
NUP133 Controls Nuclear Pore Assembly, Transcriptome Composition, and Cytoskeleton Regulation in Podocytes

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Cell Culture
2.2. CRISPR/Cas9
2.3. Expression Plasmids
2.4. Immunoprecipitatio
2.5. Western Blot Analysis
2.6. Antibodies and Fluorescent Dyes
2.7. Immunofluorescence
2.8. Microscopy
2.9. Nuclear IF Quantification
2.10. Integrin Adhesion Complex Analysis
2.11. Cell Spreading Assay
2.12. RNA Sequencing
2.13. 3D Structure Model
2.14. Statistical Analysis
3. Results
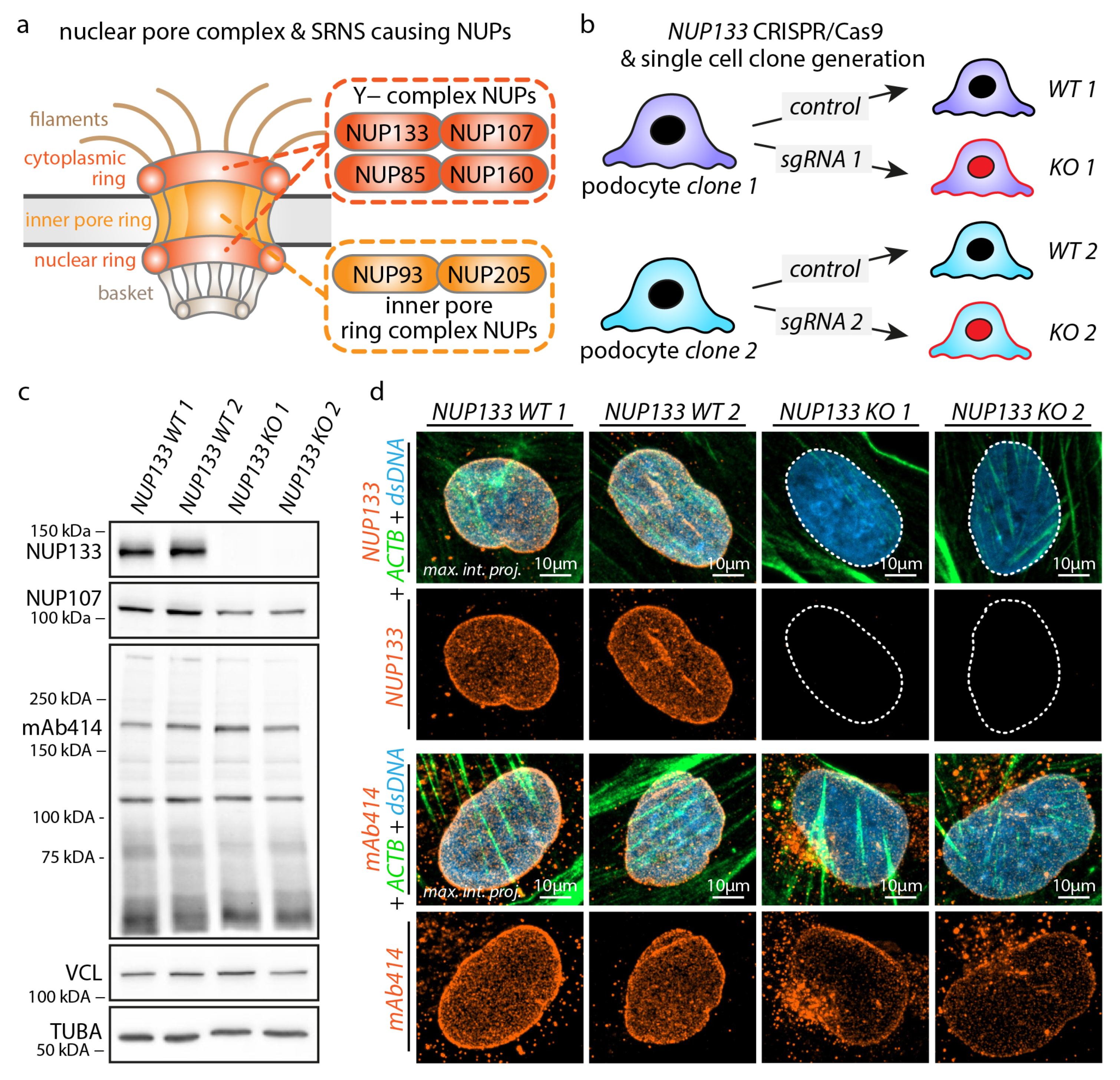
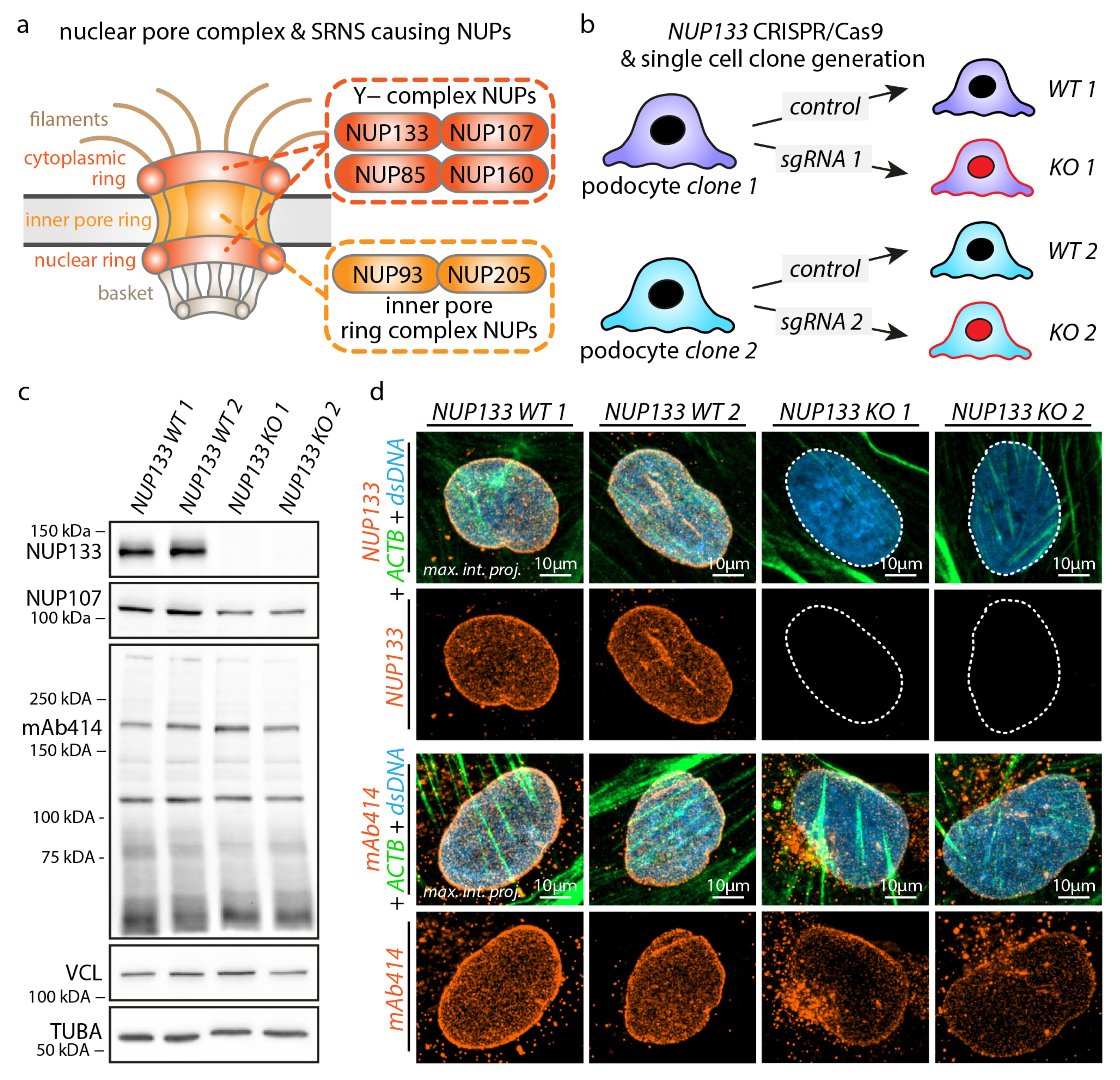
3.1. Knockout of NUP133 Impedes Nuclear Pore Complex Formation in Podocytes
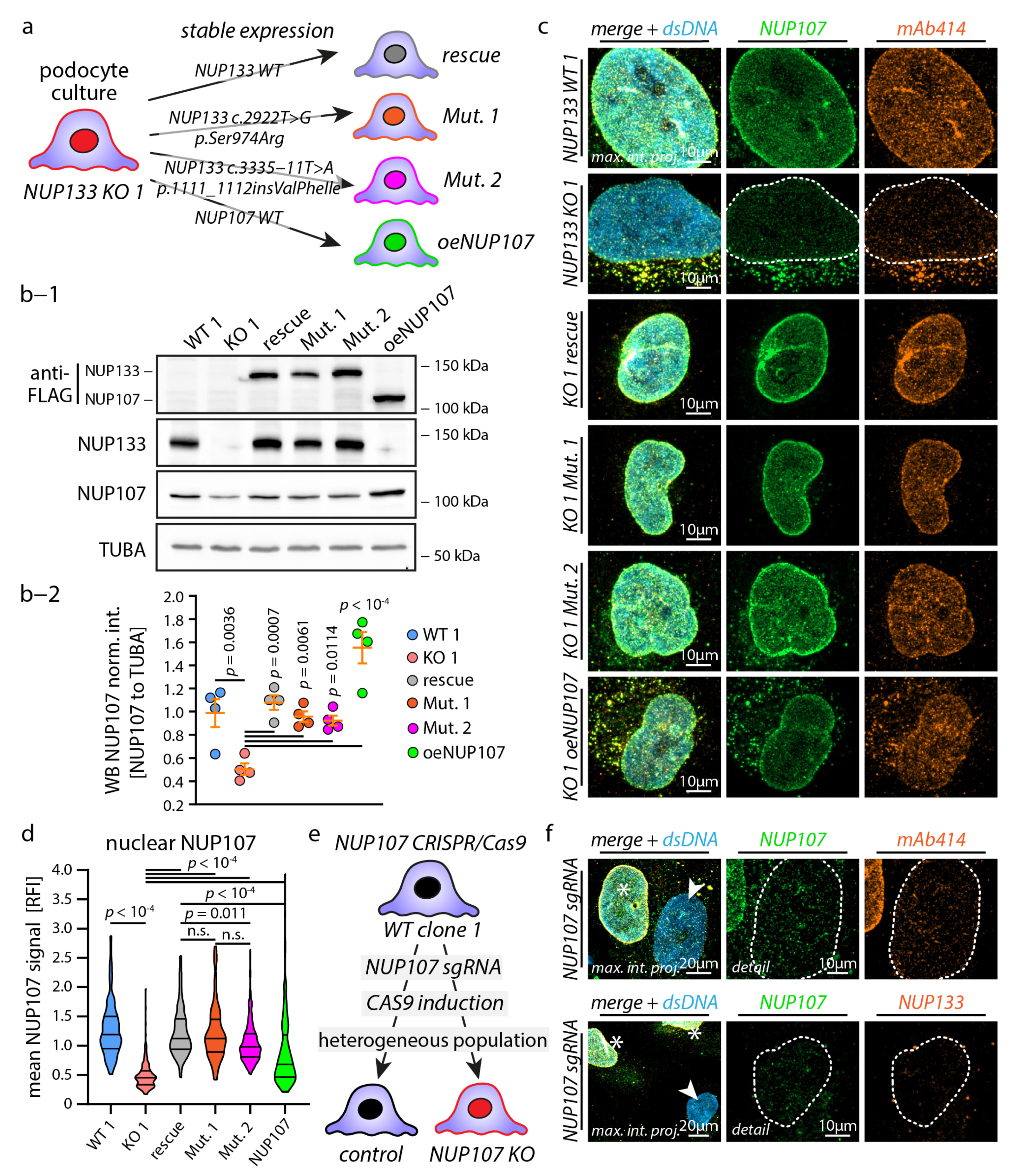
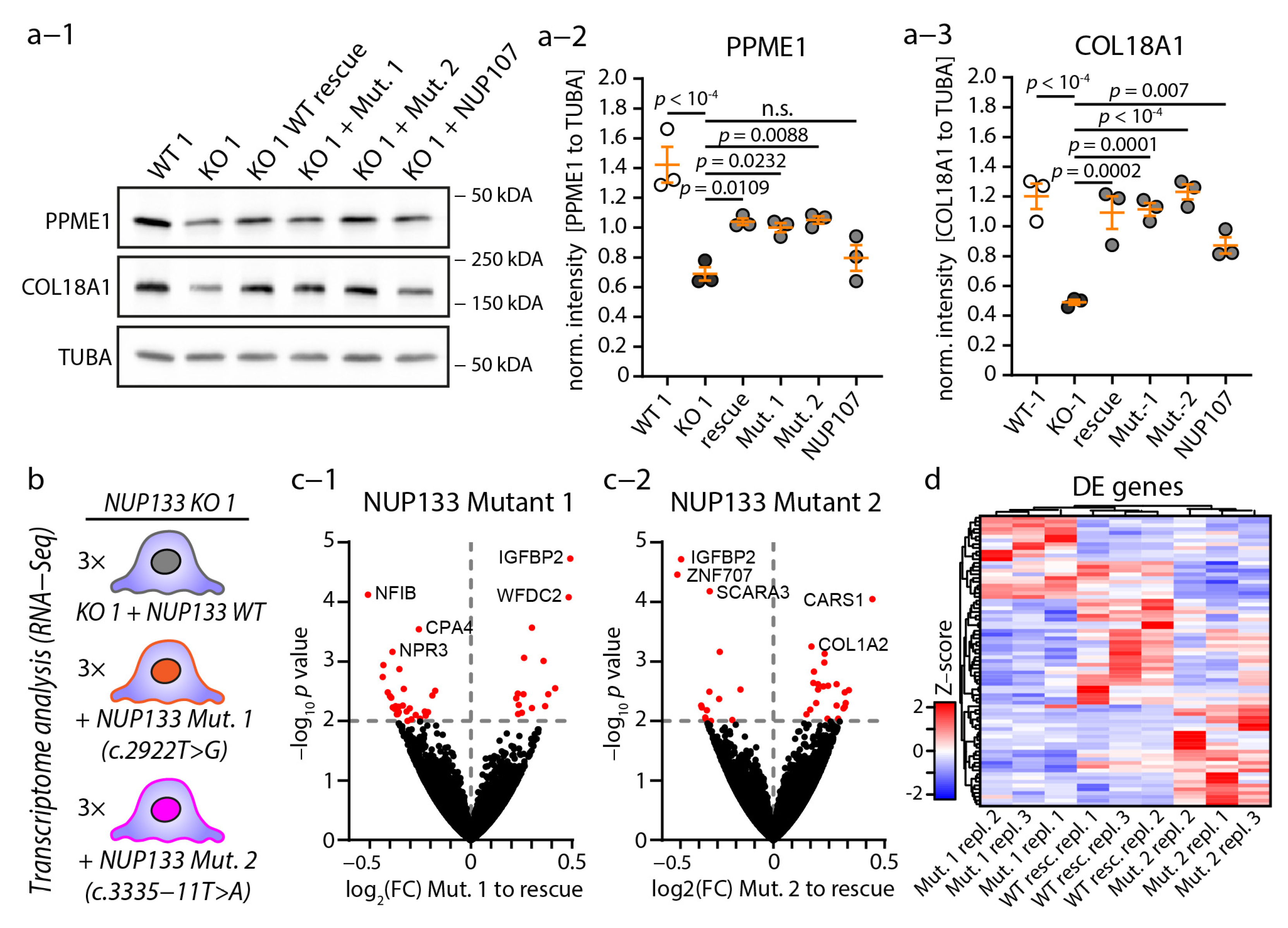
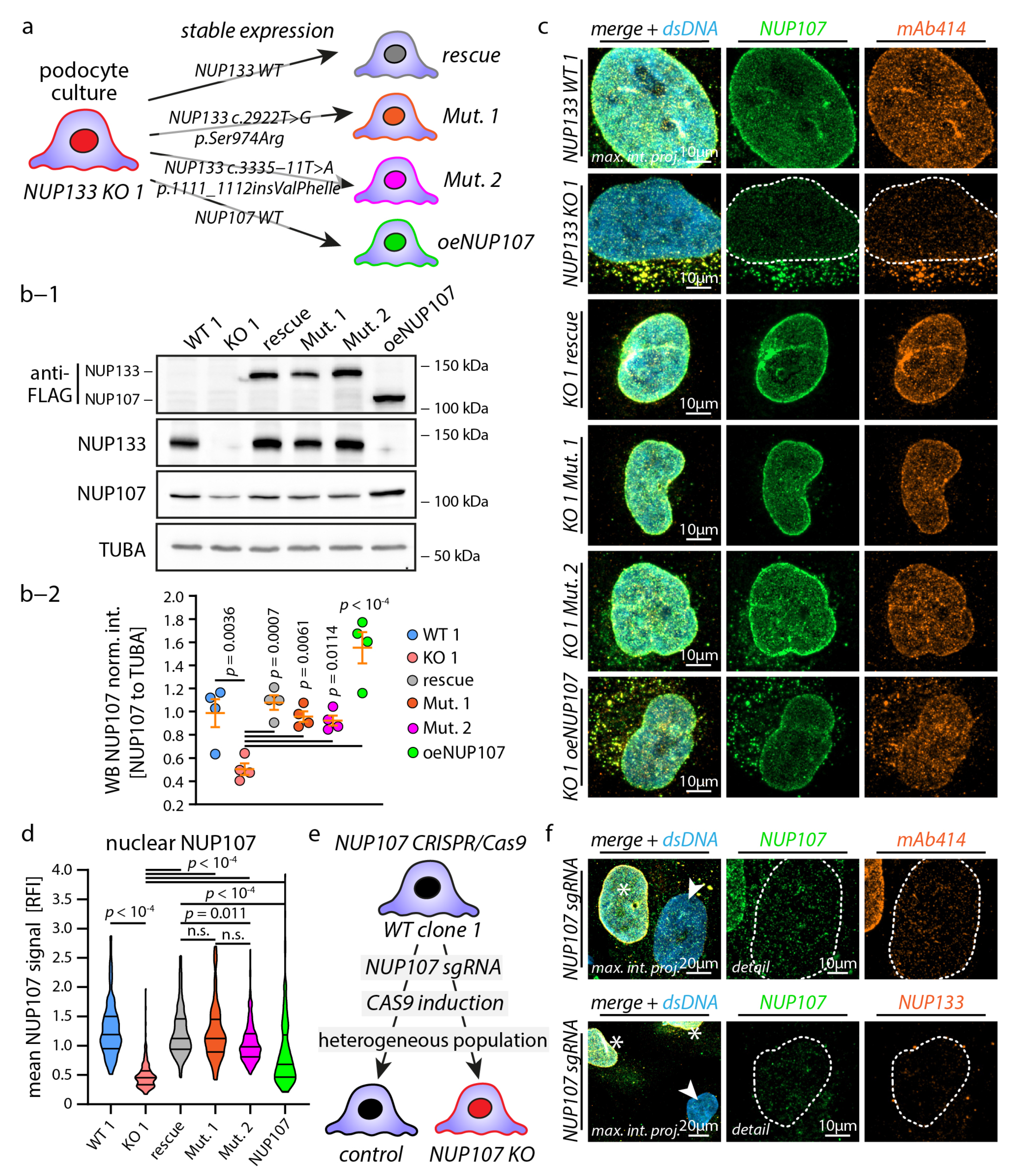
3.2. Causative NUP133 Mutants Restore Y-Complex and NPC Assembly
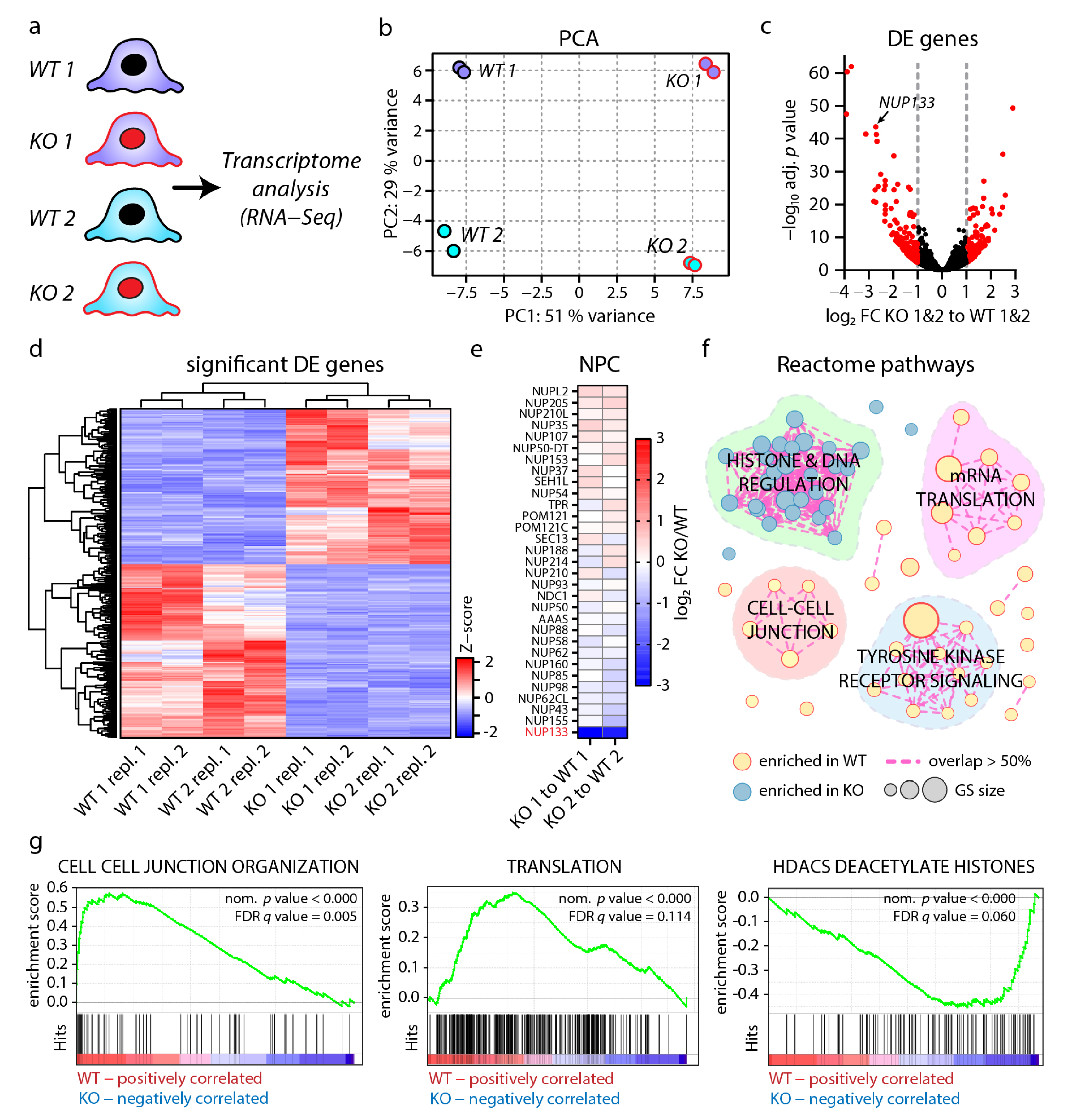
3.3. Loss of NUP133 Remodels the Transcriptome of Specific Regulatory Pathways
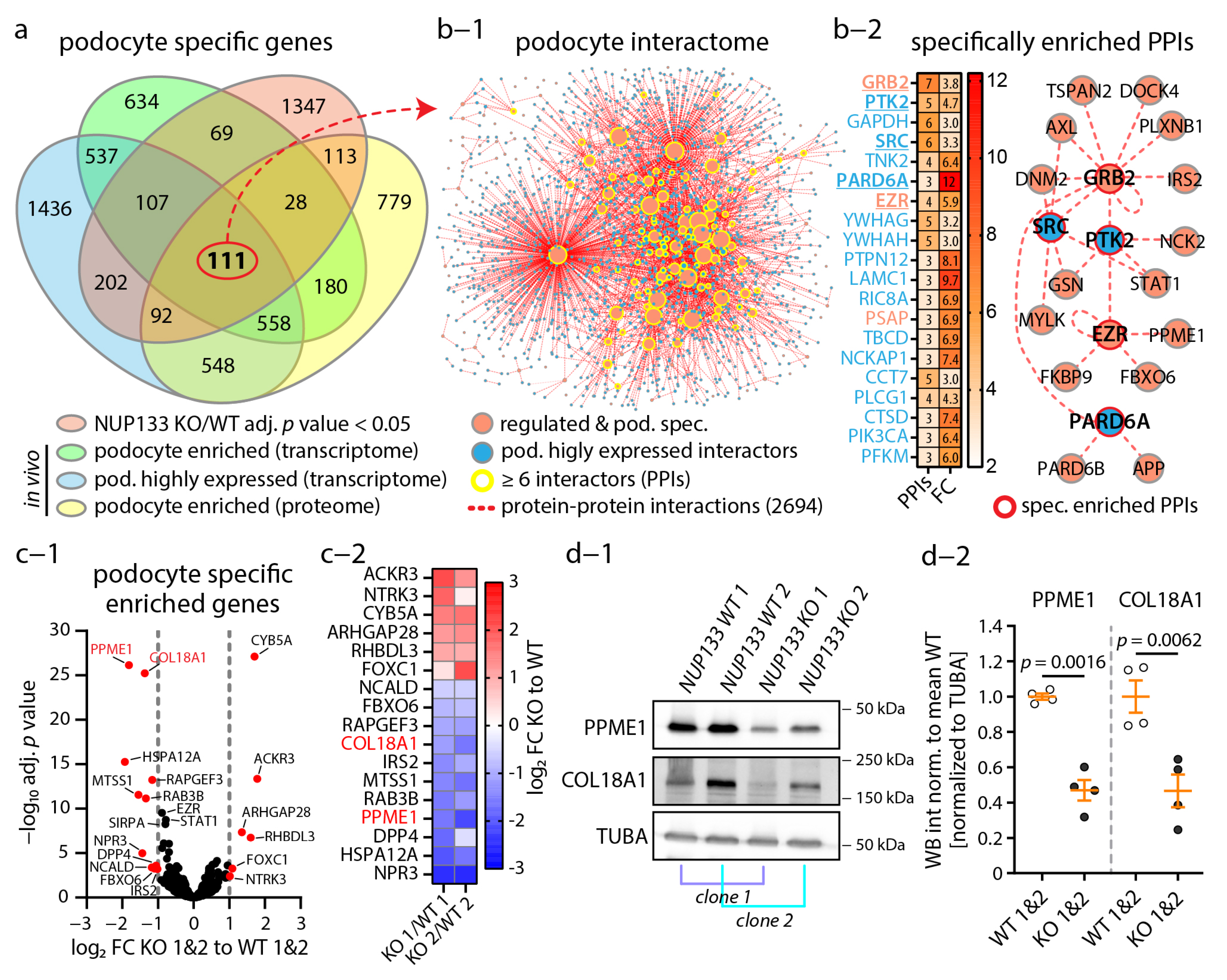
3.4. The Podocyte-Specific Transcriptome Is Disturbed by Loss of NUP133
3.5. Causative NUP133 Variants Do Not Significantly Alter Transcriptome Composition
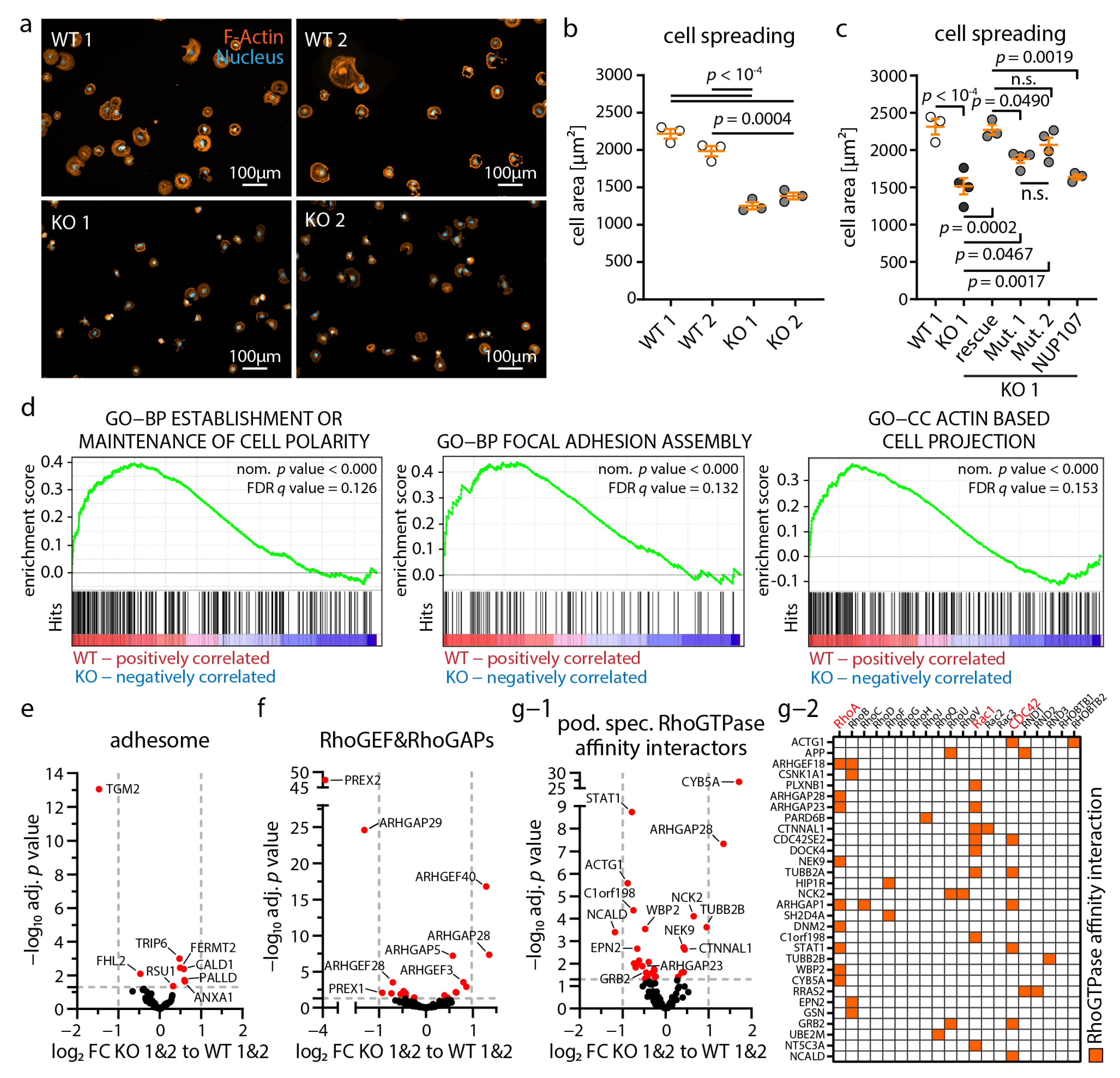
3.6. Loss of NUP133 Disrupts Dynamic Cytoskeleton Regulation in Podocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benzing, T.; Salant, D. Insights into glomerular filtration and albuminuria. N. Engl. J. Med. 2021, 384, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Grahammer, F.; Schell, C.; Huber, T.B. The podocyte slit diaphragm—From a thin grey line to a complex signalling hub. Nat. Rev. Nephrol. 2013, 9, 587–598. [Google Scholar] [CrossRef] [PubMed]
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the global burden of disease study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Levin, A.; Tonelli, M.; Bonventre, J.; Coresh, J.; Donner, J.A.; Fogo, A.B.; Fox, C.S.; Gansevoort, R.T.; Heerspink, H.J.L.; Jardine, M.; et al. Global kidney health 2017 and beyond: A roadmap for closing gaps in care, research, and policy. Lancet 2017, 390, 1888–1917. [Google Scholar] [CrossRef]
- Wiggins, R.C. The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 2007, 71, 1205–1214. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.I.; Rogg, M.; Helmstadter, M.; Sammarco, A.; Walz, G.; Werner, M.; Schell, C. A novel model for nephrotic syndrome reveals associated dysbiosis of the gut microbiome and extramedullary hematopoiesis. Cells 2021, 10, 1509. [Google Scholar] [CrossRef]
