
MicroRNA-like snoRNA-Derived RNAs (sdRNAs) Promote Castration-Resistant Prostate Cancer


 add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
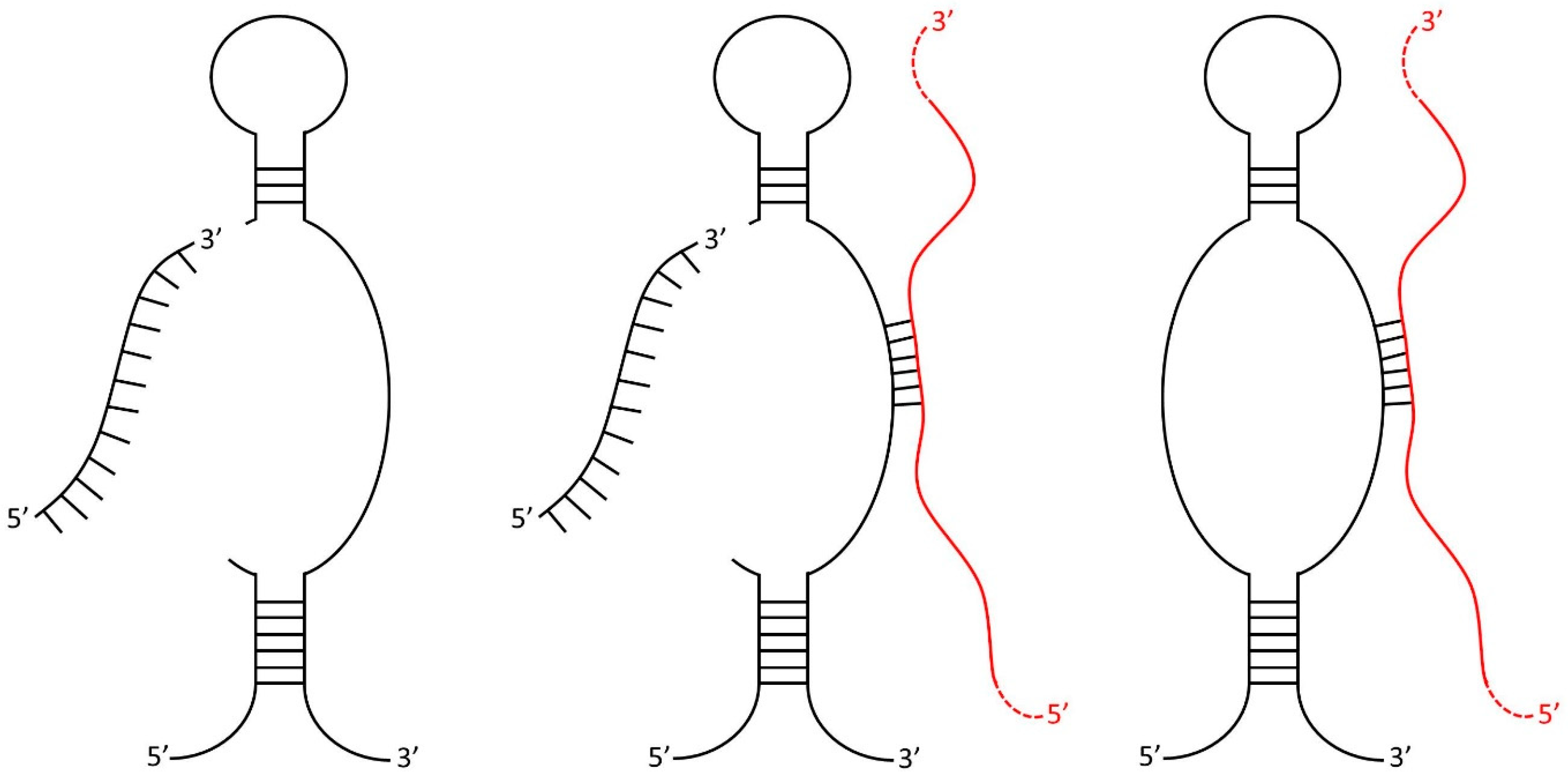
2.1. SURFR Alignment and Data Analysis
2.2. Validation of sdRNA Expression via Quantitative RT-PCR
2.3. Manipulating sdRNA-D19b and -A24 levels
2.4. Phenotypic Assays
2.5. Vector Construction
2.6. Luciferase Assays
2.7. Statistical Analyses
3. Results
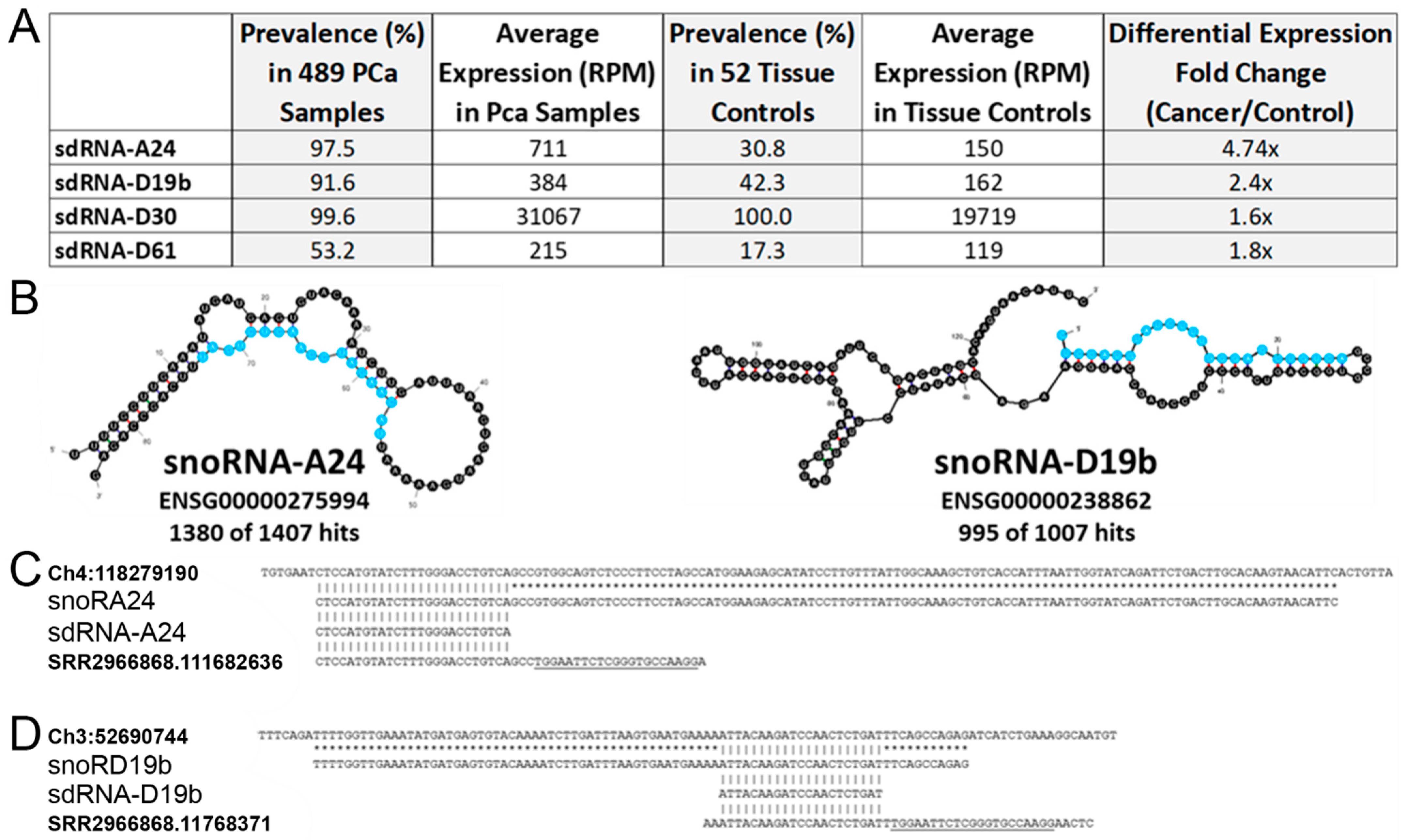
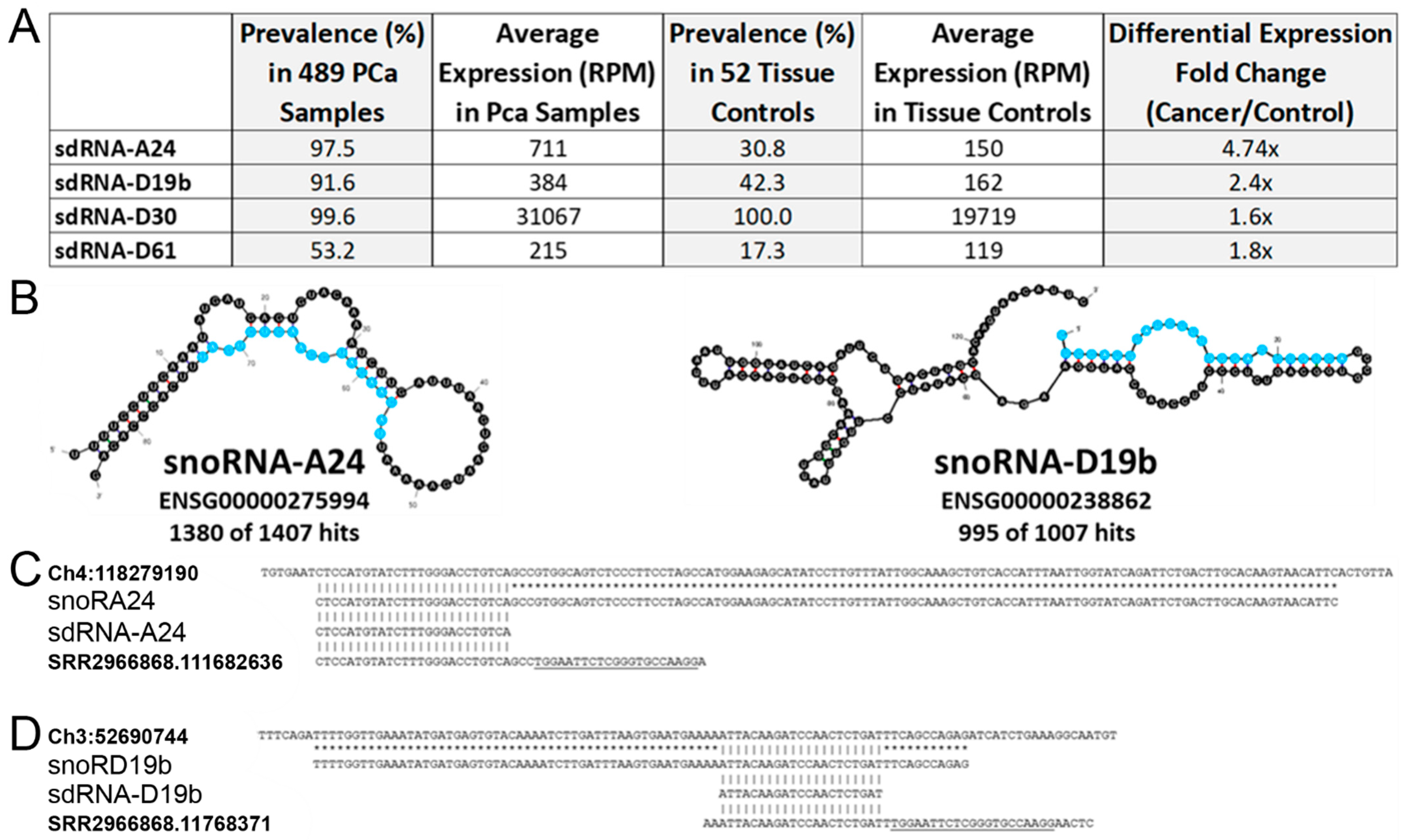
3.1. In Silico Identification of PCa-Overexpressed sdRNAs
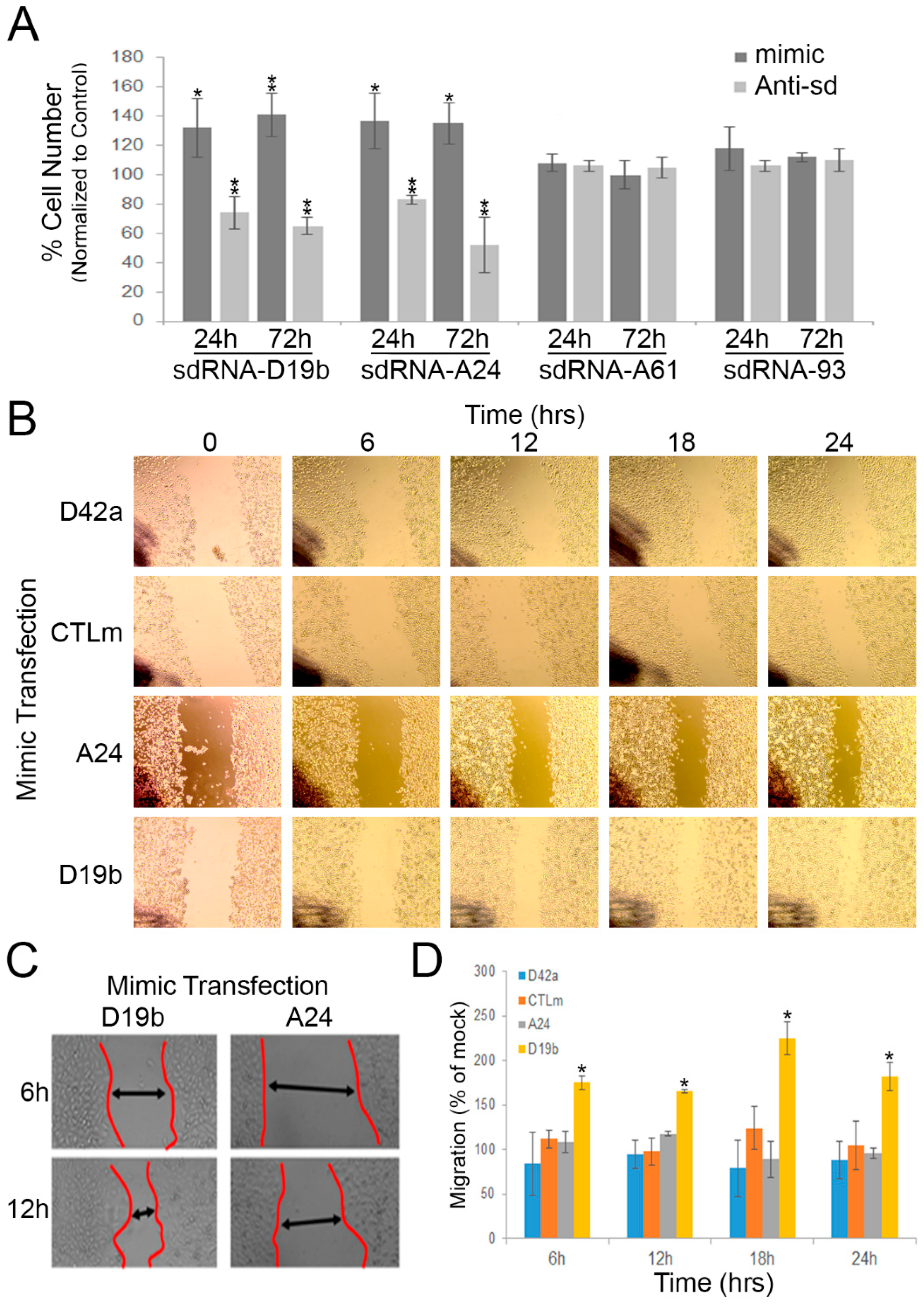
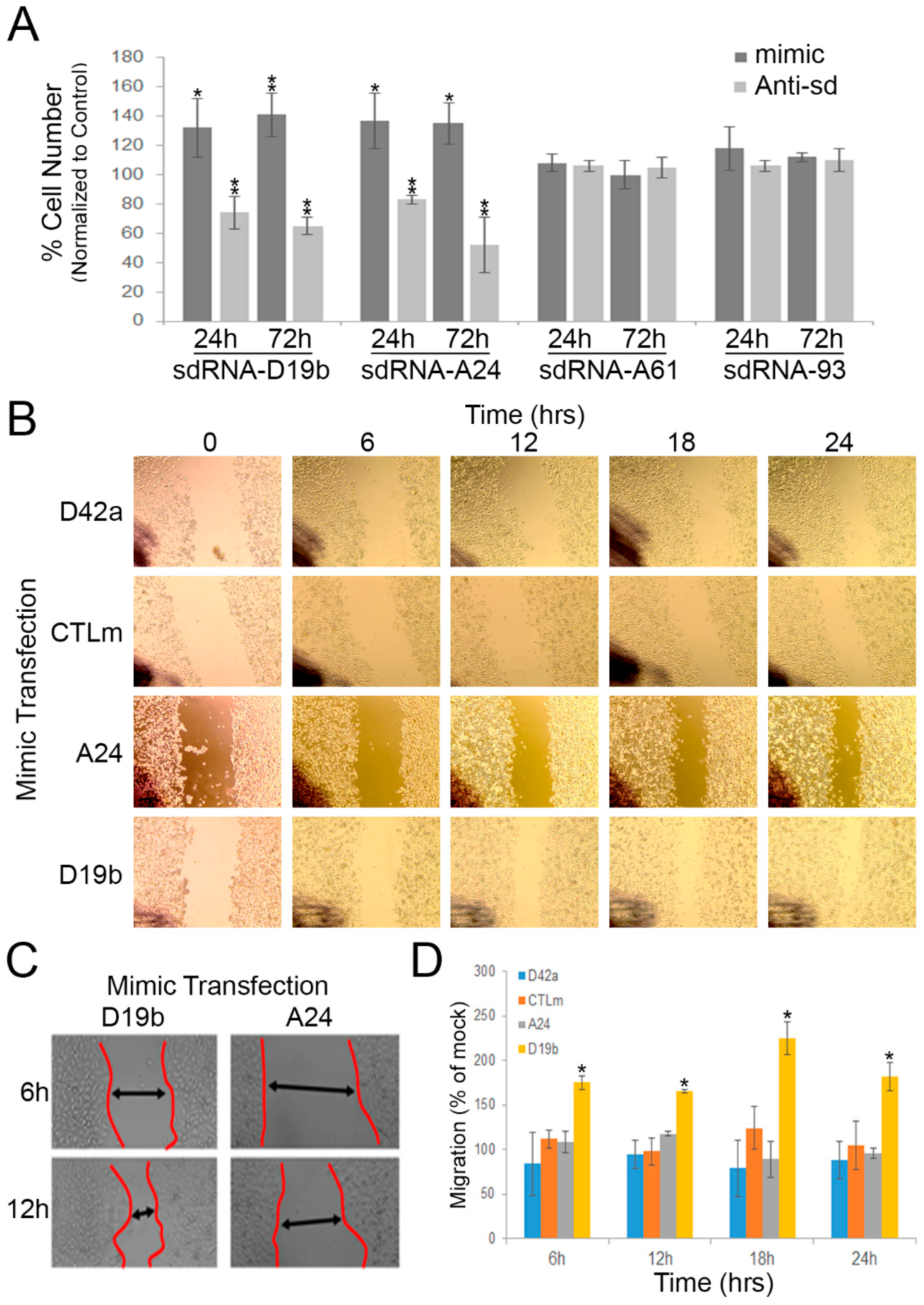
3.2. sdRNA-D19b and sdRNA-A24 Expressions Directly Affect PC3 Cell Proliferation
3.3. sdRNA-D19b Overexpression Enhances PC3 Cell Migration
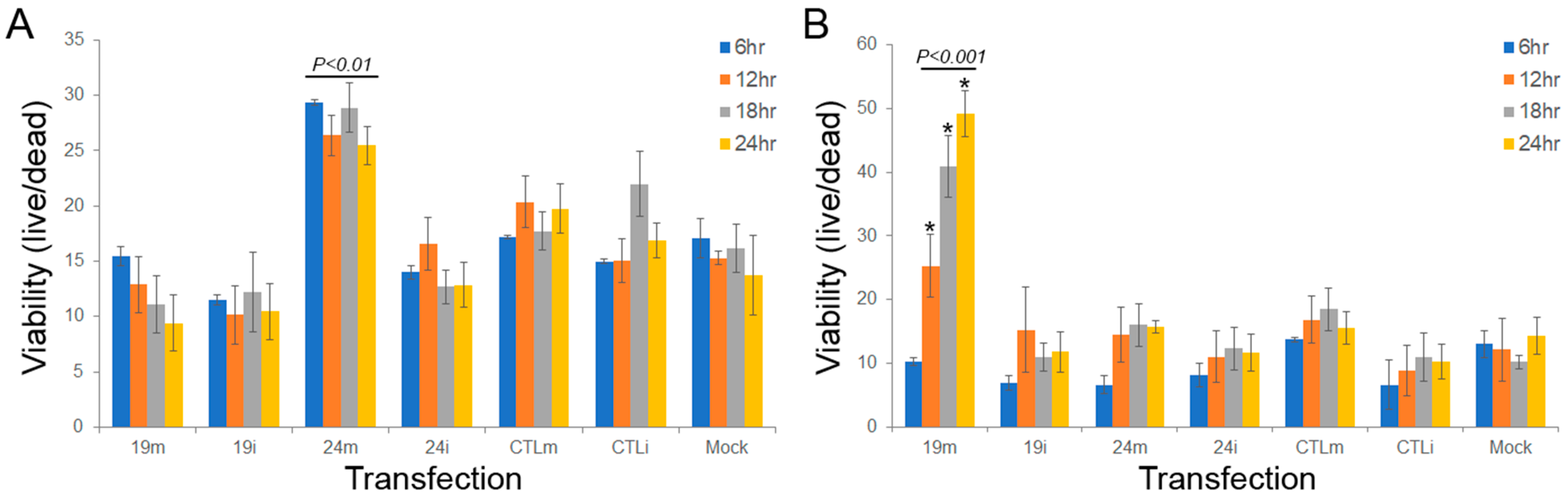
3.4. sdRNA-D19b and sdRNA-A24 Manipulations Alter Drug Sensitivities In Vitro
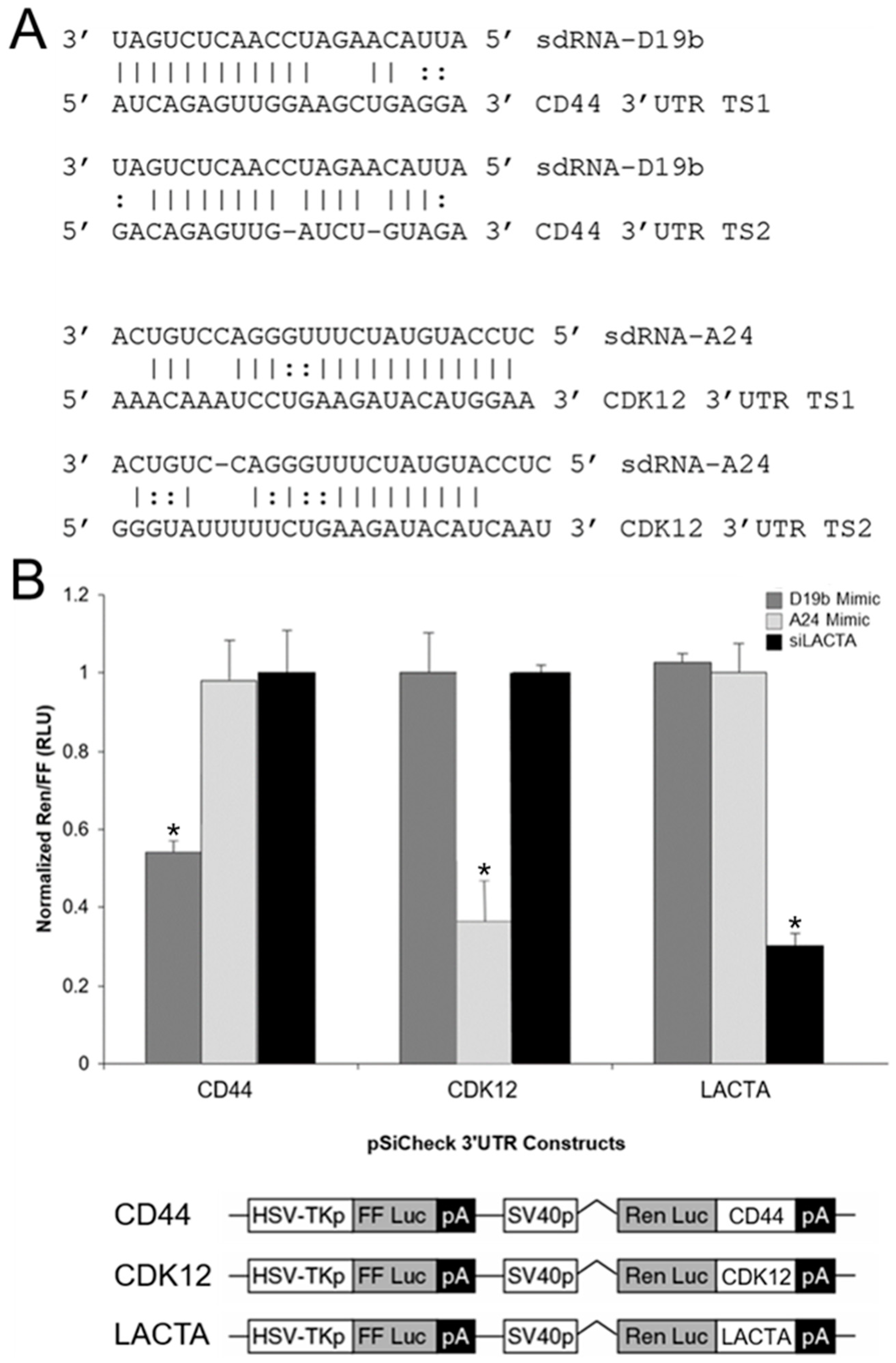
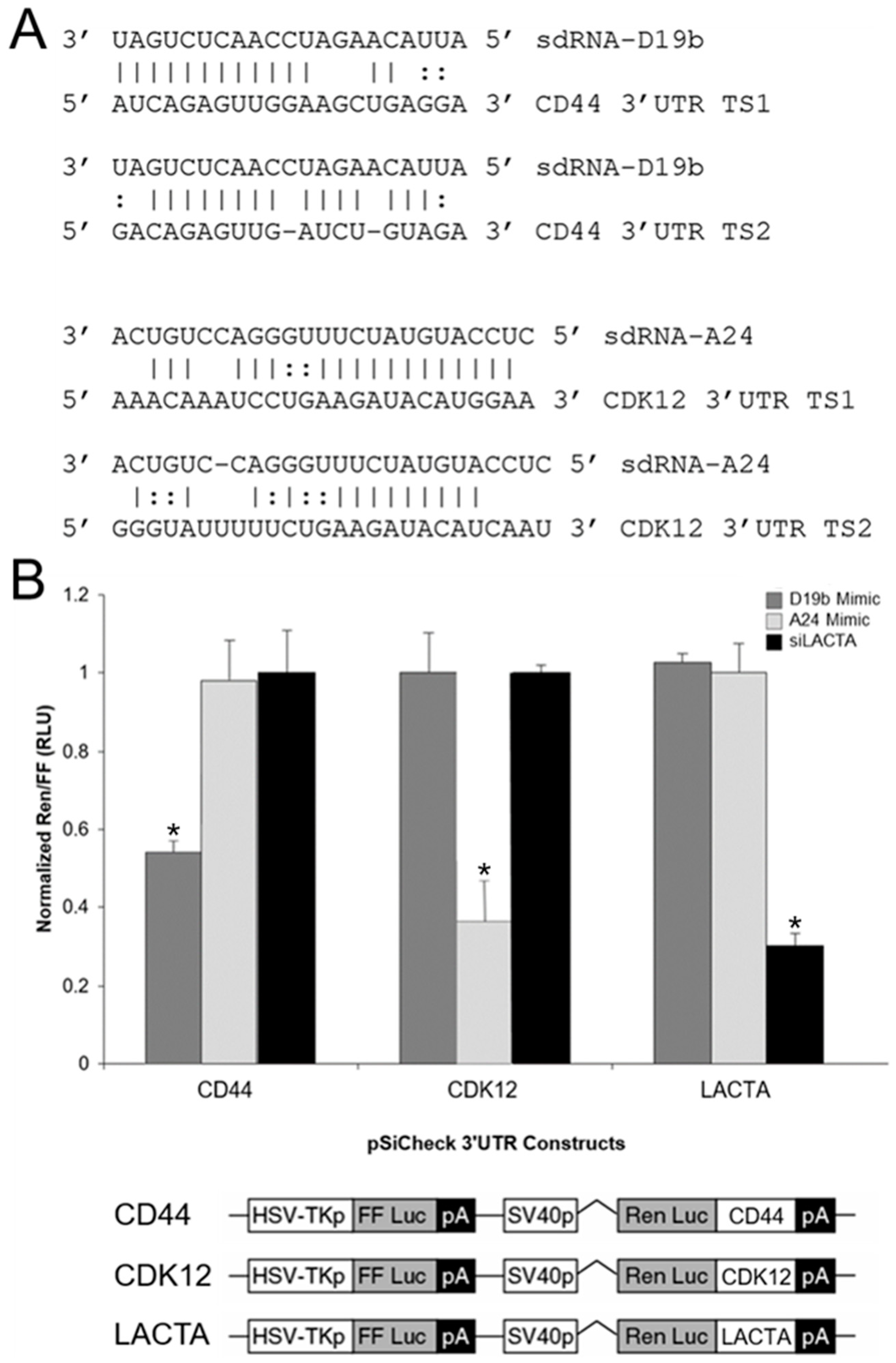
3.5. sdRNA-D19b and sdRNA-A24 Target the 3′UTRs of CD44 and CDK12, Respectively
