
Chaperone-Mediated Autophagy in Neurodegenerative Diseases and Acute Neurological Insults in the Central Nervous System



{kind=link}
Abstract
1. Introduction
2. General Characteristics of CMA
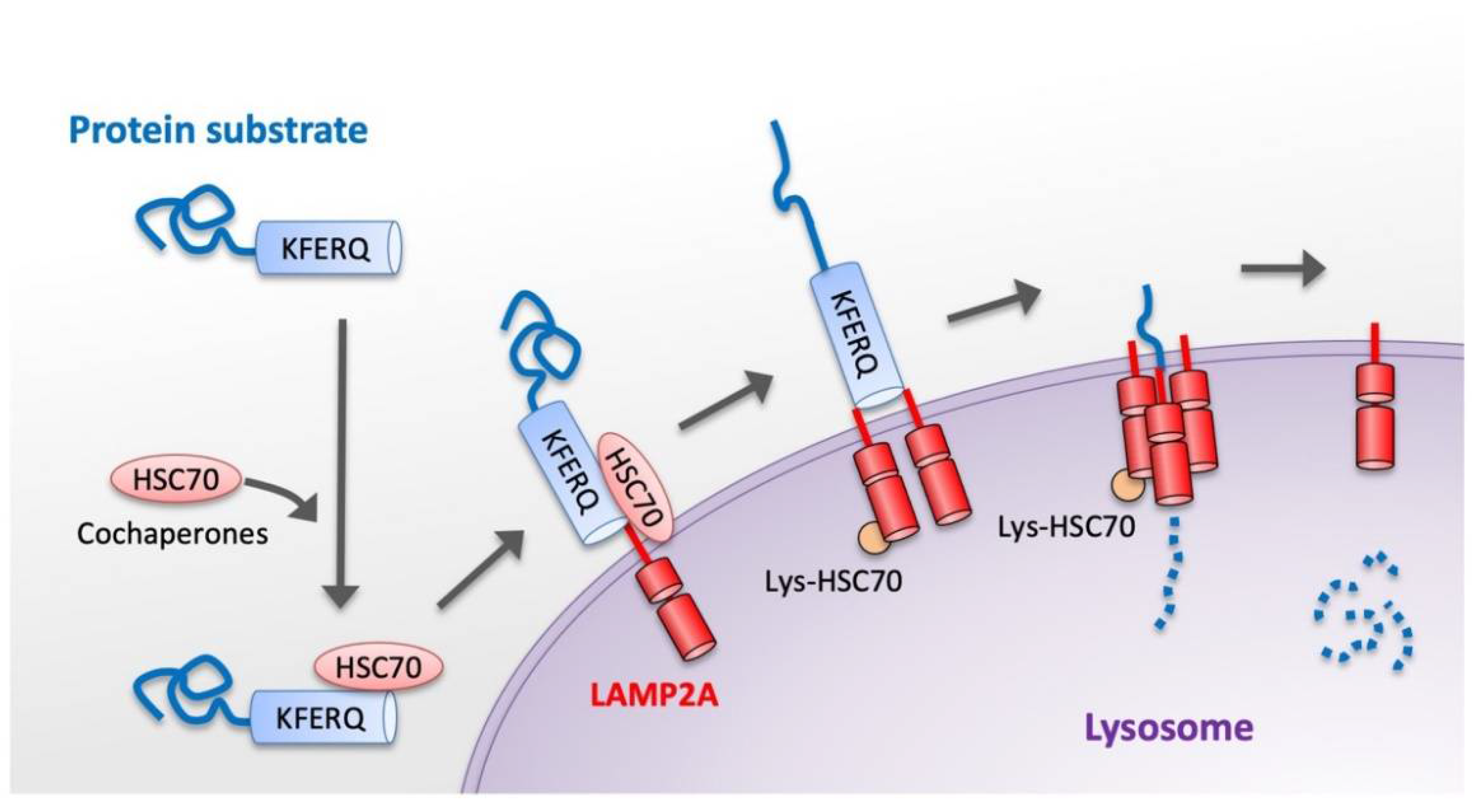
3. Basic Molecular Mechanism of CMA
4. Experimental Research Tools to Assay CMA Activity
4.1. Useful Analyses for Monitoring CMA Activity
4.2. Functional Assays
4.2.1. Intracellular Protein Degradation Assessment
4.2.2. Photoconvertible CMA Reporters
4.2.3. In Vitro Analyses of CMA Using Isolated Lysosomes
5. Neurodegenerative Diseases and CMA
5.1. Parkinson’s Disease
5.2. Alzheimer’s Disease
5.3. Huntington’s Disease
5.4. Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
6. Acute Neurological Insults and CMA
6.1. Traumatic Brain Injury
6.2. Cerebral Ischemia
6.3. Spinal Cord Injury
7. Therapeutic Potential of CMA for Neurodegenerative Diseases
8. Therapeutic Potential of CMA for Acute Neurological Insults
9. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Li, W.; Nie, T.; Xu, H.; Yang, J.; Yang, Q.; Mao, Z. Chaperone-mediated autophagy: Advances from bench to bedside. Neurobiol. Dis. 2019, 122, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Amanullah, A.; Upadhyay, A.; Joshi, V.; Mishra, R.; Jana, N.R.; Mishra, A. Progressing neurobiological strategies against proteostasis failure: Challenges in neurodegeneration. Prog. Neurobiol. 2017, 159, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Hekmatimoghaddam, S.; Zare-Khormizi, M.R.; Pourrajab, F. Underlying mechanisms and chemical/biochemical therapeutic approaches to ameliorate protein misfolding neurodegenerative diseases. BioFactors 2016, 43, 737–759. [Google Scholar] [CrossRef]
- Auzmendi-Iriarte, J.; Matheu, A. Impact of Chaperone-Mediated Autophagy in Brain Aging: Neurodegenerative Diseases and Glioblastoma. Front. Aging Neurosci. 2021, 12, 630743. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular selfdigestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.-L.; Terlecky, S.R.; Plant, C.P.; Dice, J.F. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989, 246, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2013, 24, 92–104. [Google Scholar] [CrossRef]
- Dice, J.F. Chaperone-mediated autophagy. Autophagy 2007, 3, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.N.A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. Physiol. 1995, 269, C1200–C1208. [Google Scholar] [CrossRef] [PubMed]
- Hubbi, M.E.; Hu, H.; Kshitiz, F.; Ahmed, I.; Levchenko, A.; Semenza, G.L. Chaperone-mediated Autophagy Targets Hypoxia-inducible Factor-1α (HIF-1α) for Lysosomal Degradation. J. Biol. Chem. 2013, 288, 10703–10714. [Google Scholar] [CrossRef]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of Chaperone-mediated Autophagy during Oxidative Stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-Mediated Autophagy Is Required for Tumor Growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Wang, B.; Liu, W.; Xu, Q.; Hou, L.; Song, J.; Guo, Q.; Li, N. Dysfunction of chaperone-mediated autophagy in human diseases. Mol. Cell. Biochem. 2021, 476, 1439–1454. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Song, J.-X.; Wang, S.-F.; Cai, C.-Z.; Li, M.; Lu, J.-H. Selective autophagy: The new player in the fight against neurodegenerative diseases? Brain Res. Bull. 2018, 137, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Liu, C.; Luo, T.; Dietrich, W.D.; Bramlett, H.; Hu, B. Chaperone-Mediated Autophagy after Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1449–1457. [Google Scholar] [CrossRef]
