
RNA Editing in Glioma as a Sexually Dimorphic Prognostic Factor That Affects mRNA Abundance in Fatty Acid Metabolism and Inflammation Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Information and Status of Known Glioma Biomarkers
2.2. Prediction of Mutation Status of IDH and 1p/19q in TCGA Samples
2.3. Characterization of Editing Events of TCGA and CGGA Samples
2.4. Dimension Reduction, Clustering, and Visualization of Editing Profiles
2.5. Survival Analysis
2.6. Comparisons of Editing and Gene Expression between High-Risk and Low-Risk Cases
2.7. Evaluation of the Gene Overlap
2.8. Development and Validation of Editing-Based Classification Models
2.9. Over-Representation Analyses on Genes with DESs and DEGs
2.10. Regression Analysis
2.11. Analysis of U87 ADAR1 KD RNA-Seq Data
3. Results
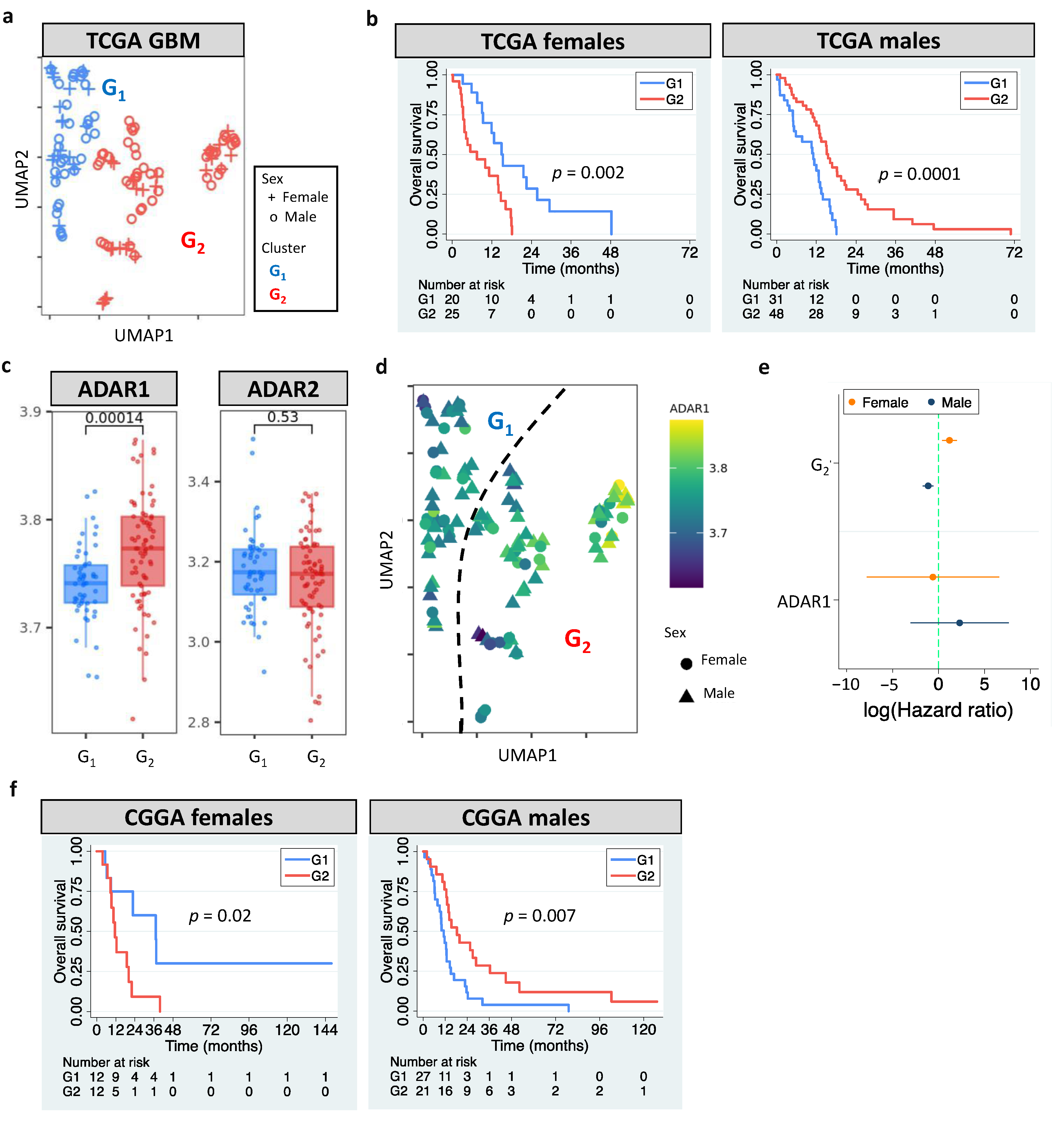
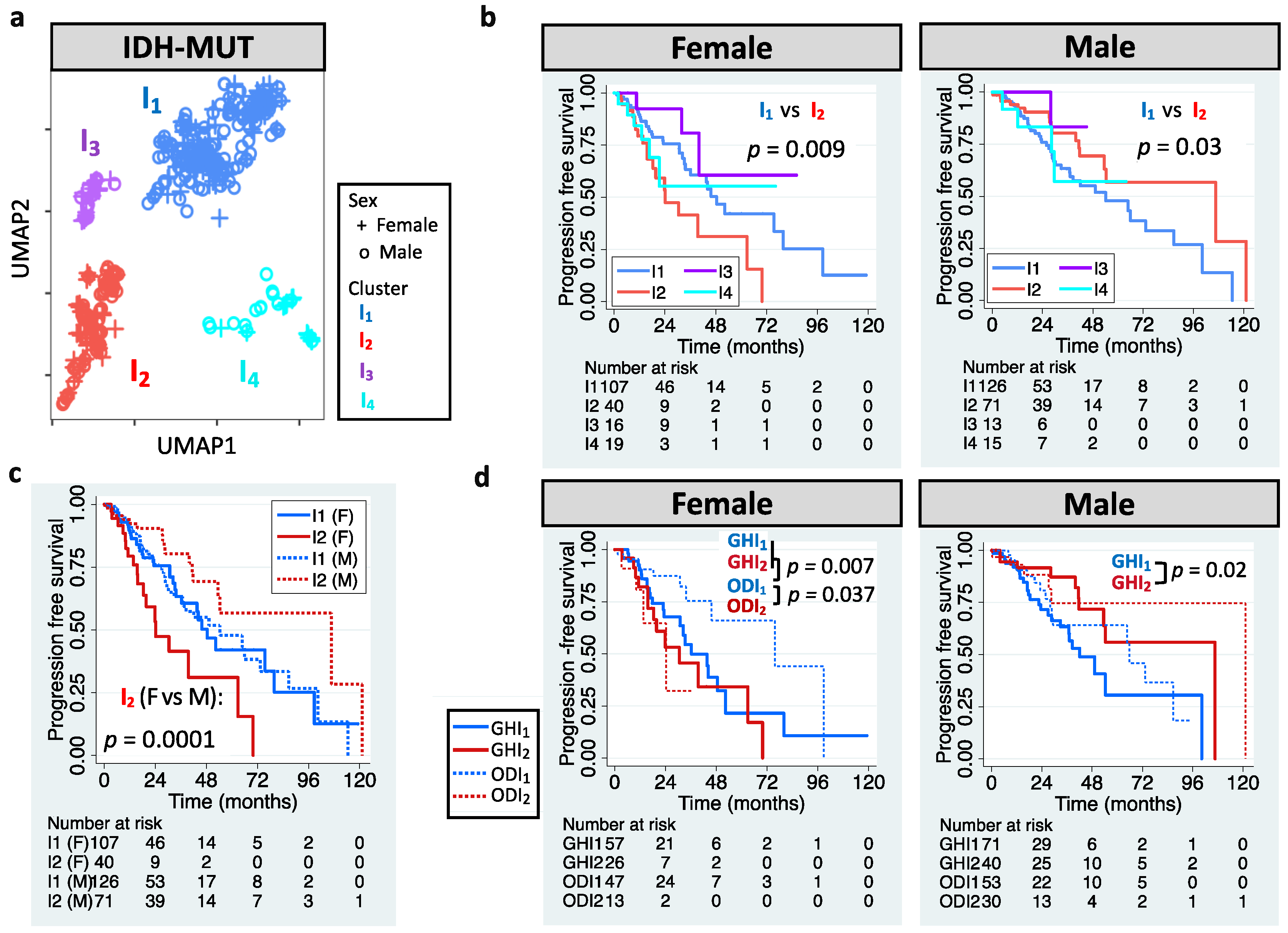
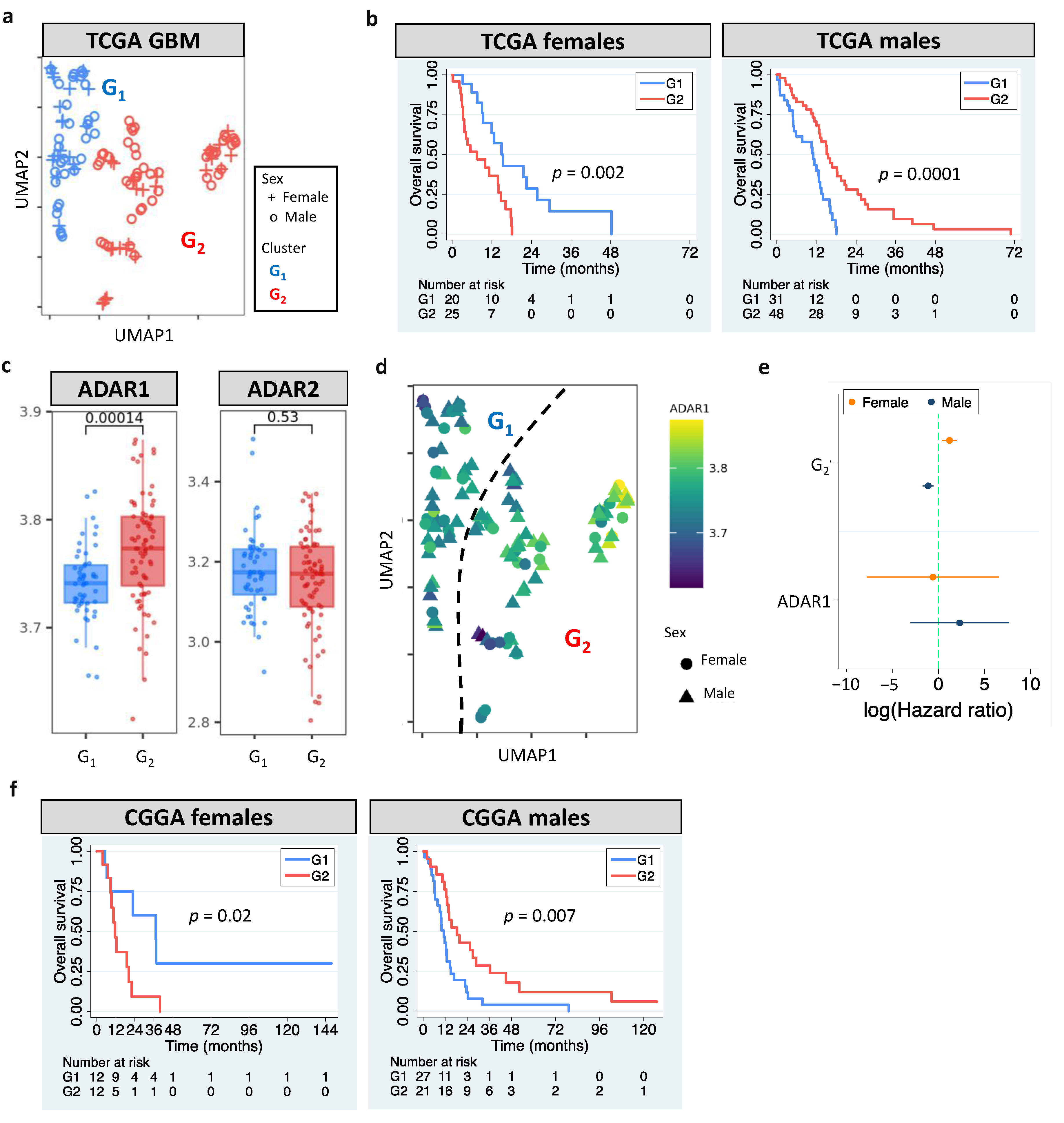
3.1. Sexually Dimorphic Association between Editing Profiles and Patient Survival
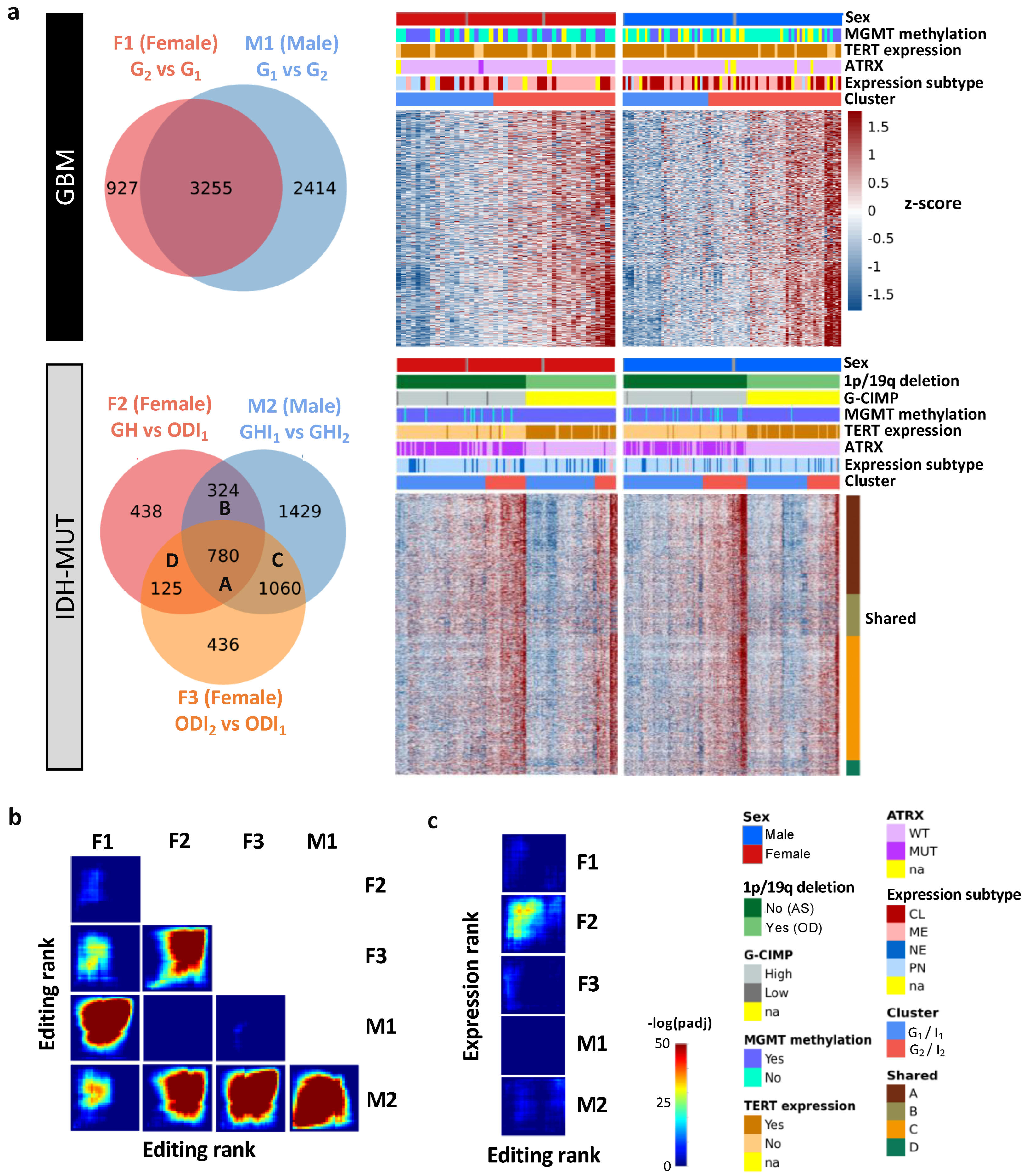
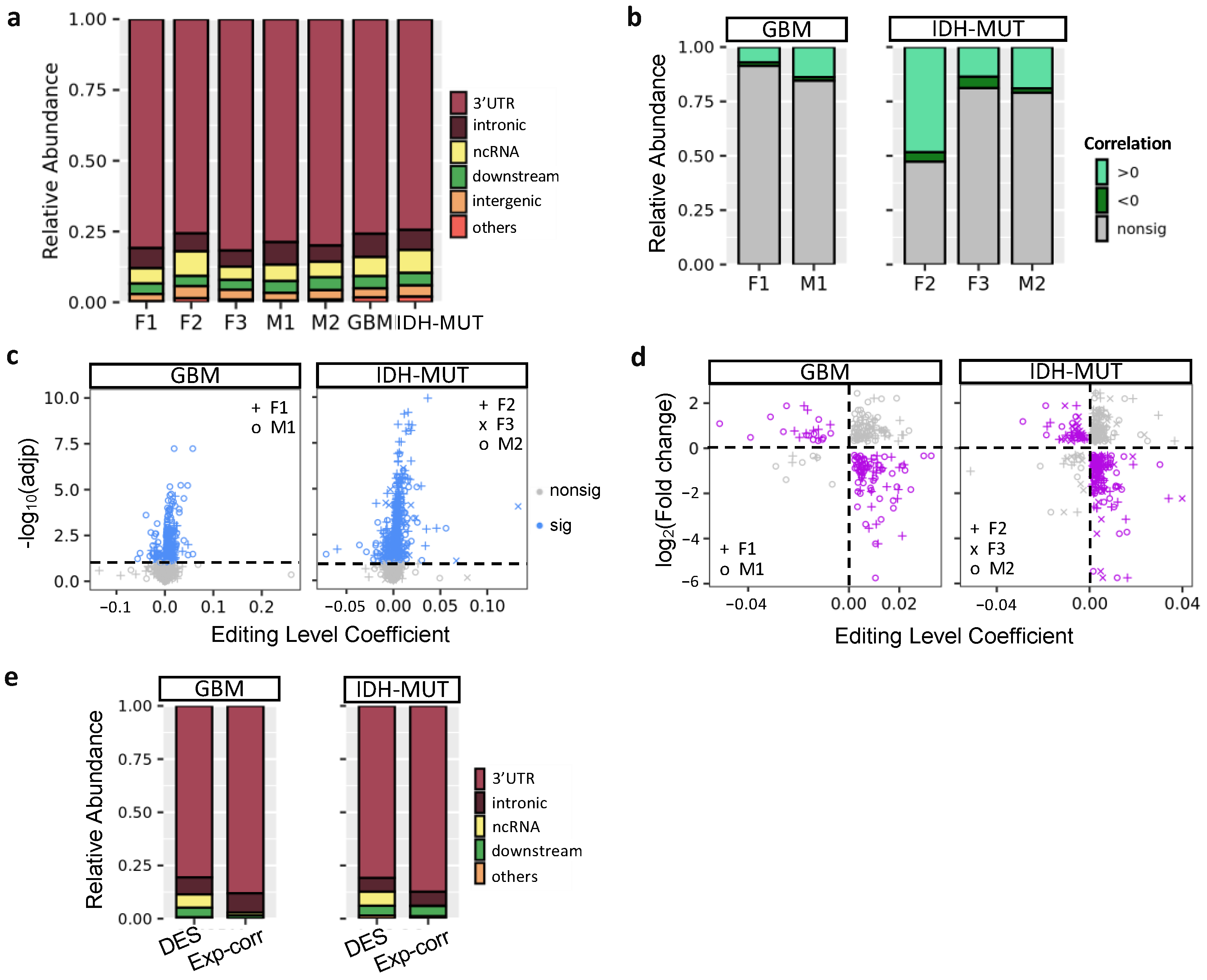
3.2. Editing Changes Are Shared between Sexes and Subtypes and Distinct from Differential Expression
3.3. Clinical Utility of RNA Editing for GBM Prognosis Is Independently Validated
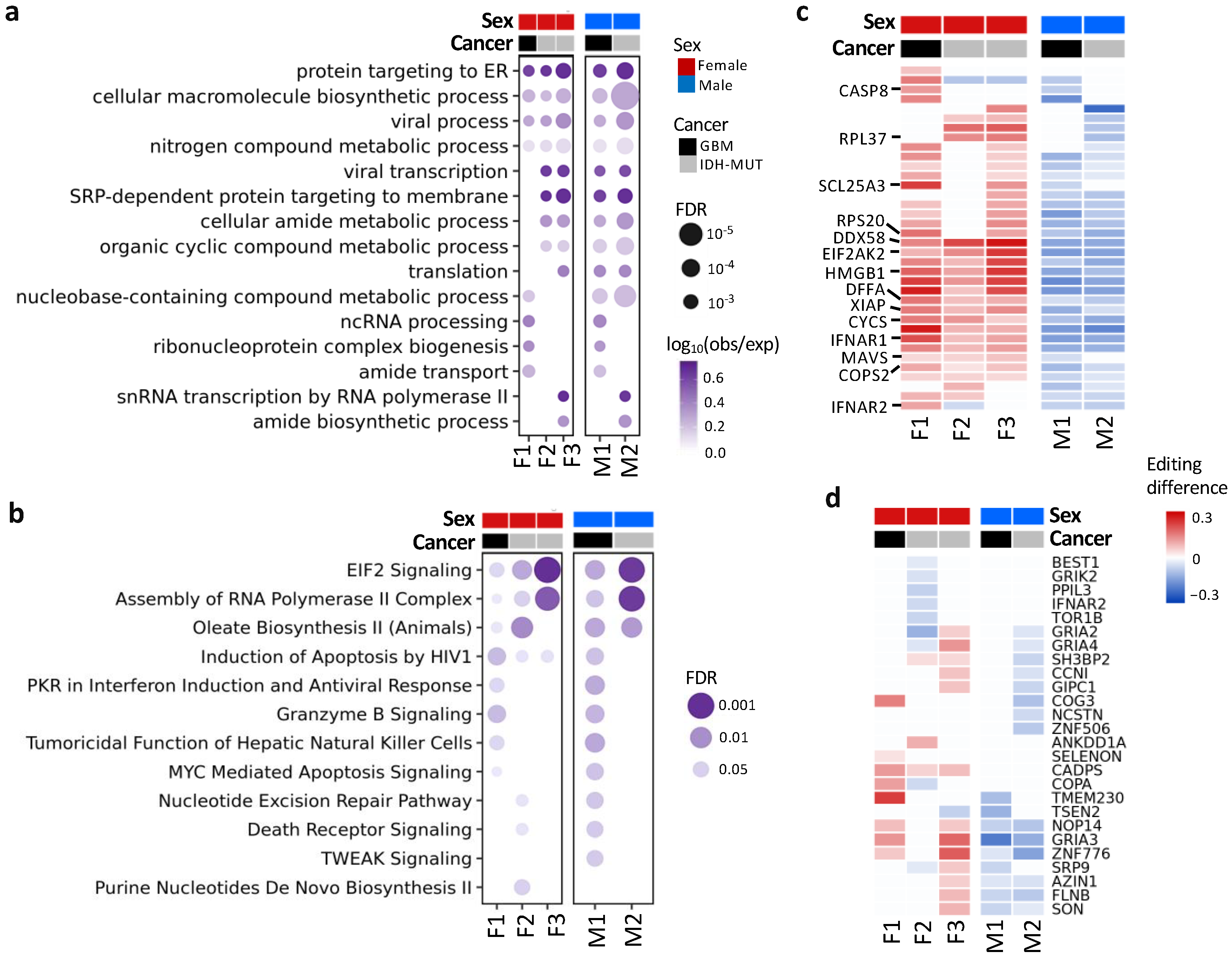
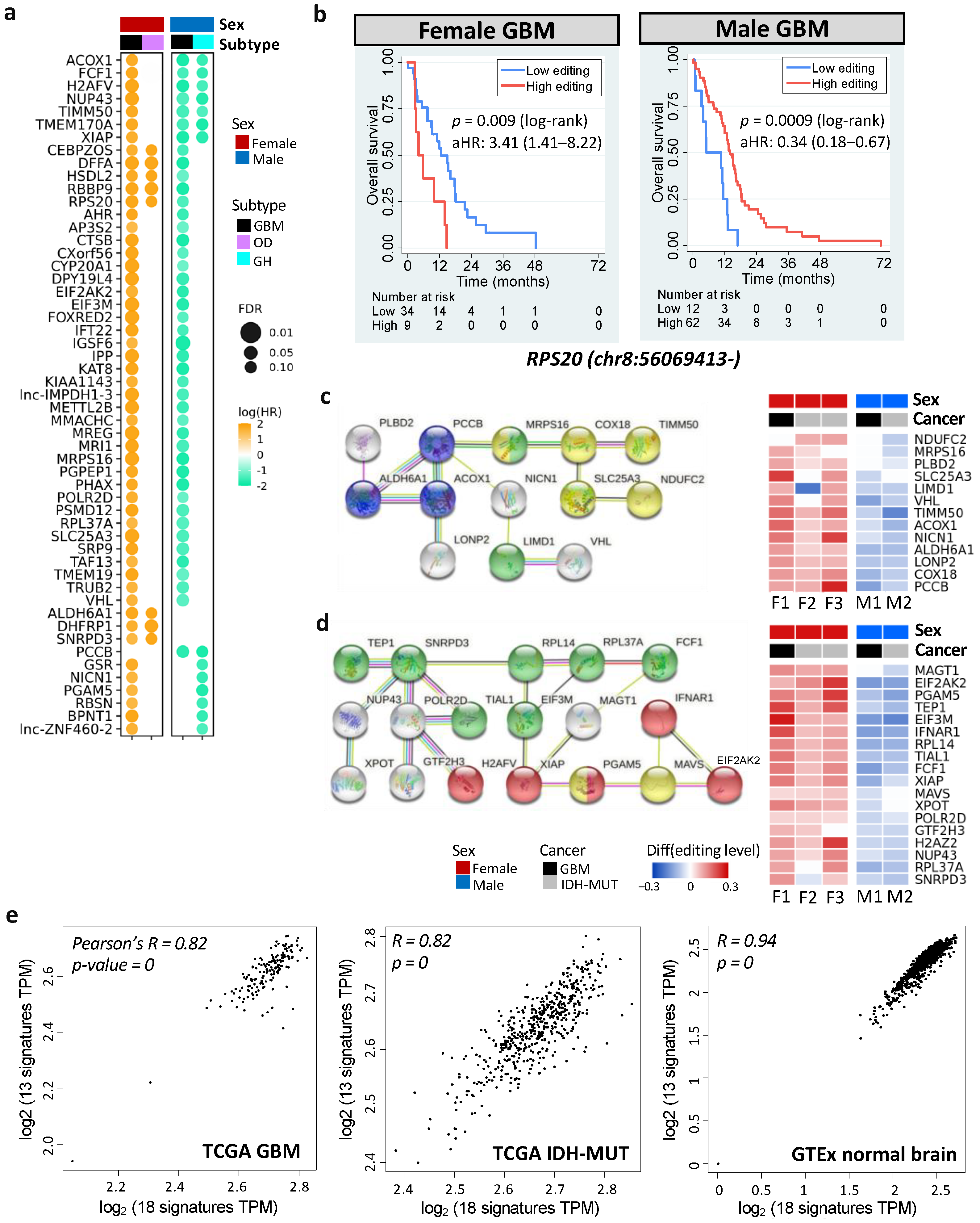
3.4. Differentially Edited Genes Are Associated with Immune Regulation and Cancer Progression
3.5. Noncoding Editing Events Impact mRNA Abundance of Their Host Genes
3.6. Necroptosis and Propanoate Metabolism Genes Are Editing-Regulated and Associated with Glioma Progression
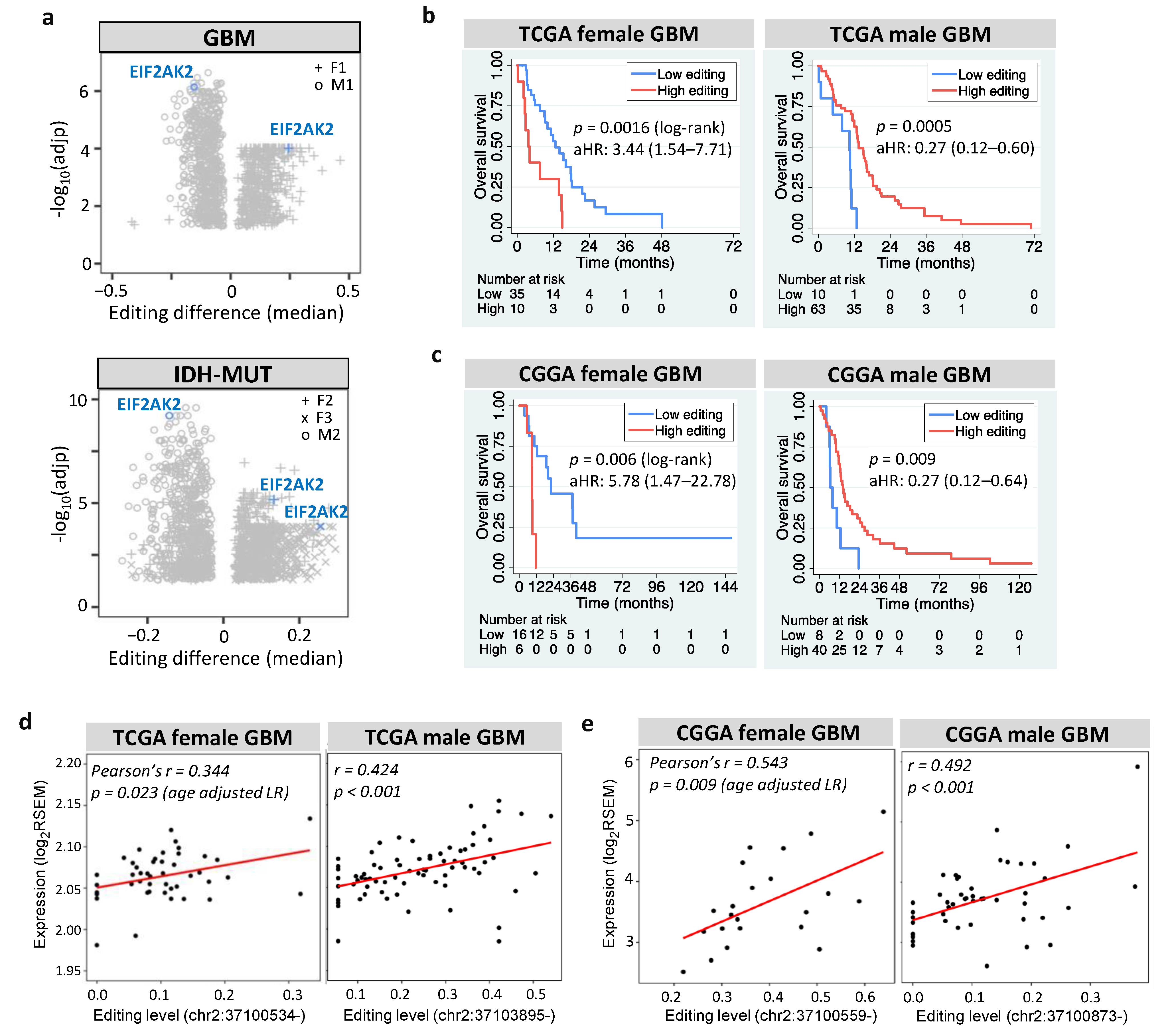
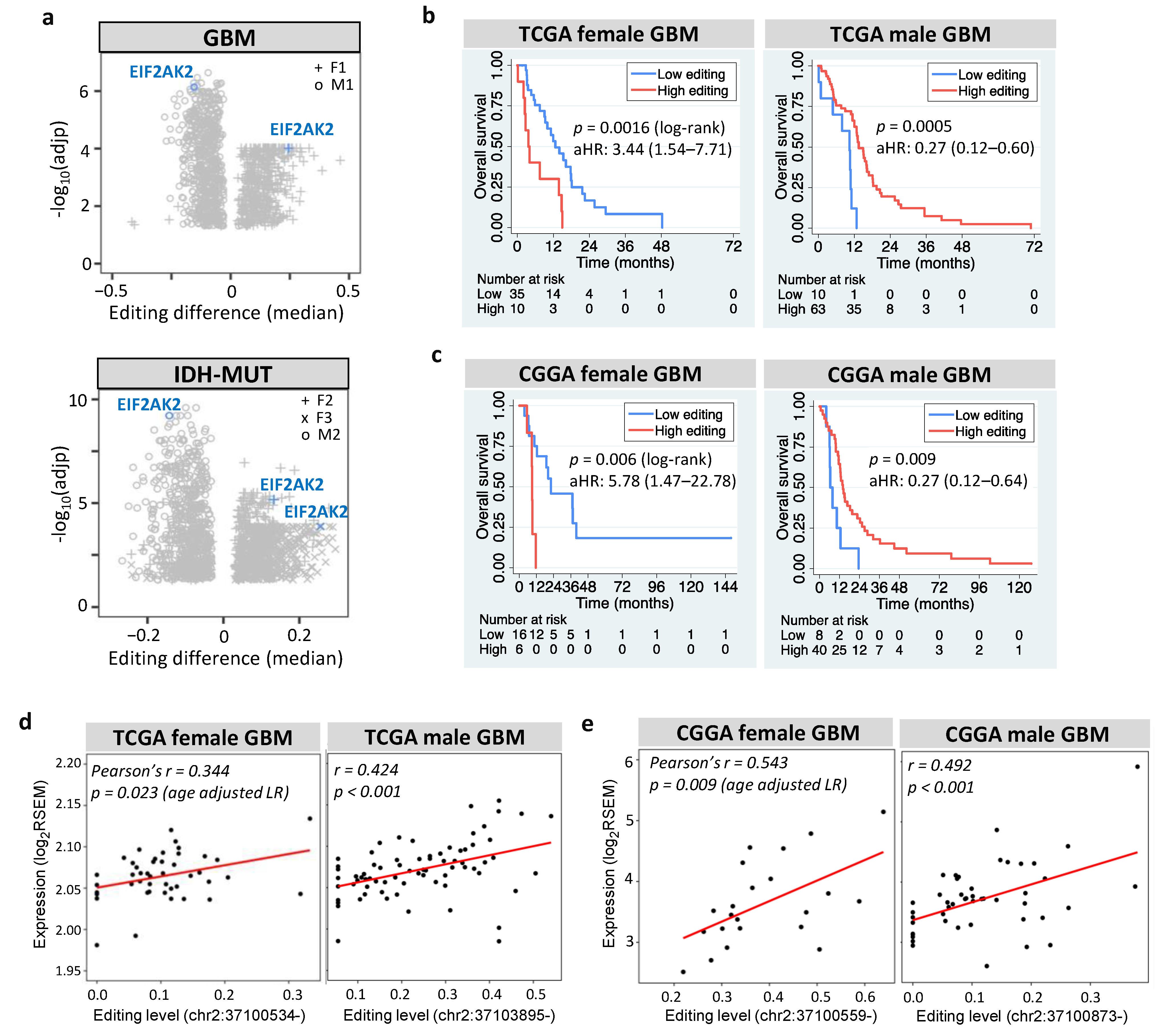
3.7. EIF2AK2 Shows the Maximum Editing Difference between High- and Low-Risk Gliomas
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2020, 18, 170–186. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-Oncol. 2015, 17, iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [Green Version]
- Sidaway, P. CNS cancer: Glioblastoma subtypes revisited. Nat. Rev. Clin. Oncol. 2017, 14, 587. [Google Scholar]
- Gupta, T.; Sarin, R. Poor-prognosis high-grade gliomas: Evolving an evidence-based standard of care. Lancet Oncol. 2002, 3, 557–564. [Google Scholar] [CrossRef]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.A.; Karajannis, M.A.; Harter, D.H. Glioblastoma multiforme: State of the art and future therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Silvestris, D.A.; Picardi, E.; Cesarini, V.; Fosso, B.; Mangraviti, N.; Massimi, L.; Martini, M.; Pesole, G.; Locatelli, F.; Gallo, A. Dynamic inosinome profiles reveal novel patient stratification and gender-specific differences in glioblastoma. Genome Biol. 2019, 20, 33. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.M.; Liang, H. When MicroRNAs Meet RNA Editing in Cancer: A Nucleotide Change Can Make a Difference. Bioessays 2018, 40, 1700188. [Google Scholar] [CrossRef]
- Han, L.; Diao, L.X.; Yu, S.X.; Xu, X.Y.; Li, J.; Zhang, R.; Yang, Y.; Werner, H.M.J.; Eterovic, A.K.; Yuan, Y.; et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 2015, 28, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.L.; Li, Y.; Lin, C.H.; Chan, T.H.M.; Chow, R.K.K.; Song, Y.Y.; Liu, M.; Yuan, Y.F.; Fu, L.; Kong, K.L.; et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat. Med. 2013, 19, 209–216. [Google Scholar] [CrossRef]