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kawaji, H.; Nakamura, M.; Takahashi, Y.; Sandelin, A.; Katayama, S.; Fukuda, S.; Daub, C.O.; Kai, C.; Kawai, J.; Yasuda, J.; et al. Hidden layers of human small RNAs. BMC Genom. 2008, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Taft, R.J.; Glazov, E.A.; Lassmann, T.; Hayashizaki, Y.; Carninci, P.; Mattick, J.S. Small RNAs derived from snoRNAs. RNA 2009, 15, 1233–1240. [Google Scholar] [CrossRef] [Green Version]
- Cole, C.; Sobala, A.; Lu, C.; Thatcher, S.R.; Bowman, A.; Brown, J.W.S.; Green, P.J.; Barton, G.J.; Hutvagner, G. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA 2009, 15, 2147–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Shibata, Y.; Malhotra, A.; Dutta, A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 2009, 23, 2639–2649. [Google Scholar] [CrossRef] [Green Version]
- Haussecker, D.; Huang, Y.; Lau, A.; Parameswaran, P.; Fire, A.Z.; Kay, M.A. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA 2010, 16, 673–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tollervey, D.; Kiss, T. Function and synthesis of small nucleolar RNAs. Curr. Opin. Cell Biol. 1997, 9, 337–342. [Google Scholar] [CrossRef]