- Kopp, J.B.; Anders, H.J.; Susztak, K.; Podesta, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat. Rev. Dis. Primers 2020, 6, 68. [Google Scholar] [CrossRef]
- Warejko, J.K.; Tan, W.; Daga, A.; Schapiro, D.; Lawson, J.A.; Shril, S.; Lovric, S.; Ashraf, S.; Rao, J.; Hermle, T.; et al. Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2018, 13, 53–62. [Google Scholar] [CrossRef]
- Li, A.S.; Ingham, J.F.; Lennon, R. Genetic disorders of the glomerular filtration barrier. Clin. J. Am. Soc. Nephrol. 2020, 15, 1818–1828. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.A.; Sadowski, C.E.; Kohl, S.; Lovric, S.; Astrinidis, S.A.; Pabst, W.L.; Gee, H.Y.; Ashraf, S.; Lawson, J.A.; Shril, S.; et al. Mutations in nuclear pore genes nup93, nup205 and xpo5 cause steroid-resistant nephrotic syndrome. Nat. Genet. 2016, 48, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.A.; Lovric, S.; Schapiro, D.; Schneider, R.; Marquez, J.; Asif, M.; Hussain, M.S.; Daga, A.; Widmeier, E.; Rao, J.; et al. Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome. J. Clin. Investig. 2018, 128, 4313–4328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, A.; Tsukaguchi, H.; Koshimizu, E.; Nakazato, H.; Itoh, K.; Kuraoka, S.; Komohara, Y.; Shiina, M.; Nakamura, S.; Kitajima, M.; et al. Homozygous splicing mutation in nup133 causes galloway-mowat syndrome. Ann. Neurol. 2018, 84, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Tsukaguchi, H.; Koshimizu, E.; Shono, A.; Matsunaga, S.; Shiina, M.; Mimura, Y.; Imamura, S.; Hirose, T.; Okudela, K.; et al. Biallelic mutations in nuclear pore complex subunit nup107 cause early-childhood-onset steroid-resistant nephrotic syndrome. Am. J. Hum. Genet. 2015, 97, 555–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.A.; et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Beck, M.; Hurt, E. The nuclear pore complex: Understanding its function through structural insight. Nat. Rev. Mol. Cell. Biol. 2017, 18, 73–89. [Google Scholar] [CrossRef]
- Raices, M.; D’Angelo, M.A. Nuclear pore complex composition: A new regulator of tissue-specific and developmental functions. Nat. Rev. Mol. Cell. Biol. 2012, 13, 687–699. [Google Scholar] [CrossRef]