- Bass, B.L.; Nishikura, K.; Keller, W.; Seeburg, P.H.; Emeson, R.B.; OConnell, M.A.; Samuel, C.E.; Herbert, A. A standardized nomenclature for adenosine deaminases that act on RNA. RNA 1997, 3, 947–949. [Google Scholar]
- Xu, X.; Wang, Y.; Liang, H. The role of A-to-I RNA editing in cancer development. Curr. Opin. Genet. Dev. 2018, 48, 51–56. [Google Scholar] [CrossRef]
- Fumagalli, D.; Gacquer, D.; Rothe, F.; Lefort, A.; Libert, F.; Brown, D.; Kheddoumi, N.; Shlien, A.; Konopka, T.; Salgado, R.; et al. Principles Governing A-to-I RNA Editing in the Breast Cancer Transcriptome. Cell Rep. 2015, 13, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Paz-Yaacov, N.; Bazak, L.; Buchumenski, L.; Porath, H.T.; Danan-Gotthold, M.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep. 2015, 13, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Okugawa, Y.; Toiyama, Y.; Shigeyasu, K.; Yamamoto, A.; Shigemori, T.; Yin, C.; Ichikawa, T.; Yasuda, H.; Fujikawa, H.; Yoshiyama, S.; et al. Enhanced AZIN1 RNA editing and overexpression of its regulatory enzyme ADAR1 are important prognostic biomarkers in gastric cancer. J. Transl. Med. 2018, 16, 366. [Google Scholar] [CrossRef]
- Baysal, B.E.; Sharma, S.; Hashemikhabir, S.; Janga, S.C. RNA Editing in Pathogenesis of Cancer. Cancer Res. 2017, 77, 3733–3739. [Google Scholar] [CrossRef] [Green Version]
- Galeano, F.; Rossetti, C.; Tomaselli, S.; Cifaldi, L.; Lezzerini, M.; Pezzullo, M.; Boldrini, R.; Massimi, L.; Di Rocco, C.M.; Locatelli, F.; et al. ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene 2013, 32, 998–1009. [Google Scholar] [CrossRef] [Green Version]
- Patil, V.; Pal, J.; Mahalingam, K.; Somasundaram, K. Global RNA editome landscape discovers reduced RNA editing in glioma: Loss of editing of gamma-amino butyric acid receptor alpha subunit 3 (GABRA3) favors glioma migration and invasion. PeerJ 2020, 8, e9755. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Qin, Y.R.; Ming, X.Y.; Zuo, X.B.; Diao, Y.W.; Zhang, L.Y.; Ai, J.; Liu, B.L.; Huang, T.X.; Cao, T.T.; et al. RNA editing of SLC22A3 drives early tumor invasion and metastasis in familial esophageal cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E4631–E4640. [Google Scholar] [CrossRef] [Green Version]
- Han, S.W.; Kim, H.P.; Shin, J.Y.; Jeong, E.G.; Lee, W.C.; Kim, K.Y.; Park, S.Y.; Lee, D.W.; Won, J.K.; Jeong, S.Y.; et al. RNA editing in RHOQ promotes invasion potential in colorectal cancer. J. Exp. Med. 2014, 211, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.W.; Fu, T.; Bahn, J.H.; Jun, H.I.; Lee, J.H.; Quinones-Valdez, G.; Cheng, C.H.; Xiao, X.S. RNA editing in cancer impacts mRNA abundance in immune response pathways. Genome Biol. 2020, 21, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Fu, A.Q.; Bolt, M.J.; White, K.P. Clinical Relevance of Noncoding Adenosine-to-Inosine RNA Editing in Multiple Human Cancers. JCO Clin. Cancer Inform. 2019, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sharpnack, M.F.; Chen, B.; Aran, D.; Kosti, I.; Sharpnack, D.D.; Carbone, D.P.; Mallick, P.; Huang, K. Global Transcriptome Analysis of RNA Abundance Regulation by ADAR in Lung Adenocarcinoma. EBioMedicine 2018, 27, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416e411. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Zhang, K.N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Genom. Proteom. Bioinform. 2021, 19, 1–12. [Google Scholar] [CrossRef]