- Dohi, E.; Tanaka, S.; Seki, T.; Miyagi, T.; Hide, I.; Takahashi, T.; Matsumoto, M.; Sakai, N. Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem. Int. 2012, 60, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Handa, K.; Kanno, H.; Matsuda, M.; Sugaya, T.; Murakami, T.; Prudnikova, M.M.; Ozawa, H.; Itoi, E. Chaperone-Mediated Autophagy after Spinal Cord Injury. J. Neurotrauma 2020, 37, 1687–1695. [Google Scholar] [CrossRef]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of Cytosolic Proteins by Late Endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef]
- Schuck, S. Microautophagy–distinct molecular mechanisms handle cargoes of many sizes. J. Cell Sci. 2020, 133, jcs246322. [Google Scholar] [CrossRef] [PubMed]
- Marzella, L.; Ahlberg, J.; Glaumann, H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1981, 36, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Gough, N.R.; Hatem, C.L.; Fambrough, D.M. The family of LAMP-2 proteins arises by alternative splicing from a single gene: Characterization of the avian LAMP-2 gene and identification of mammalian homologs of LAMP-2b and LAMP-2c. DNA Cell Biol. 1995, 14, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, S.J.; Kuo, S.-H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef]
- Cannizzo, E.S.; Clement, C.C.; Morozova, K.; Valdor, R.; Kaushik, S.; Almeida, L.N.; Follo, C.; Sahu, R.; Cuervo, A.M.; Macian, F.; et al. Age-related oxidative stress compromises endosomal proteostasis. Cell Rep. 2012, 2, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Muela, N.; Koga, H.; García-Ledo, L.; de la Villa, P.; de la Rosa, E.J.; Cuervo, A.M.; Boya, P. Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 2013, 12, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cuervo, A.M. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 2008, 14, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Hu, W.; Lim, B.; Dice, J.F. IκB is a substrate for a selective pathway of lysosomal proteolysis. Mol. Biol. Cell 1998, 9, 1995–2010. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Hildebrand, H.; Bomhard, E.M.; Dice, J.F. Direct lysosomal uptake of α2-microglobulin contributes to chemically induced nephropathy. Kidney Int. 1999, 55, 529–545. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Finn, P.F.; Dice, J.F. Ketone bodies stimulate chaperone-mediatedautophagy. J. Biol. Chem. 2005, 280, 25864–25870. [Google Scholar] [CrossRef] [PubMed]
- Wing, S.S.; Chiang, H.L.; Goldberg, A.L.; Dice, J.F. Proteins containing peptide sequences related to Lys-Phe-Glu-Arg-Gln are selectively depleted in liver and heart, but not skeletal muscle, of fasted rats. Biochem. J. 1991, 275, 165–169. [Google Scholar] [CrossRef]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Kshitiz; Ahmed, I.; Semenza, G.L. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1α to promote cell-cycle progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3325–E3334. [Google Scholar] [CrossRef]
- Yang, Q.; She, H.; Gearing, M.; Colla, E.; Lee, M.; Shacka, J.J.; Mao, Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 2009, 323, 124–127. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef]
- Salvador, N.; Aguado, C.; Horst, M.; Knecht, E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J. Biol. Chem. 2000, 275, 27447–27456. [Google Scholar] [CrossRef]
- Park, C.; Cuervo, A.M. Selective autophagy: Talking with the UPS. Cell Biophys. 2013, 67, 3–13. [Google Scholar] [CrossRef]