- Patterson, D.G.; Roberts, J.T.; King, V.M.; Houserova, D.; Barnhill, E.C.; Crucello, A.; Polska, C.J.; Brantley, L.W.; Kaufman, G.C.; Nguyen, M.; et al. Human snoRNA-93 is processed into a microRNA-like RNA that promotes breast cancer cell invasion. NPJ Breast Cancer 2017, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Ender, C.; Krek, A.; Friedländer, M.R.; Beitzinger, M.; Weinmann, L.; Chen, W.; Pfeffer, S.; Rajewsky, N.; Meister, G. A Human snoRNA with MicroRNA-like Functions. Mol. Cell 2008, 32, 519–528. [Google Scholar] [CrossRef]
- Brameier, M.; Herwig, A.; Reinhardt, R.; Walter, L.; Gruber, J. Human box C/D snoRNAs with miRNA like functions: Expanding the range of regulatory RNAs. Nucleic Acids Res. 2010, 39, 675–686. [Google Scholar] [CrossRef]
- Martens-Uzunova, E.S.; Olvedy, M.; Jenster, G. Beyond microRNA—Novel RNAs derived from small non-coding RNA and their implication in cancer. Cancer Lett. 2013, 340, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Martens-Uzunova, E.S.; Hoogstrate, Y.; Kalsbeek, A.; Pigmans, B.; Vredenbregt-van den Berg, M.; Dits, N.; Nielsen, S.J.; Baker, A.; Visakorpi, T.; Bangma, C.; et al. C/D-box snoRNA-derived RNA production is associated with malignant transformation and metastatic progression in prostate cancer. Oncotarget 2015, 6, 17430–17444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falaleeva, M.; Stamm, S. Processing of snoRNAs as a new