- Wilson, S.; Fitzsimons, M.; Ferguson, M.; Heath, A.; Jensen, M.; Miller, J.; Murphy, M.W.; Porter, J.; Sahni, H.; Staudt, L.; et al. Developing Cancer Informatics Applications and Tools Using the NCI Genomic Data Commons API. Cancer Res 2017, 77, e15–e18. [Google Scholar] [CrossRef] [Green Version]
- Touleimat, N.; Tost, J. Complete pipeline for Infinium((R)) Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics 2012, 4, 325–341. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.C.; Lo, C.M.; Wang, S.H.; Su, E.C. RNA editing-based classification of diffuse gliomas: Predicting isocitrate dehydrogenase mutation and chromosome 1p/19q codeletion. BMC Bioinform. 2019, 20, 659. [Google Scholar] [CrossRef]
- Lin, C.H.; Chen, S.C.C. The Cancer Editome Atlas: A Resource for Exploratory Analysis of the Adenosine-to-Inosine RNA Editome in Cancer. Cancer Res. 2019, 79, 3001–3006. [Google Scholar] [CrossRef] [Green Version]
- Lo Giudice, C.; Tangaro, M.A.; Pesole, G.; Picardi, E. Investigating RNA editing in deep transcriptome datasets with REDItools and REDIportal. Nat. Protoc. 2020, 15, 1098–1131. [Google Scholar] [CrossRef]
- Picardi, E.; D’Erchia, A.M.; Lo Giudice, C.; Pesole, G. REDIportal: A comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res. 2017, 45, D750–D757. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Meng, F.; Wang, W.; Wang, Z.; Zhang, C.; Jiang, T. Comprehensive RNA-seq transcriptomic profiling in the malignant progression of gliomas. Sci. Data 2017, 4, 170024. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A Toolkit for Quality Control of Next Generation Sequencing Data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- National Cancer Institute Genomic Data Commons. TCGA mRNA Analysis Pipeline. Available online: https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/ (accessed on 10 January 2020).
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. J. Open Source Softw. 2018, 6, 03426. [Google Scholar]
- Campello, R.J.G.B.; Moulavi, D.; Zimek, A.; Sander, J. Hierarchical Density Estimates for Data Clustering, Visualization, and Outlier Detection. ACM T Knowl. Discov. D 2015, 10, 1–51. [Google Scholar] [CrossRef]
- Zhang, X.C.; Liu, H.; Zhang, X.T. Novel density-based and hierarchical density-based clustering algorithms for uncertain data. Neural Netw. 2017, 93, 240–255. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Plaisier, S.B.; Taschereau, R.; Wong, J.A.; Graeber, T.G. Rank-rank hypergeometric overlap: Identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res. 2010, 38, e169. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Bahn, J.H.; Lee, J.H.; Li, G.; Greer, C.; Peng, G.; Xiao, X. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res. 2012, 22, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Carrano, A.; Juarez, J.J.; Incontri, D.; Ibarra, A.; Guerrero Cazares, H. Sex-Specific Differences in Glioblastoma. Cells 2021, 10, 1783. [Google Scholar] [CrossRef]