- Wu, H.; Chen, S.; Ammar, A.-B.; Xu, J.; Wu, Q.; Pan, K.; Zhang, J.; Hong, Y. Crosstalk Between Macroautophagy and Chaperone-Mediated Autophagy: Implications for the Treatment of Neurological Diseases. Mol. Neurobiol. 2014, 52, 1284–1296. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Massey, A.C.; Mizushima, N.; Cuervo, A.M. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell 2008, 19, 2179–2192. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.; Peter, M. Substrate recognition in selective autophagy and the ubiquitin–proteasome system. Biochim. Biophys. Acta 2014, 1843, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Nedelsky, N.B.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2008, 1782, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, P.; Song, W.; Sun, X. Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J. 2009, 23, 3383–3392. [Google Scholar] [CrossRef]
- Patel, B.; Cuervo, A.M. Methods to study chaperone-mediated autophagy. Methods 2015, 75, 133–140. [Google Scholar] [CrossRef]
- Arias, E. Methods to study chaperone-mediated autophagy. Methods Enzymol. 2017, 588, 283–305. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.; Knecht, E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J. Biol. Chem. 1997, 272, 5606–5615. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chapter 19 Methods to Monitor Chaperone-Mediated Autophagy. Methods Enzymol. 2009, 452, 297–324. [Google Scholar] [CrossRef]
- Koga, H.; Martinez-Vicente, M.; Macian, F.; Verkhusha, V.V.; Cuervo, A.M. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun. 2011, 2, 386. [Google Scholar] [CrossRef] [PubMed]
- Storrie, B.; Amadden, E. [16] Isolation of subcellular organelles. Methods Enzymol. 1990, 182, 203–225. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The Chaperone-Mediated Autophagy Receptor Organizes in Dynamic Protein Complexes at the Lysosomal Membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Kaushik, S.; Zeng, M.; Bandyopadhyay, U.; Zhang, C.; Massey, A.C.; Martinez-Vicente, M.; Cuervo, A.M. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J. Cell Sci. 2007, 120, 782–791. [Google Scholar] [CrossRef]
- Cuervo, A.; Dice, J. Regulation of Lamp2a levels in the lysosomal membrane. Traffic 2000, 1, 570–583. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Cuervo, A.M. Entering the lysosome through a transient gate by chaperone-mediated autophagy. Autophagy 2008, 4, 1101–1103. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.-M.; Cuervo, A.M.; Mandelkow, E. Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy 2010, 6, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y.; et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010, 28, 256–263. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Arosio, A.; Cristofani, R.; Pansarasa, O.; Crippa, V.; Riva, C.; Sirtori, R.; Rodriguez-Menendez, V.; Riva, N.; Gerardi, F.; Lunetta, C.; et al. HSC70 expression is reduced in lymphomonocytes of sporadic ALS patients and contributes to TDP-43 accumulation. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 51–62. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Xilouri, M.; Stefanis, L. Chaperone mediated autophagy to the rescue: A new-fangled target for the treatment of neurodegenerative diseases. Mol. Cell. Neurosci. 2015, 66, 29–36. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.-C.; Follenzi, A.; et al. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [CrossRef]
