- Forsprecher, J.; Wang, Z.; Nelea, V.; Kaartinen, M.T. Enhanced osteoblast adhesion on transglutaminase 2-crosslinked fibronectin. Amino Acids 2009, 36, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Chau, D.Y.; Collighan, R.J.; Verderio, E.A.; Addy, V.L.; Griffin, M. The cellular response to transglutaminase-cross-linked collagen. Biomaterials 2005, 26, 6518–6529. [Google Scholar] [CrossRef] [PubMed]
- Akimov, S.S.; Krylov, D.; Fleischman, L.F.; Belkin, A.M. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 2000, 148, 825–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, K.; Fok, J.Y.; Mangala, L.S. Tissue transglutaminase: From biological glue to cell survival cues. Front. Biosci. 2006, 11, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Verderio, E.A.; Telci, D.; Okoye, A.; Melino, G.; Griffin, M. A novel RGD-independent cell adhesion pathway mediated by fibronectin-bound tissue transglutaminase rescues cells from anoikis. J. Biol. Chem. 2003, 278, 42604–42614. [Google Scholar] [CrossRef] [Green Version]
- Zemskov, E.A.; Janiak, A.; Hang, J.; Waghray, A.; Belkin, A.M. The role of tissue transglutaminase in cell-matrix interactions. Front. Biosci. 2006, 11, 1057–1076. [Google Scholar] [CrossRef] [Green Version]
- Eckert, R.L. Transglutaminase 2 takes center stage as a cancer cell survival factor and therapy target. Mol. Carcinog. 2019, 58, 837–853. [Google Scholar] [CrossRef]
- Huang, L.; Xu, A.M.; Liu, W. Transglutaminase 2 in cancer. Am. J. Cancer Res. 2015, 5, 2756–2776. [Google Scholar]
- Fok, J.Y.; Ekmekcioglu, S.; Mehta, K. Implications of tissue transglutaminase expression in malignant melanoma. Mol. Cancer Ther. 2006, 5, 1493–1503. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Oh, Y.T.; Kim, J.Y.; Kim, S.S.; Choi, E.; Kim, T.H.; Hong, J.H.; Chang, N.; Cho, H.J.; Sa, J.K.; et al. Transglutaminase 2 Inhibition Reverses Mesenchymal Transdifferentiation of Glioma Stem Cells by Regulating C/EBPbeta Signaling. Cancer Res. 2017, 77, 4973–4984. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Sun, Z.; Liu, Z. Silencing of TGM2 reverses epithelial to mesenchymal transition and modulates the chemosensitivity of breast cancer to docetaxel. Exp. Ther. Med. 2015, 10, 1413–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Petrusca, D.N.; Satpathy, M.; Nakshatri, H.; Petrache, I.; Matei, D. Tissue transglutaminase protects epithelial ovarian cancer cells from cisplatin-induced apoptosis by promoting cell survival signaling. Carcinogenesis 2008, 29, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Tandler, N.; Mosch, B.; Pietzsch, J. Protein and non-protein biomarkers in melanoma: A critical update. Amino Acids 2012, 43, 2203–2230. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Lin, H.X.; Lin, H.; Guo, L.B.; Ge, N.; Cai, X.Y.; Sun, R.; Chen, W.K.; Li, Q.L.; Hu, W.H. Expression TGM2 and BNIP3 have prognostic significance in laryngeal cancer patients receiving surgery and postoperative radiotherapy: A retrospective study. J. Transl. Med. 2012, 10, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Lee, K.B.; Son, Y.H.; Shin, J.; Lee, J.H.; Kim, H.J.; Hong, A.Y.; Bae, H.W.; Kwon, M.A.; Lee, W.J.; et al. Transglutaminase 2 mediates UV-induced skin inflammation by enhancing inflammatory cytokine production. Cell Death Dis. 2017, 8, e3148. [Google Scholar] [CrossRef]