- D’Angelo, M.A. Nuclear pore complexes as hubs for gene regulation. Nucleus 2018, 9, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Kalverda, B.; Pickersgill, H.; Shloma, V.V.; Fornerod, M. Nucleoporins directly stimulate expression of developmental and cell-cycle genes inside the nucleoplasm. Cell 2010, 140, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Capelson, M.; Liang, Y.; Schulte, R.; Mair, W.; Wagner, U.; Hetzer, M.W. Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell 2010, 140, 372–383. [Google Scholar] [CrossRef] [Green Version]
- Dickmanns, A.; Kehlenbach, R.H.; Fahrenkrog, B. Nuclear pore complexes and nucleocytoplasmic transport: From structure to function to disease. Int. Rev. Cell. Mol. Biol. 2015, 320, 171–233. [Google Scholar]
- Nofrini, V.; Di Giacomo, D.; Mecucci, C. Nucleoporin genes in human diseases. Eur. J. Hum. Genet. 2016, 24, 1388–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg-Shukron, A.; Renbaum, P.; Kalifa, R.; Zeligson, S.; Ben-Neriah, Z.; Dreifuss, A.; Abu-Rayyan, A.; Maatuk, N.; Fardian, N.; Rekler, D.; et al. A mutation in the nucleoporin-107 gene causes xx gonadal dysgenesis. J. Clin. Investig. 2015, 125, 4295–4304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Castonguay, P.; Sidhom, E.H.; Clark, A.R.; Dvela-Levitt, M.; Kim, S.; Sieber, J.; Wieder, N.; Jung, J.Y.; Andreeva, S.; et al. A small-molecule inhibitor of trpc5 ion channels suppresses progressive kidney disease in animal models. Science 2017, 358, 1332–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atmaca, M.; Gulhan, B.; Korkmaz, E.; Inozu, M.; Soylemezoglu, O.; Candan, C.; Bayazit, A.K.; Elmaci, A.M.; Parmaksiz, G.; Duzova, A.; et al. Follow-up results of patients with adck4 mutations and the efficacy of coq10 treatment. Pediatr. Nephrol. 2017, 32, 1369–1375. [Google Scholar] [CrossRef]
- Schijvens, A.M.; van de Kar, N.C.; Bootsma-Robroeks, C.M.; Cornelissen, E.A.; van den Heuvel, L.P.; Schreuder, M.F. Mitochondrial disease and the kidney with a special focus on coq10 deficiency. Kidney Int. Rep. 2020, 5, 2146–2159. [Google Scholar] [CrossRef]
- Daehn, I.S.; Duffield, J.S. The glomerular filtration barrier: A structural target for novel kidney therapies. Nat. Rev. Drug Discov. 2021, 20, 770–788. [Google Scholar] [CrossRef]