source of regulatory non-coding RNAs snoRNA fragments form a new class of functional RNAs. BioEssays 2013, 35, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogstrate, Y.; Jenster, G.; Martens-Uzunova, E.S. FlaiMapper: Computational annotation of small ncRNA-derived fragments using RNA-seq high-throughput data. Bioinformatics 2015, 31, 665–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasukurthi, M.V.; Li, S.; Borchert, G.M.; Huang, J.; Zhang, D.; Housevera, M.; Huang, Y.; Tan, S.; Ma, B.; Li, D.; et al. SURFr: Algorithm for identification and analysis of ncRNA-derived RNAs. In Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 18–21 November 2019; pp. 1504–1507. [Google Scholar] [CrossRef]
- Kasukurthi, M.V.; Houserova, D.; Huang, Y.; Barchie, A.A.; Roberts, J.T.; Li, D.; Wu, B.; Huang, J.; Borchert, G.M. SALTS—SURFR (sncRNA) And LAGOOn (lncRNA) Transcriptomics Suite. bioRxiv 2021. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acunzo, M.; Romano, G.; Wernicke, D.; Croce, C.M. MicroRNA and cancer—A brief overview. Adv. Biol. Regul. 2015, 57, 1–9. [Google Scholar] [CrossRef]
- Zaheer, U.; Faheem, M.; Qadri, I.; Begum, N.; Yassine, H.M.; Al Thani, A.A.; Mathew, S. Expression profile of MicroRNA: An Emerging Hallmark of Cancer. Curr. Pharm. Des. 2019, 25, 642–653. [Google Scholar] [CrossRef]