- Sancar, F. Quick Uptakes: Sex-Specific Tumor Signatures Help Explain Differences in Glioblastoma Survival. JAMA 2019, 321, 824–825. [Google Scholar] [CrossRef]
- Song, C.; Sakurai, M.; Shiromoto, Y.; Nishikura, K. Functions of the RNA Editing Enzyme ADAR1 and Their Relevance to Human Diseases. Genes 2016, 7, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, M.M.; Fogg, R.L.; Emeson, R.B.; Stanwood, G.D. ADAR1 and ADAR2 expression and editing activity during forebrain development. Dev. NeuroSci. 2009, 31, 223–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinones-Valdez, G.; Tran, S.S.; Jun, H.I.; Bahn, J.H.; Yang, E.W.; Zhan, L.; Brummer, A.; Wei, X.; Van Nostrand, E.L.; Pratt, G.A.; et al. Regulation of RNA editing by RNA-binding proteins in human cells. Commun. Biol. 2019, 2, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malta, T.M.; de Souza, C.F.; Sabedot, T.S.; Silva, T.C.; Mosella, M.S.; Kalkanis, S.N.; Snyder, J.; Castro, A.V.B.; Noushmehr, H. Glioma CpG island methylator phenotype (G-CIMP): Biological and clinical implications. Neuro Oncol. 2018, 20, 608–620. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef]
- Ren, Z.; Ding, T.; Zuo, Z.; Xu, Z.; Deng, J.; Wei, Z. Regulation of MAVS Expression and Signaling Function in the Antiviral Innate Immune Response. Front. Immunol. 2020, 11, 1030. [Google Scholar] [CrossRef]
- Zhou, B.; Wu, Q.; Chen, G.; Zhang, T.P.; Zhao, Y.P. NOP14 promotes proliferation and metastasis of pancreatic cancer cells. Cancer Lett. 2012, 322, 195–203. [Google Scholar] [CrossRef]
- Miller, S.; Rogers, H.A.; Lyon, P.; Rand, V.; Adamowicz-Brice, M.; Clifford, S.C.; Hayden, J.T.; Dyer, S.; Pfister, S.; Korshunov, A.; et al. Genome-wide molecular characterization of central nervous system primitive neuroectodermal tumor and pineoblastoma. Neuro. Oncol. 2011, 13, 866–879. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.Y.; An, O.; Ren, X.; Chan, T.H.M.; Tay, D.J.T.; Tang, S.J.; Han, J.; Hong, H.Q.; Ng, V.H.E.; Ke, X.Y.; et al. RNA editing mediates the functional switch of COPA in a novel mechanism of hepatocarcinogenesis. J. Hepatol. 2021, 74, 135–147. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Fritsche, J.; Roszik, J.; Williams, L.J.; Peng, X.X.; Chiu, Y.L.; Tsou, C.C.; Hoffgaard, F.; Goldfinger, V.; Schoor, O.; et al. RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.R.; Qiao, J.J.; Chan, T.H.M.; Zhu, Y.H.; Li, F.F.; Liu, H.B.; Fei, J.; Li, Y.; Guan, X.Y.; Chen, L.L. Adenosine-to-Inosine RNA Editing Mediated by ADARs in Esophageal Squamous Cell Carcinoma. Cancer Res. 2014, 74, 840–851. [Google Scholar] [CrossRef] [Green Version]
- Picardi, E.; Manzari, C.; Mastropasqua, F.; Aiello, I.; D’Erchia, A.M.; Pesole, G. Profiling RNA editing in human tissues: Towards the inosinome Atlas. Sci. Rep. 2015, 5, 14941. [Google Scholar] [CrossRef] [Green Version]