- Guedes, L.C.; Ferreira, J.J.; Rosa, M.M.; Coelho, M.; Bonifati, V.; Sampaio, C. Worldwide frequency of G2019S LRRK2 mutation in Parkinson’s disease: A systematic review. Park. Relat. Disord. 2010, 16, 237–242. [Google Scholar] [CrossRef]
- Ho, P.W.-L.; Leung, C.-T.; Liu, H.; Pang, S.Y.-Y.; Lam, C.S.-C.; Xian, J.; Li, L.; Kung, M.H.-W.; Ramsden, D.B.; Ho, S.-L. Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: Role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy 2020, 16, 347–370. [Google Scholar] [CrossRef] [PubMed]
- Agarraberes, F.A.; Dice, J.F. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 2001, 114, 2491–2499. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Kabuta, T.; Furuta, A.; Aoki, S.; Furuta, K.; Wada, K. Aberrant Interaction between Parkinson disease-associated Mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J. Biol. Chem. 2008, 283, 23731–23738. [Google Scholar] [CrossRef]
- Wang, B.; Cai, Z.; Tao, K.; Zeng, W.; Lu, F.; Yang, R.; Feng, D.; Gao, G.; Yang, Q. Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy 2016, 12, 1215–1228. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Xu, C.-Y.; Kang, W.-Y.; Chen, Y.-M.; Jiang, T.-F.; Zhang, J.; Zhang, L.-N.; Ding, J.-Q.; Liu, J.; Chen, S.-D. DJ-1 Inhibits α-Synuclein Aggregation by Regulating Chaperone-Mediated Autophagy. Front. Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.-M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef]
- Hayden, E.Y.; Teplow, D.B. Amyloid β-protein oligomers and Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 60. [Google Scholar] [CrossRef]
- Klein, W.L. Synaptotoxic Amyloid-β Oligomers: A molecular basis for the cause, diagnosis, and treatment of Alzheimer’s Disease? J. Alzheimers Dis. 2012, 33, S49–S65. [Google Scholar] [CrossRef]
- Dou, J.; Su, P.; Xu, C.; Wen, Z.; Mao, Z.; Li, W. Targeting Hsc70-based autophagy to eliminate amyloid β oligomers. Biochem. Biophys. Res. Commun. 2020, 524, 923–928. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Hamdane, M.; Gompel, M.; Bégard, S.; Drobecq, H.; Ghestem, A.; Grosjean, M.-E.; Kostanjevecki, V.; Grognet, P.; Vanmechelen, E.; et al. Phosphorylation of amyloid precursor carboxy-terminal fragments enhances their processing by a gamma-secretase-dependent mechanism. Neurobiol. Dis. 2005, 20, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Kim, D.-H.; Yoon, S.-Y. Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 2016, 49, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, Y.; Cen, X.; Shan, B.; Zhao, Q.; Xie, T.; Wang, Z.; Hou, T.; Xue, Y.; Zhang, M.; et al. Metformin activates chaperone-mediated autophagy and improves disease pathologies in an Alzheimer disease mouse model. Protein Cell 2021, 12, 769–787. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.D.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef]
- Alonso, A.D.C.; Grundke-Iqbal, I.; Barra, H.S.; Iqbal, K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA 1997, 94, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.D.; Ermak, G.; Davies, K.J.A. RCAN1-1L is overexpressed in neurons of Alzheimer’s disease patients. FEBS J. 2007, 274, 1715–1724. [Google Scholar] [CrossRef]
- Qi, L.; Zhang, X.-D.; Wu, J.-C.; Lin, F.; Wang, J.; DiFiglia, M.; Qin, Z.-H. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS ONE 2012, 7, e46834. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.-H.; Wang, Y.; Kegel, K.B.; Kazantsev, A.; Apostol, B.L.; Thompson, L.M.; Yoder, J.; Aronin, N.; DiFiglia, M. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 2003, 12, 3231–3244. [Google Scholar] [CrossRef]