- Kent, O.A.; Mendell, J.T.; Rottapel, R. Transcriptional Regulation of miR-31 by Oncogenic KRAS Mediates Metastatic Phenotypes by Repressing RASA1. Mol. Cancer Res. 2016, 14, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Lin, H.; Luo, X.; Luo, X.; Wang, Z. miR-605joins p53 network to form a p53:miR-605:Mdm2 positive feedback loop in response to stress. EMBO J. 2011, 30, 524–532. [Google Scholar] [CrossRef]
- Zhou, Y.-J.; Yang, H.-Q.; Xia, W.; Cui, L.; Xu, R.-F.; Lu, H.; Xue, Z.; Zhang, B.; Tian, Z.-N.; Cao, Y.-J.; et al. Down-regulation of miR-605 promotes the proliferation and invasion of prostate cancer cells by up-regulating EN2. Life Sci. 2017, 190, 7–14. [Google Scholar] [CrossRef]
- Alhasan, A.H.; Scott, A.W.; Wu, J.J.; Feng, G.; Meeks, J.J.; Thaxton, C.S.; Mirkin, C.A. Circulating microRNA signature for the diagnosis of very high-risk prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 10655–10660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.-P.; Lévesque, E.; Guillemette, C.; Yu, C.-C.; Huang, C.-Y.; Lin, V.C.; Chung, I.-C.; Chen, L.