- Mansi, L.; Tangaro, M.A.; Lo Giudice, C.; Flati, T.; Kopel, E.; Schaffer, A.A.; Castrignano, T.; Chillemi, G.; Pesole, G.; Picardi, E. REDIportal: Millions of novel A-to-I RNA editing events from thousands of RNAseq experiments. Nucleic Acids Res. 2021, 49, D1012–D1019. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Yong, W.H.; Shabihkhani, M.; Telesca, D.; Yang, S.; Tso, J.L.; Menjivar, J.C.; Wei, B.; Lucey, G.M.; Mareninov, S.; Chen, Z.; et al. Ribosomal Proteins RPS11 and RPS20, Two Stress-Response Markers of Glioblastoma Stem Cells, Are Novel Predictors of Poor Prognosis in Glioblastoma Patients. PLoS ONE 2015, 10, e0141334. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Solomon, O.; Di Segni, A.; Cesarkas, K.; Porath, H.T.; Marcu-Malina, V.; Mizrahi, O.; Stern-Ginossar, N.; Kol, N.; Farage-Barhom, S.; Glick-Saar, E.; et al. RNA editing by ADAR1 leads to context-dependent transcriptome-wide changes in RNA secondary structure. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Prabhu, A.; Kesarwani, P.; Kant, S.; Graham, S.F.; Chinnaiyan, P. Histologically defined intratumoral sequencing uncovers evolutionary cues into conserved molecular events driving gliomagenesis. Neuro Oncol. 2017, 19, 1599–1606. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wang, Y.Y.; Xu, K.B.; Wang, Z.; Fan, X.; Zhang, C.B.; Li, S.W.; Qiu, X.G.; Jiang, T. Relationship between necrotic patterns in glioblastoma and patient survival: Fractal dimension and lacunarity analyses using magnetic resonance imaging. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005, 7, 134–153. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Tang, Z.; Zhang, W.; Ye, Z.; Liu, F. GEPIA2021: Integrating multiple deconvolution-based analysis into GEPIA. Nucleic Acids Res 2021, 49, W242–W246. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.Y.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451. [Google Scholar] [CrossRef]
- Tian, X.L.; Hellman, J.; Horswill, A.R.; Crosby, H.A.; Francis, K.P.; Prakash, A. Elevated Gut Microbiome-Derived Propionate Levels Are Associated With Reduced Sterile Lung Inflammation and Bacterial Immunity in Mice. Front. Microbiol. 2019, 10, 159. [Google Scholar]
- Negroni, A.; Colantoni, E.; Cucchiara, S.; Stronati, L. Necroptosis in Intestinal Inflammation and Cancer: New Concepts and Therapeutic Perspectives. Biomolecules 2020, 10, 1431. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. NeuroSci. 2018, 11, 480. [Google Scholar] [CrossRef] [Green Version]
- Achyut, B.R.; Angara, K.; Jain, M.; Borin, T.F.; Rashid, M.H.; Iskander, A.S.M.; Ara, R.; Kolhe, R.; Howard, S.; Venugopal, N.; et al. Canonical NF kappa B signaling in myeloid cells is required for the glioblastoma growth. Sci. Rep. 2017, 7, 1–12. [Google Scholar]
- Bell, E.H.; Zhang, P.; Shaw, E.G.; Buckner, J.C.; Barger, G.R.; Bullard, D.E.; Mehta, M.P.; Gilbert, M.R.; Brown, P.D.; Stelzer, K.J.; et al. Comprehensive Genomic Analysis in NRG Oncology/RTOG 9802: A Phase III Trial of Radiation Versus Radiation Plus Procarbazine, Lomustine (CCNU), and Vincristine in High-Risk Low-Grade Glioma. J. Clin. Oncol. 2020, 38, 3407. [Google Scholar] [CrossRef] [PubMed]
- Buckner, J.C.; Shaw, E.G.; Pugh, S.L.; Chakravarti, A.; Gilbert, M.R.; Barger, G.R.; Coons, S.; Ricci, P.; Bullard, D.; Brown, P.D.; et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N. Engl. J. Med. 2016, 374, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Geurts, M.; van den Bent, M.J. On high-risk, low-grade glioma: What distinguishes high from low? Cancer 2019, 125, 174–176. [Google Scholar] [CrossRef]