- Berg, T.J.; Marques, C.; Pantazopoulou, V.; Johansson, E.; von Stedingk, K.; Lindgren, D.; Jeannot, P.; Pietras, E.J.; Bergström, T.; Swartling, F.J.; et al. The Irradiated Brain Microenvironment Supports Glioma Stemness and Survival via Astrocyte-Derived Transglutaminase 2. Cancer Res. 2021, 81, 2101–2115. [Google Scholar] [CrossRef]
- Doss, L.L.; Memula, N. The radioresponsiveness of melanoma. Int. J. Radiat. Oncol. Biol. Phys. 1982, 8, 1131–1134. [Google Scholar]
- Pak, B.J.; Lee, J.; Thai, B.L.; Fuchs, S.Y.; Shaked, Y.; Ronai, Z.; Kerbel, R.S.; Ben-David, Y. Radiation resistance of human melanoma analysed by retroviral insertional mutagenesis reveals a possible role for dopachrome tautomerase. Oncogene 2004, 23, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef] [Green Version]
- Lentini, A.; Provenzano, B.; Caraglia, M.; Shevchenko, A.; Abbruzzese, A.; Beninati, S. Impairment of the metastatic activity of melanoma cells by transglutaminase-catalyzed incorporation of polyamines into laminin and Matrigel. Amino Acids 2008, 34, 251–256. [Google Scholar] [CrossRef]
- Yang, L.; Friedland, S.; Corson, N.; Xu, L. GPR56 inhibits melanoma growth by internalizing and degrading its ligand TG2. Cancer Res. 2014, 74, 1022–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, N.; Michen, S.; Tietze, S.; Töpfer, K.; Schulte, A.; Lamszus, K.; Schmitz, M.; Schackert, G.; Pastan, I.; Temme, A. Engineering NK Cells Modified With an EGFRvIII-specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1alpha-secreting Glioblastoma. J. Immunother. 2015, 38, 197–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wodtke, R.; Hauser, C.; Ruiz-Gomez, G.; Jäckel, E.; Bauer, D.; Lohse, M.; Wong, A.; Pufe, J.; Ludwig, F.A.; Fischer, S.; et al. N(epsilon)-Acryloyllysine Piperazides as Irreversible Inhibitors of Transglutaminase 2: Synthesis, Structure-Activity Relationships, and Pharmacokinetic Profiling. J. Med. Chem. 2018, 61, 4528–4560. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.; Wodtke, R.; Tondera, C.; Wodtke, J.; Neffe, A.T.; Hampe, J.; Lendlein, A.; Löser, R.; Pietzsch, J. Characterization of Tissue Transglutaminase as a Potential Biomarker for Tissue Response toward Biomaterials. ACS Biomater. Sci. Eng. 2019, 5, 5979–5989. [Google Scholar] [CrossRef]
- Wolf, S.; Haase-Kohn, C.; Lenk, J.; Hoppmann, S.; Bergmann, R.; Steinbach, J.; Pietzsch, J. Expression, purification and fluorine-18 radiolabeling of recombinant S100A4: A potential probe for molecular imaging of receptor for advanced glycation endproducts in vivo? Amino Acids 2011, 41, 809–820. [Google Scholar] [CrossRef]
- Hauser, C.; Wodtke, R.; Löser, R.; Pietsch, M. A fluorescence anisotropy-based assay for determining the activity of tissue transglutaminase. Amino Acids 2017, 49, 567–583. [Google Scholar] [CrossRef]
- Wityak, J.; Prime, M.E.; Brookfield, F.A.; Courtney, S.M.; Erfan, S.; Johnsen, S.; Johnson, P.D.; Li, M.; Marston, R.W.; Reed, L.; et al. SAR Development of Lysine-Based Irreversible Inhibitors of Transglutaminase 2 for Huntington’s Disease. ACS Med. Chem. Lett. 2012, 3, 1024–1028. [Google Scholar] [CrossRef] [Green Version]
- Hsu, K.H.; Friedman, H. Dimethyl sulfoxide-induced transglutaminase activity in murine-derived Friend erythroleukemia cells. J. Natl. Cancer Inst. 1983, 70, 965–969. [Google Scholar]