-C.; Laverdière, I.; Lacombe, L.; et al. Genetic variants in microRNAs and microRNA target sites predict biochemical recurrence after radical prostatectomy in localized prostate cancer. Int. J. Cancer 2014, 135, 2661–2667. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.S.; Avolio, F.; Ono, M.; Lamond, A.I.; Barton, G.J. Human miRNA Precursors with Box H/ACA snoRNA Features. PLoS Comput. Biol. 2009, 5, e1000507. [Google Scholar] [CrossRef] [Green Version]
- Martens-Uzunova, E.S.; Jalava, S.E.; Dits, N.F.; van Leenders, G.J.L.H.; Møller, S.; Trapman, J.; Bangma, C.H.; Litman, T.; Visakorpi, T.; Jenster, G. Diagnostic and prognostic signatures from the small non-coding RNA transcriptome in prostate cancer. Oncogene 2012, 31, 978–991. [Google Scholar] [CrossRef] [Green Version]
- Blenkiron, C.; Hurley, D.G.; Fitzgerald, S.; Print, C.G.; Lasham, A. Links between the Oncoprotein YB-1 and Small Non-Coding RNAs in Breast Cancer. PLoS ONE 2013, 8, e80171. [Google Scholar] [CrossRef]
- Yu, F.; Bracken, C.P.; Pillman, K.A.; Lawrence, D.M.; Goodall, G.J.; Callen, D.F.; Neilsen, P.M. p53 Represses the Oncogenic Sno-MiR-28 Derived from a SnoRNA. PLoS ONE 2015, 10, e0129190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Chen, X.; Zhang, X.; Duan, X.; Pan, T.; Hu, Q.; Zhang, Y.; Zhong, F.; Liu, J.; Zhang, H.; et