- Fritzell, K.; Xu, L.D.; Lagergren, J.; Ohman, M. ADARs and editing: The role of A-to-I RNA modification in cancer progression. Semin. Cell Dev. Biol. 2018, 79, 123–130. [Google Scholar] [CrossRef]
- Kurkowiak, M.; Arcimowicz, L.; Chrusciel, E.; Urban-Wojciuk, Z.; Papak, I.; Keegan, L.; O’Connell, M.; Kowalski, J.; Hupp, T.; Marek-Trzonkowska, N. The effects of RNA editing in cancer tissue at different stages in carcinogenesis. RNA Biol. 2021, 18, 1524–1539. [Google Scholar] [CrossRef]
- Anadon, C.; Guil, S.; Simo-Riudalbas, L.; Moutinho, C.; Setien, F.; Martinez-Cardus, A.; Moran, S.; Villanueva, A.; Calaf, M.; Vidal, A.; et al. Gene amplification-associated overexpression of the RNA editing enzyme ADAR1 enhances human lung tumorigenesis. Oncogene 2016, 35, 4407–4413. [Google Scholar] [CrossRef]
- Nakano, M.; Fukami, T.; Gotoh, S.; Nakajima, M. A-to-I RNA Editing Up-regulates Human Dihydrofolate Reductase in Breast Cancer. J. Biol. Chem. 2017, 292, 4873–4884. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Moya, J.; Baker, A.R.; Slack, F.J.; Santisteban, P. ADAR1-mediated RNA editing is a novel oncogenic process in thyroid cancer and regulates miR-200 activity. Oncogene 2020, 39, 3738–3753. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Moya, J.; Miliotis, C.; Baker, A.R.; Gregory, R.I.; Slack, F.J.; Santisteban, P. An ADAR1-dependent RNA editing event in the cyclin-dependent kinase CDK13 promotes thyroid cancer hallmarks. Mol. Cancer 2021, 20, 115. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Cui, Y.; Zhang, X. Suppression of RNA editing by miR-17 inhibits the stemness of melanoma stem cells. Mol. Ther. Nucleic Acids 2022, 27, 439–455. [Google Scholar] [CrossRef]
- Zipeto, M.A.; Court, A.C.; Sadarangani, A.; Delos Santos, N.P.; Balaian, L.; Chun, H.J.; Pineda, G.; Morris, S.R.; Mason, C.N.; Geron, I.; et al. ADAR1 Activation Drives Leukemia Stem Cell Self-Renewal by Impairing Let-7 Biogenesis. Cell Stem Cell 2016, 19, 177–191. [Google Scholar] [CrossRef]
- Yang, W.; Warrington, N.M.; Taylor, S.J.; Whitmire, P.; Carrasco, E.; Singleton, K.W.; Wu, N.; Lathia, J.D.; Berens, M.E.; Kim, A.H.; et al. Sex differences in GBM revealed by analysis of patient imaging, transcriptome, and survival data. Sci. Transl. Med. 2019, 11, eaao5253. [Google Scholar] [CrossRef] [Green Version]
- Rubin, J.B.; Lagas, J.S.; Broestl, L.; Sponagel, J.; Rockwell, N.; Rhee, G.; Rosen, S.F.; Chen, S.; Klein, R.S.; Imoukhuede, P.; et al. Sex differences in cancer mechanisms. Biol. Sex. Differ. 2020, 11, 17. [Google Scholar] [CrossRef] [Green Version]
- Ippolito, J.E.; Yim, A.K.; Luo, J.; Chinnaiyan, P.; Rubin, J.B. Sexual dimorphism in glioma glycolysis underlies sex differences in survival. JCI Insight 2017, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Lenz, K.M.; McCarthy, M.M. A Starring Role for Microglia in Brain Sex Differences. Neuroscientist 2015, 21, 306–321. [Google Scholar] [CrossRef] [Green Version]