- Koga, H.; Martinez-Vicente, M.; Arias, E.; Kaushik, S.; Sulzer, D.; Cuervo, A.M. Constitutive Upregulation of Chaperone-Mediated Autophagy in Huntington’s Disease. J. Neurosci. 2011, 31, 18492–18505. [Google Scholar] [CrossRef]
- Thompson, L.M.; Aiken, C.T.; Kaltenbach, L.S.; Agrawal, N.; Illes, K.; Khoshnan, A.; Martinez-Vicente, M.; Arrasate, M.; O’Rourke, J.G.; Khashwji, H.; et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 2009, 187, 1083–1099. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.-B.; Almeida, S.; Lopez-Gonzalez, R. Dysregulated molecular pathways in amyotrophic lateral sclerosis–frontotemporal dementia spectrum disorder. EMBO J. 2017, 36, 2931–2950. [Google Scholar] [CrossRef] [PubMed]
- Ormeño, F.; Hormazabal, J.; Moreno, J.; Riquelme, F.; Rios, J.; Criollo, A.; Albornoz, A.; Alfaro, I.E.; Budini, M. Chaperone Mediated Autophagy Degrades TDP-43 Protein and Is Affected by TDP-43 Aggregation. Front. Mol. Neurosci. 2020, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-C.; Bose, J.K.; Majumder, P.; Lee, K.-H.; Huang, J.-T.J.; Huang, J.; Shen, C.-K.J. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J. Cell Sci. 2014, 127, 3024–3038. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Chen, S.; Dietrich, D.; Hu, B.R. Changes in autophagy after traumatic brain injury. J. Cereb. Blood Flow Metab. 2008, 28, 674–683. [Google Scholar] [CrossRef]
- Sadasivan, S.; Dunn, W.A., Jr.; Hayes, R.L.; Wang, K.K. Changes in autophagy proteins in a rat model of controlled cortical impact induced brain injury. Biochem. Biophys. Res. Commun. 2008, 373, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Erlich, S.; Alexandrovich, A.; Shohami, E.; Pinkas-Kramarski, R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol. Dis. 2007, 26, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lipinski, M.M. Autophagy in Neurotrauma: Good, Bad, or Dysregulated. Cells 2019, 8, 693. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.; Wu, X.; Zhang, Y.; Lu, J.; Li, D. Sirt1 Attenuates Astrocyte Activation Via Modulating Dnajb1 and Chaperone-Mediated Autophagy after Closed Head Injury. Cereb. Cortex 2022. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; An, D.; Xu, J.; Shao, B.; Li, X.; Shi, J. Ac2-26 Induces IKKβ Degradation Through Chaperone-Mediated Autophagy via HSPB1 in NCM-Treated Microglia. Front. Mol. Neurosci. 2018, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Girelli, S.; Scopa, C.; Buonocore, G.; Longini, M.; Balduini, W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy 2010, 6, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia–ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Kitagawa, K.; Ohtsuki, T.; Yagita, Y.; Takasawa, K.; Hori, M.; Matsumoto, M. Synergistic induction of HSP40 and HSC70 in the mouse hippocampal neurons after cerebral ischemia and ischemic tolerance in gerbil hippocampus. J. Neurosci. Res. 2001, 67, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Kume, A.; Li, M.; Doyu, M.; Hata, M.; Ohtsuka, K.; Sobue, G. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J. Biol. Chem. 2000, 275, 8772–8778. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-F.; Wang, J.; Deng, M.-F.; Chi, B.; Wei, N.; Chen, J.-G.; Liu, D.; Yin, X.; Lu, Y.; Zhu, L.-Q. The Peptide-Directed Lysosomal Degradation of CDK5 Exerts Therapeutic Effects against Stroke. Aging Dis. 2019, 10, 1140–1145. [Google Scholar] [CrossRef]