al. An Lnc RNA (GAS5)/SnoRNA-derived piRNA induces activation of TRAIL gene by site-specifically recruiting MLL/COMPASS-like complexes. Nucleic Acids Res. 2015, 43, 3712–3725. [Google Scholar] [CrossRef] [PubMed]
- Stepanov, G.A.; Filippova, J.A.; Nushtaeva, A.A.; Kuligina, E.V.; Koval, O.A.; Richter, V.A.; Semenov, D.V. Artificial Analogues of Circulating Box C/D RNAs Induce Strong Innate Immune Response and MicroRNA Activation in Human Adenocarcinoma Cells. Adv. Exp. Med. Biol. 2016, 924, 121–125. [Google Scholar] [CrossRef]
- Saranyutanon, S.; Srivastava, S.K.; Pai, S.; Singh, S.; Singh, A.P. Therapies Targeted to Androgen Receptor Signaling Axis in Prostate Cancer: Progress, Challenges, and Hope. Cancers 2019, 12, 51. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Men Health 2019, 37, 288–295. [Google Scholar] [CrossRef]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Targeting molecular resistance in castration-resistant prostate cancer. BMC Med. 2015, 13, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; He, B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Rstudio: Integrated Development Environment for R. Available online: http://www.rstudio.org (accessed on 14 January 2021).