- Loram, L.C.; Sholar, P.W.; Taylor, F.R.; Wiesler, J.L.; Babb, J.A.; Strand, K.A.; Berkelhammer, D.; Day, H.E.W.; Maier, S.F.; Watkins, L.R. Sex and estradiol influence glial pro-inflammatory responses to lipopolysaccharide in rats. Psychoneuroendocrino 2012, 37, 1688–1699. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Wahl, D.R. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers 2019, 11, 1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Zhang, P.; Shen, L.; Niu, L.; Tan, Y.; Chen, L.; Zhao, Y.; Bai, L.; Hao, X.; Li, X.; et al. Short-Chain Fatty Acids and Their Association with Signalling Pathways in Inflammation, Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2020, 21, 6356. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, B.T.; Zhu, L.; Eckel, R.H.; Stafford, J.M. Sex differences in lipid and lipoprotein metabolism. Mol. Metab. 2018, 15, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kwon, O.; Ryu, T.Y.; Jung, C.R.; Kim, J.; Min, J.K.; Kim, D.S.; Son, M.Y.; Cho, H.S. Propionate of a microbiota metabolite induces cell apoptosis and cell cycle arrest in lung cancer. Mol. Med. Rep. 2019, 20, 1569–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dynamic nutrient microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef] [Green Version]
- McKelvey, K.J.; Wilson, E.B.; Short, S.; Melcher, A.A.; Biggs, M.; Diakos, C.I.; Howell, V.M. Glycolysis and Fatty Acid Oxidation Inhibition Improves Survival in Glioblastoma. Front. Oncol. 2021, 11, 633210. [Google Scholar] [CrossRef]
- Duman, C.; Yaqubi, K.; Hoffmann, A.; Acikgoz, A.A.; Korshunov, A.; Bendszus, M.; Herold-Mende, C.; Liu, H.K.; Alfonso, J. Acyl-CoA-Binding Protein Drives Glioblastoma Tumorigenesis by Sustaining Fatty Acid Oxidation. Cell Metab. 2019, 30, 274–289. [Google Scholar] [CrossRef]
- Robinson, N.; McComb, S.; Mulligan, R.; Dudani, R.; Krishnan, L.; Sad, S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 2012, 13, 954–962. [Google Scholar] [CrossRef]
- Martin, S.J. Cell death and inflammation: The case for IL-1 family cytokines as the canonical DAMPs of the immune system. FEBS J. 2016, 283, 2599–2615. [Google Scholar] [CrossRef]
- Bayik, D.; Zhou, Y.; Park, C.; Hong, C.; Vail, D.; Silver, D.J.; Lauko, A.; Roversi, G.; Watson, D.C.; Lo, A.; et al. Myeloid-Derived Suppressor Cell Subsets Drive Glioblastoma Growth in a Sex-Specific Manner. Cancer Discov. 2020, 10, 1210–1225. [Google Scholar] [CrossRef] [Green Version]
- Kearney, C.J.; Martin, S.J. An Inflammatory Perspective on Necroptosis. Mol. Cell 2017, 65, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Nelson, L.H.; Lenz, K.M. The immune system as a novel regulator of sex differences in brain and behavioral development. J. NeuroSci. Res. 2017, 95, 447–461. [Google Scholar] [CrossRef]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 34, 1115–1120. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. Mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar] [CrossRef] [Green Version]
- George, C.X.; Ramaswami, G.; Li, J.B.; Samuel, C.E. Editing of Cellular Self-RNAs by Adenosine Deaminase ADAR1 Suppresses Innate Immune Stress Responses. J. Biol. Chem. 2016, 291, 6158–6168. [Google Scholar] [CrossRef] [Green Version]
- Nakahama, T.; Kato, Y.; Kim, J.I.; Vongpipatana, T.; Suzuki, Y.; Walkley, C.R.; Kawahara, Y. ADAR1-mediated RNA editing is required for thymic self-tolerance and inhibition of autoimmunity. EMBO Rep. 2018, 19, e46303. [Google Scholar] [CrossRef]
- Kawahara, Y.; Grimberg, A.; Teegarden, S.; Mombereau, C.; Liu, S.; Bale, T.L.; Blendy, J.A.; Nishikura, K. Dysregulated editing of serotonin 2C receptor mRNAs results in energy dissipation and loss of fat mass. J. NeuroSci. 2008, 28, 12834–12844. [Google Scholar] [CrossRef] [Green Version]