- Saleem, M.A.; O’Hare, M.J.; Reiser, J.; Coward, R.J.; Inward, C.D.; Farren, T.; Xing, C.Y.; Ni, L.; Mathieson, P.W.; Mundel, P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 2002, 13, 630–638. [Google Scholar] [CrossRef]
- Schell, C.; Rogg, M.; Suhm, M.; Helmstadter, M.; Sellung, D.; Yasuda-Yamahara, M.; Kretz, O.; Kuttner, V.; Suleiman, H.; Kollipara, L.; et al. The ferm protein epb41l5 regulates actomyosin contractility and focal adhesion formation to maintain the kidney filtration barrier. Proc. Natl. Acad. Sci. USA 2017, 114, E4621–E4630. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.I.; Rogg, M.; Helmstadter, M.; Sammarco, A.; Schilling, O.; Sabass, B.; Miner, J.H.; Dengjel, J.; Walz, G.; Werner, M.; et al. Epb41l5 controls podocyte extracellular matrix assembly by adhesome-dependent force transmission. Cell Rep. 2021, 34, 108883. [Google Scholar] [CrossRef]
- Rogg, M.; Maier, J.I.; Van Wymersch, C.; Helmstadter, M.; Sammarco, A.; Lindenmeyer, M.; Zareba, P.; Montanez, E.; Walz, G.; Werner, M.; et al. Alpha-parvin defines a specific integrin adhesome to maintain the glomerular filtration barrier. J. Am. Soc. Nephrol. 2022, 33, 786–808. [Google Scholar] [CrossRef]
- Rogg, M.; Maier, J.I.; Dotzauer, R.; Artelt, N.; Kretz, O.; Helmstadter, M.; Abed, A.; Sammarco, A.; Sigle, A.; Sellung, D.; et al. Srgap1 controls small rho gtpases to regulate podocyte foot process maintenance. J. Am. Soc. Nephrol. 2021, 32, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. Qupath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schell, C.; Sabass, B.; Helmstaedter, M.; Geist, F.; Abed, A.; Yasuda-Yamahara, M.; Sigle, A.; Maier, J.I.; Grahammer, F.; Siegerist, F.; et al. Arp3 controls the podocyte architecture at the kidney filtration barrier. Dev. Cell 2018, 47, 741–757.e748. [Google Scholar] [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one fastq preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. Genepattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef] [PubMed]
- Rinschen, M.M.; Godel, M.; Grahammer, F.; Zschiedrich, S.; Helmstadter, M.; Kretz, O.; Zarei, M.; Braun, D.A.; Dittrich, S.; Pahmeyer, C.; et al. A multi-layered quantitative in vivo expression atlas of the podocyte unravels kidney disease candidate genes. Cell Rep. 2018, 23, 2495–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The biogrid database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Horton, E.R.; Byron, A.; Askari, J.A.; Ng, D.H.J.; Millon-Fremillon, A.; Robertson, J.; Koper, E.J.; Paul, N.R.; Warwood, S.; Knight, D.; et al. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat. Cell Biol. 2015, 17, 1577–1587. [Google Scholar] [CrossRef] [Green Version]