- Kanno, H.; Ozawa, H.; Sekiguchi, A.; Yamaya, S.; Itoi, E. Induction of autophagy and autophagic cell death in damaged neural tissue after acute spinal cord injury in mice. Spine 2011, 36, E1427–E1434. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Ozawa, H.; Sekiguchi, A.; Itoi, E. The role of autophagy in spinal cord injury. Autophagy 2009, 5, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, A.; Kanno, H.; Ozawa, H.; Yamaya, S.; Itoi, E. Rapamycin promotes autophagy and reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. J. Neurotrauma 2012, 29, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Tateda, S.; Kanno, H.; Ozawa, H.; Sekiguchi, A.; Yahata, K.; Yamaya, S.; Itoi, E. Rapamycin suppresses microglial activation and reduces the development of neuropathic pain after spinal cord injury. J. Orthop. Res. 2017, 35, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Ozawa, H.; Sekiguchi, A.; Yamaya, S.; Tateda, S.; Yahata, K.; Itoi, E. The role of mTOR signaling pathway in spinal cord injury. Cell Cycle 2012, 11, 3175–3179. [Google Scholar] [CrossRef]
- Yong, C.; Arnold, P.M.; Zoubine, M.N.; Citron, B.A.; Watanabe, I.; Berman, N.E.J.; Festoff, B.W. Apoptosis in Cellular Compartments of rat spinal cord after severe contusion injury. J. Neurotrauma 1998, 15, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Ozawa, H.; Handa, K.; Murakami, T.; Itoi, E. Changes in Expression of Receptor-Interacting Protein Kinase 1 in Secondary Neural Tissue Damage Following Spinal Cord Injury. Neurosci. Insights 2020, 15, 2633105520906402. [Google Scholar] [CrossRef] [PubMed]
- Citron, B.A.; Arnold, P.M.; Sebastian, C.; Qin, F.; Malladi, S.; Ameenuddin, S.; Landis, M.E.; Festoff, B.W. Rapid upregulation of caspase-3 in rat spinal cord after injury: mRNA, protein, and cellular localization correlates with apoptotic cell death. Exp. Neurol. 2000, 166, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Yang, X.; Li, Z.; Le, W.; Hao, Y.; Song, Y.; Wang, F.; Guan, Y. HDAC6-mediated Hsp90 deacetylation reduces aggregation and toxicity of the protein alpha-synuclein by regulating chaperone-mediated autophagy. Neurochem. Int. 2021, 149, 105141. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Guan, H.; Zhang, F.; Gao, Y.; Teng, X.; Yang, W. HDAC6 Regulates the Chaperone-Mediated Autophagy to Prevent Oxidative Damage in Injured Neurons after Experimental Spinal Cord Injury. Oxidative Med. Cell. Longev. 2016, 2016, 7263736. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Kirik, D.; Stefanis, L. LAMP2A as a therapeutic target in Parkinson disease. Autophagy 2013, 9, 2166–2168. [Google Scholar] [CrossRef] [PubMed]
- Pedrozo, Z.; Torrealba, N.; Fernández, C.; Gatica, D.; Toro, B.; Quiroga, C.; Rodriguez, A.E.; Sanchez, G.; Gillette, T.G.; Hill, J.A.; et al. Cardiomyocyte ryanodine receptor degradation by chaperone-mediated autophagy. Cardiovasc. Res. 2013, 98, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Finn, P.F.; Mesires, N.T.; Vine, M.; Dice, J.F. Effects of small molecules on chaperone-mediated autophagy. Autophagy 2005, 1, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.K.; Rasheed, M.S.U.; Mishra, A.K.; Patel, D.K.; Singh, M.P. Silymarin Protects Against Impaired Autophagy Associated with 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-Induced Parkinsonism. J. Mol. Neurosci. 2019, 70, 276–283. [Google Scholar] [CrossRef]