- Isaac, J.T.; Ashby, M.C.; McBain, C.J. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 2007, 54, 859–871. [Google Scholar] [CrossRef] [Green Version]
- Brusa, R.; Zimmermann, F.; Koh, D.S.; Feldmeyer, D.; Gass, P.; Seeburg, P.H.; Sprengel, R. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science 1995, 270, 1677–1680. [Google Scholar] [CrossRef]
- Hideyama, T.; Yamashita, T.; Suzuki, T.; Tsuji, S.; Higuchi, M.; Seeburg, P.H.; Takahashi, R.; Misawa, H.; Kwak, S. Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J. NeuroSci. 2010, 30, 11917–11925. [Google Scholar] [CrossRef]
- Davidson, N.O.; Shelness, G.S. APOLIPOPROTEIN B: mRNA editing, lipoprotein assembly, and presecretory degradation. Ann. Rev. Nutr. 2000, 20, 169–193. [Google Scholar] [CrossRef]
- Davidson, N.O. RNA editing of the apolipoprotein B gene A mechanism to regulate the atherogenic potential of intestinal lipoproteins? Trends Cardiovasc. Med. 1994, 4, 231–235. [Google Scholar] [CrossRef]
- Meurs, E.F.; Galabru, J.; Barber, G.N.; Katze, M.G.; Hovanessian, A.G. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1993, 90, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Gil, J.; Esteban, M. Induction of apoptosis by the dsRNA-dependent protein kinase (PKR): Mechanism of action. Apoptosis 2000, 5, 107–114. [Google Scholar] [CrossRef]
- Delgado Andre, N.; De Lucca, F.L. Knockdown of PKR expression by RNAi reduces pulmonary metastatic potential of B16-F10 melanoma cells in mice: Possible role of NF-kappaB. Cancer Lett. 2007, 258, 118–125. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Pikarsky, E.; Ben-Neriah, Y. NF-kappaB inhibition: A double-edged sword in cancer? Eur. J. Cancer 2006, 42, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Shir, A.; Levitzki, A. Inhibition of glioma growth by tumor-specific activation of double-stranded RNA-dependent protein kinase PKR. Nat. Biotechnol. 2002, 20, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Wakimoto, H. Oncolytic herpes simplex virus-based strategies: Toward a breakthrough in glioblastoma therapy. Front. Microbiol. 2014, 5, 303. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.-H.; Chen, S.C.-C. RNA Editing in Glioma as a Sexually Dimorphic Prognostic Factor That Affects mRNA Abundance in Fatty Acid Metabolism and Inflammation Pathways. Cells 2022, 11, 1231. https://doi.org/10.3390/cells11071231
Lin S-H, Chen SC-C. RNA Editing in Glioma as a Sexually Dimorphic Prognostic Factor That Affects mRNA Abundance in Fatty Acid Metabolism and Inflammation Pathways. Cells. 2022; 11(7):1231. https://doi.org/10.3390/cells11071231
Chicago/Turabian StyleLin, Sheng-Hau, and Sean Chun-Chang Chen. 2022. "RNA Editing in Glioma as a Sexually Dimorphic Prognostic Factor That Affects mRNA Abundance in Fatty Acid Metabolism and Inflammation Pathways" Cells 11, no. 7: 1231. https://doi.org/10.3390/cells11071231
APA StyleLin, S.-H., & Chen, S. C.-C. (2022). RNA Editing in Glioma as a Sexually Dimorphic Prognostic Factor That Affects mRNA Abundance in Fatty Acid Metabolism and Inflammation Pathways. Cells, 11(7), 1231. https://doi.org/10.3390/cells11071231