- Boehmer, T.; Jeudy, S.; Berke, I.C.; Schwartz, T.U. Structural and functional studies of nup107/nup133 interaction and its implications for the architecture of the nuclear pore complex. Mol. Cell 2008, 30, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodova, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koca, J.; Rose, A.S. Mol* viewer: Modern web app for 3d visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Davis, L.I.; Blobel, G. Nuclear pore complex contains a family of glycoproteins that includes p62: Glycosylation through a previously unidentified cellular pathway. Proc. Natl. Acad. Sci. USA 1987, 84, 7552–7556. [Google Scholar] [CrossRef] [Green Version]
- Mimura, Y.; Takemoto, S.; Tachibana, T.; Ogawa, Y.; Nishimura, M.; Yokota, H.; Imamoto, N. A statistical image analysis framework for pore-free islands derived from heterogeneity distribution of nuclear pore complexes. Sci. Rep. 2017, 7, 16315. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho gtpases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. Gefs and gaps: Critical elements in the control of small g proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boerries, M.; Grahammer, F.; Eiselein, S.; Buck, M.; Meyer, C.; Goedel, M.; Bechtel, W.; Zschiedrich, S.; Pfeifer, D.; Laloe, D.; et al. Molecular fingerprinting of the podocyte reveals novel gene and protein regulatory networks. Kidney Int. 2013, 83, 1052–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kann, M.; Ettou, S.; Jung, Y.L.; Lenz, M.O.; Taglienti, M.E.; Park, P.J.; Schermer, B.; Benzing, T.; Kreidberg, J.A. Genome-wide analysis of wilms’ tumor 1-controlled gene expression in podocytes reveals key regulatory mechanisms. J. Am. Soc. Nephrol. 2015, 26, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Koehler, S.; Odenthal, J.; Ludwig, V.; Unnersjo Jess, D.; Hohne, M.; Jungst, C.; Grawe, F.; Helmstadter, M.; Janku, J.L.; Bergmann, C.; et al. Scaffold polarity proteins par3a and par3b share redundant functions while par3b acts independent of atypical protein kinase c/par6 in podocytes to maintain the kidney filtration barrier. Kidney Int. 2021, 101, 733–751. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Navarro, S.; Fischer, T.; Luo, M.J.; Antunez, O.; Brettschneider, S.; Lechner, J.; Perez-Ortin, J.E.; Reed, R.; Hurt, E. Sus1, a functional component of the saga histone acetylase complex and the nuclear pore-associated mrna export machinery. Cell 2004, 116, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Lupu, F.; Alves, A.; Anderson, K.; Doye, V.; Lacy, E. Nuclear pore composition regulates neural stem/progenitor cell differentiation in the mouse embryo. Dev. Cell 2008, 14, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Kinnunen, A.I.; Sormunen, R.; Elamaa, H.; Seppinen, L.; Miller, R.T.; Ninomiya, Y.; Janmey, P.A.; Pihlajaniemi, T. Lack of collagen xviii long isoforms affects kidney podocytes, whereas the short form is needed in the proximal tubular basement membrane. J. Biol. Chem. 2011, 286, 7755–7764. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Ye, Y.; Xu, C.; Gao, C.; Zhang, Y.; Zhou, J.; Lin, W.; Mao, J. Protein phosphatase 2a modulates podocyte maturation and glomerular functional integrity in mice. Cell Commun. Signal. 2019, 17, 91. [Google Scholar] [CrossRef] [Green Version]
- Gozalo, A.; Duke, A.; Lan, Y.; Pascual-Garcia, P.; Talamas, J.A.; Nguyen, S.C.; Shah, P.P.; Jain, R.; Joyce, E.F.; Capelson, M. Core components of the nuclear pore bind distinct states of chromatin and contribute to polycomb repression. Mol. Cell 2020, 77, 67–81.e67. [Google Scholar] [CrossRef]
- Loiodice, I.; Alves, A.; Rabut, G.; Van Overbeek, M.; Ellenberg, J.; Sibarita, J.B.; Doye, V. The entire nup107-160 complex, including three new members, is targeted as one entity to kinetochores in mitosis. Mol. Biol. Cell 2004, 15, 3333–3344. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Martinez, J.; Phillips, J.; Sekedat, M.D.; Diaz-Avalos, R.; Velazquez-Muriel, J.; Franke, J.D.; Williams, R.; Stokes, D.L.; Chait, B.T.; Sali, A.; et al. Structure-function mapping of a heptameric module in the nuclear pore complex. J. Cell Biol. 2012, 196, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doye, V.; Wepf, R.; Hurt, E.C. A novel nuclear pore protein nup133p with distinct roles in poly(a)+ rna transport and nuclear pore distribution. EMBO J. 1994, 13, 6062–6075. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, L.F.; Rout, M.P.; Blobel, G. Disruption of the nucleoporin gene nup133 results in clustering of nuclear pore complexes. Proc. Natl. Acad. Sci. USA 1995, 92, 1187–1191. [Google Scholar] [CrossRef] [Green Version]