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Tai, S.; Sun, Y.; Squires, J.M.; Zhang, H.; Oh, W.K.; Liang, C.-Z.; Huang, J. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 2011, 71, 1668–1679. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Li, W.; Qian, L.; Lin, J.; Huang, G.; Hao, N.; Wei, X.; Wang, W.; Liang, J. CD44 regulates prostate cancer proliferation, invasion and migration via PDK1 and PFKFB4. Oncotarget 2017, 8, 65143–65151. [Google Scholar] [CrossRef] [Green Version]
- Schweizer, M.T.; Ha, G.; Gulati, R.; Brown, L.C.; McKay, R.R.; Dorff, T.; Hoge, A.C.H.; Reichel, J.; Vats, P.; Kilari, D.; et al. CDK12-Mutated Prostate Cancer: Clinical Outcomes with Standard Therapies and Immune Checkpoint Blockade. JCO Precis. Oncol. 2020, 4, 382–392. [Google Scholar] [CrossRef]
- Giacinti, S.; Poti, G.; Roberto, M.; Macrini, S.; Bassanelli, M.; Di Pietro, F.; Aschelter, A.M.; Ceribelli, A.; Ruggeri, E.M.; Marchetti, P. Molecular Basis of Drug Resistance and Insights for New Treatment Approaches in mCRPC. Anticancer Res. 2018, 38, 6029–6039. [Google Scholar] [CrossRef] [PubMed]
- Seruga, B.; Ocana, A.; Tannock, I.F. Drug resistance in metastatic castration-resistant prostate cancer. Nat. Rev. Clin. Oncol. 2010, 8, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Lee, Y.H.; Koo, K.C. Current Status and Future Perspectives of Androgen Receptor Inhibition Therapy for Prostate Cancer: A Comprehensive Review. Biomolecules 2021, 11, 492. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Torres, J.; José, E.S. Src Tyrosine Kinase Inhibitors: New Perspectives on Their Immune, Antiviral, and Senotherapeutic Potential. Front. Pharmacol. 2019, 10, 1011. [Google Scholar] [CrossRef]
- Peterson, S.M.; Thompson, J.A.; Ufkin, M.L.; Sathyanarayana, P.; Liaw, L.; Congdon, C.B. Common features of microRNA target prediction tools. Front. Genet. 2014, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Sethupathy, P.; Corda, B.; Hatzigeorgiou, A.G. TarBase: A comprehensive database of experimentally supported animal microRNA targets. RNA 2005, 12, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; Da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, D146–D152. [Google Scholar] [CrossRef]
- Iczkowski, K.A. Cell adhesion molecule CD44: Its functional roles in prostate cancer. Am. J. Transl. Res. 2011, 3, 1–7. [Google Scholar]
- Wu, Y.-M.; Cieślik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782.e14. [Google Scholar] [CrossRef] [Green Version]
- Mei, J.; Liu, Y.; Yu, X.; Hao, L.; Ma, T.; Zhan, Q.; Zhang, Y.; Zhu, Y. YWHAZ interacts with DAAM1 to promote cell migration in breast cancer. Cell Death Discov. 2021, 7, 221. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Contreras, A.; Holm, M.; Uechi, T.; Forester, C.M.; Pang, X.; Jackson, C.; Calvert, M.E.; Chen, B.; Quigley, D.A.; et al. A single H/ACA small nucleolar RNA mediates tumor suppression downstream of oncogenic RAS. eLife 2019, 8, 48847. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Lee, S.-S. Therapeutic advances of miRNAs: A preclinical and clinical update. J. Adv. Res. 2021, 28, 127–138. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coley, A.B.; Stahly, A.N.; Kasukurthi, M.V.; Barchie, A.A.; Hutcheson, S.B.; Houserova, D.; Huang, Y.; Watters, B.C.; King, V.M.; Dean, M.A.; et al. MicroRNA-like snoRNA-Derived RNAs (sdRNAs) Promote Castration-Resistant Prostate Cancer. Cells 2022, 11, 1302. https://doi.org/10.3390/cells11081302
Coley AB, Stahly AN, Kasukurthi MV, Barchie AA, Hutcheson SB, Houserova D, Huang Y, Watters BC, King VM, Dean MA, et al. MicroRNA-like snoRNA-Derived RNAs (sdRNAs) Promote Castration-Resistant Prostate Cancer. Cells. 2022; 11(8):1302. https://doi.org/10.3390/cells11081302
Chicago/Turabian StyleColey, Alexander B., Ashlyn N. Stahly, Mohan V. Kasukurthi, Addison A. Barchie, Sam B. Hutcheson, Dominika Houserova, Yulong Huang, Brianna C. Watters, Valeria M. King, Meghan A. Dean, and et al. 2022. "MicroRNA-like snoRNA-Derived RNAs (sdRNAs) Promote Castration-Resistant Prostate Cancer" Cells 11, no. 8: 1302. https://doi.org/10.3390/cells11081302
APA StyleColey, A. B., Stahly, A. N., Kasukurthi, M. V., Barchie, A. A., Hutcheson, S. B., Houserova, D., Huang, Y., Watters, B. C., King, V. M., Dean, M. A., Roberts, J. T., DeMeis, J. D., Amin, K. V., McInnis, C. H., Godang, N. L., Wright, R. M., Haider, D. F., Piracha, N. B., Brown, C. L., ... Borchert, G. M. (2022). MicroRNA-like snoRNA-Derived RNAs (sdRNAs) Promote Castration-Resistant Prostate Cancer. Cells, 11(8), 1302. https://doi.org/10.3390/cells11081302