- Szondy, Z.; Sarang, Z.; Molnar, P.; Nemeth, T.; Piacentini, M.; Mastroberardino, P.G.; Falasca, L.; Aeschlimann, D.; Kovacs, J.; Kiss, I.; et al. Transglutaminase 2-/- mice reveal a phagocytosis-associated crosstalk between macrophages and apoptotic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 7812–7817. [Google Scholar] [CrossRef] [Green Version]
- Schärer, O.D. Chemistry and biology of DNA repair. Angew. Chem. Int. Ed. Engl. 2003, 42, 2946–2974. [Google Scholar] [CrossRef]
- Odii, B.O.; Coussons, P. Biological functionalities of transglutaminase 2 and the possibility of its compensation by other members of the transglutaminase family. Sci. World J. 2014, 2014, 714561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deasey, S.; Shanmugasundaram, S.; Nurminskaya, M. Tissue-specific responses to loss of transglutaminase 2. Amino Acids 2013, 44, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frezza, V.; Terrinoni, A.; Pitolli, C.; Mauriello, A.; Melino, G.; Candi, E. Transglutaminase 3 Protects against Photodamage. J. Investig. Dermatol. 2017, 137, 1590–1594. [Google Scholar] [CrossRef] [PubMed]
- Candi, E.; Melino, G.; Mei, G.; Tarcsa, E.; Chung, S.I.; Marekov, L.N.; Steinert, P.M. Biochemical, structural, and transglutaminase substrate properties of human loricrin, the major epidermal cornified cell envelope protein. J. Biol. Chem. 1995, 270, 26382–26390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.M.; Jeon, J.H.; Kim, C.W.; Cho, S.Y.; Kwon, J.C.; Lee, H.J.; Choi, K.H.; Park, S.C.; Kim, I.G. Cell type-specific activation of intracellular transglutaminase 2 by oxidative stress or ultraviolet irradiation: Implications of transglutaminase 2 in age-related cataractogenesis. J. Biol. Chem. 2004, 279, 15032–15039. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay Type | Proliferation | Clonal Expansion | |||
|---|---|---|---|---|---|
| Irradiation Dose | 0–4 Gy | 0 | 0.5 | 2 | 4 |
| A375 | 100,000 | 500 | 500 | 1000 | 1000 |
| MeWo | 200,000 | 1000 | 1000 | 3000 | 3000 |
| Primary Antibodies | Secondary Antibodies | ||||||
|---|---|---|---|---|---|---|---|
| Target | TG1 | TG3 | TG6 | TG2 | ß-Actin | IgG, Rabbit | IgG, Mouse |
| Source | polyclonal rabbit | monoclonal mouse | polyclonal goat | polyclonal rabbit | |||
| Company | Abcam (Cambridge, UK) | Sigma-Aldrich (Steinheim, Germany) | |||||
| Product no. | ab103814 | ab203229 | ab180959 | ab2386 | A5316 | A0545 | A9044 |
| Dilution | 1:100 | 1:500 | 1:1000 | 1:500 | 1:1000 | 1:2000 | 1:2000/10,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aepler, J.; Wodtke, J.; Wodtke, R.; Haase-Kohn, C.; Löser, R.; Pietzsch, J.; Hauser, S. The Role of Transglutaminase 2 in the Radioresistance of Melanoma Cells. Cells 2022, 11, 1342. https://doi.org/10.3390/cells11081342
Aepler J, Wodtke J, Wodtke R, Haase-Kohn C, Löser R, Pietzsch J, Hauser S. The Role of Transglutaminase 2 in the Radioresistance of Melanoma Cells. Cells. 2022; 11(8):1342. https://doi.org/10.3390/cells11081342
Chicago/Turabian StyleAepler, Julia, Johanna Wodtke, Robert Wodtke, Cathleen Haase-Kohn, Reik Löser, Jens Pietzsch, and Sandra Hauser. 2022. "The Role of Transglutaminase 2 in the Radioresistance of Melanoma Cells" Cells 11, no. 8: 1342. https://doi.org/10.3390/cells11081342
APA StyleAepler, J., Wodtke, J., Wodtke, R., Haase-Kohn, C., Löser, R., Pietzsch, J., & Hauser, S. (2022). The Role of Transglutaminase 2 in the Radioresistance of Melanoma Cells. Cells, 11(8), 1342. https://doi.org/10.3390/cells11081342