- Cianciolo Cosentino, C.; Berto, A.; Pelletier, S.; Hari, M.; Loffing, J.; Neuhauss, S.C.F.; Doye, V. Moderate nucleoporin 133 deficiency leads to glomerular damage in zebrafish. Sci. Rep. 2019, 9, 4750. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, J.; Asano-Matsuda, K.; Kitzler, T.M.; Takano, T. Rho gtpase regulatory proteins in podocytes. Kidney Int. 2021, 99, 336–345. [Google Scholar] [CrossRef]
- Schell, C.; Huber, T.B. The evolving complexity of the podocyte cytoskeleton. J. Am. Soc. Nephrol. 2017, 28, 3166–3174. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, J.; Maier, M.; Aoudjit, L.; Baldwin, C.; Takano, T. Arhgef7 (beta-pix) is required for the maintenance of podocyte architecture and glomerular function. J. Am. Soc. Nephrol. 2020, 31, 996–1008. [Google Scholar] [CrossRef]
- Blattner, S.M.; Hodgin, J.B.; Nishio, M.; Wylie, S.A.; Saha, J.; Soofi, A.A.; Vining, C.; Randolph, A.; Herbach, N.; Wanke, R.; et al. Divergent functions of the rho gtpases rac1 and cdc42 in podocyte injury. Kidney Int. 2013, 84, 920–930. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Suleiman, H.; Kim, A.H.; Miner, J.H.; Dani, A.; Shaw, A.S.; Akilesh, S. Rac1 activation in podocytes induces rapid foot process effacement and proteinuria. Mol. Cell Biol. 2013, 33, 4755–4764. [Google Scholar] [CrossRef] [Green Version]
- Raices, M.; D’Angelo, M.A. Nuclear pore complexes and regulation of gene expression. Curr. Opin. Cell Biol. 2017, 46, 26–32. [Google Scholar] [CrossRef]
- Walther, T.C.; Alves, A.; Pickersgill, H.; Loiodice, I.; Hetzer, M.; Galy, V.; Hulsmann, B.B.; Kocher, T.; Wilm, M.; Allen, T.; et al. The conserved nup107–160 complex is critical for nuclear pore complex assembly. Cell 2003, 113, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Morais, M.; Tian, P.; Lawless, C.; Murtuza-Baker, S.; Hopkinson, L.; Woods, S.; Mironov, A.; Long, D.A.; Gale, D.P.; Zorn, T.M.T.; et al. Kidney organoids recapitulate human basement membrane assembly in health and disease. eLife 2022, 11, e73486. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, L.; Chen, M.; Kampf, L.L.; Milosavljevic, J.; Lang, K.; Schneider, R.; Hildebrandt, F.; Helmstadter, M.; Walz, G.; Hermle, T. Inhibition of endoplasmic reticulum stress signaling rescues cytotoxicity of human apolipoprotein-l1 risk variants in drosophila. Kidney Int. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Butt, L.; Unnersjo-Jess, D.; Hohne, M.; Hahnfeldt, R.; Reilly, D.; Rinschen, M.M.; Plagmann, I.; Diefenhardt, P.; Brahler, S.; Brinkkotter, P.T.; et al. Super-resolution imaging of the filtration barrier suggests a role for podocin r229q in genetic predisposition to glomerular disease. J. Am. Soc. Nephrol. 2022, 33, 138–154. [Google Scholar] [CrossRef] [PubMed]
- Rubel, D.; Boulanger, J.; Craciun, F.; Xu, E.Y.; Zhang, Y.; Phillips, L.; Callahan, M.; Weber, W.; Song, W.; Ngai, N.; et al. Anti-microrna-21 therapy on top of ace inhibition delays renal failure in alport syndrome mouse models. Cells 2022, 11, 594. [Google Scholar] [CrossRef]
- Saleem, M.A.; Welsh, G.I. Podocyte rhogtpases: New therapeutic targets for nephrotic syndrome? F1000Research 2019, 8, F1000 Faculty Rev-1847. [Google Scholar] [CrossRef]
- Meyer-Schwesinger, C. The ubiquitin-proteasome system in kidney physiology and disease. Nat. Rev. Nephrol. 2019, 15, 393–411. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogg, M.; Maier, J.I.; Ehle, M.; Sammarco, A.; Schilling, O.; Werner, M.; Schell, C. NUP133 Controls Nuclear Pore Assembly, Transcriptome Composition, and Cytoskeleton Regulation in Podocytes. Cells 2022, 11, 1259. https://doi.org/10.3390/cells11081259
Rogg M, Maier JI, Ehle M, Sammarco A, Schilling O, Werner M, Schell C. NUP133 Controls Nuclear Pore Assembly, Transcriptome Composition, and Cytoskeleton Regulation in Podocytes. Cells. 2022; 11(8):1259. https://doi.org/10.3390/cells11081259
Chicago/Turabian StyleRogg, Manuel, Jasmin I. Maier, Markus Ehle, Alena Sammarco, Oliver Schilling, Martin Werner, and Christoph Schell. 2022. "NUP133 Controls Nuclear Pore Assembly, Transcriptome Composition, and Cytoskeleton Regulation in Podocytes" Cells 11, no. 8: 1259. https://doi.org/10.3390/cells11081259
APA StyleRogg, M., Maier, J. I., Ehle, M., Sammarco, A., Schilling, O., Werner, M., & Schell, C. (2022). NUP133 Controls Nuclear Pore Assembly, Transcriptome Composition, and Cytoskeleton Regulation in Podocytes. Cells, 11(8), 1259. https://doi.org/10.3390/cells11081259