- Luan, Y.; Ren, X.; Zheng, W.; Zeng, Z.; Guo, Y.; Hou, Z.; Guo, W.; Chen, X.; Li, F.; Chen, J.-F. Chronic Caffeine Treatment Protects against α-Synucleinopathy by Reestablishing Autophagy Activity in the Mouse Striatum. Front. Neurosci. 2018, 12, 301. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.-Y.; Liu, C.; Tan, X.; Ma, Z.; Wang, C.; Deng, Y.; Liu, W.; Xu, Z.-F.; Xu, B. Mn-Induced Neurocytes Injury and Autophagy Dysfunction in Alpha-Synuclein Wild-Type and Knock-Out Mice: Highlighting the Role of Alpha-Synuclein. Neurotox. Res. 2019, 36, 66–80. [Google Scholar] [CrossRef]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2018, 15, 631–651. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Deng, M.; He, Y.; Lu, S.; Liu, S.; Fang, Y. β-asarone increases MEF2D and TH levels and reduces α-synuclein level in 6-OHDA-induced rats via regulating the HSP70/MAPK/MEF2D/Beclin-1 pathway: Chaperone-mediated autophagy activation, macroautophagy inhibition and HSP70 up-expression. Behav. Brain Res. 2016, 313, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-Z.; Ardah, M.; Haikal, C.; Svanbergsson, A.; Diepenbroek, M.; Vaikath, N.N.; Li, W.; Wang, Z.-Y.; Outeiro, T.F.; El-Agnaf, O.M.; et al. Dihydromyricetin and Salvianolic acid B inhibit alpha-synuclein aggregation and enhance chaperone-mediated autophagy. Transl. Neurodegener. 2019, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Nagase, K.; Chosa, M.; Tobinai, K. Schwann cell autophagy induced by SAHA, 17-AAG, or clonazepam can reduce bortezomib-induced peripheral neuropathy. Br. J. Cancer 2010, 103, 1580–1587. [Google Scholar] [CrossRef]
- Oehme, I.; Linke, J.-P.; Böck, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M.; et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef] [PubMed]
- Obayashi, H.; Nagano, Y.; Takahashi, T.; Seki, T.; Tanaka, S.; Sakai, N.; Matsumoto, M.; Maruyama, H. Histone deacetylase 10 knockout activates chaperone-mediated autophagy and accelerates the decomposition of its substrate. Biochem. Biophys. Res. Commun. 2019, 523, 246–252. [Google Scholar] [CrossRef]
- Tamaki, Y.; Shodai, A.; Morimura, T.; Hikiami, R.; Minamiyama, S.; Ayaki, T.; Tooyama, I.; Furukawa, Y.; Takahashi, R.; Urushitani, M. Elimination of TDP-43 inclusions linked to amyotrophic lateral sclerosis by a misfolding-specific intrabody with dual proteolytic signals. Sci. Rep. 2018, 8, 6030. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Tasset, I.; Diaz, A.; Anguiano, J.; Tas, E.; Cui, L.; Kuliawat, R.; Liu, H.; Kühn, B.; Cuervo, A.M.; et al. Humanin is an endogenous activator of chaperone-mediated autophagy. J. Cell Biol. 2017, 217, 635–647. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to cns damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [PubMed]
- McConeghy, K.W.; Hatton, J.; Hughes, L.; Cook, A.M.; Cook, A.M. A review of neuroprotection pharmacology and therapies in patients with acute traumatic brain injury. CNS Drugs 2012, 26, 613–636. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Wu, J.; Faden, A.I.; Sarkar, C. Function and Mechanisms of Autophagy in Brain and Spinal Cord Trauma. Antioxid. Redox Signal. 2015, 23, 565–577. [Google Scholar] [CrossRef]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Var, S.R.; Shetty, A.V.; Grande, A.W.; Low, W.C.; Cheeran, M.C. Microglia and Macrophages in Neuroprotection, Neurogenesis, and Emerging Therapies for Stroke. Cells 2021, 10, 3555. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275 Pt 3, 316–327. [Google Scholar] [CrossRef] [PubMed]
- di Domenico, A.; Carola, G.; Calatayud, C.; Espinal, M.P.; Muñoz, J.P.; Richaud-Patin, Y.; Carasa, I.F.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Mavroeidi, P.; Arvanitaki, F.; Vetsi, M.; Becker, S.; Vlachakis, D.; Jensen, P.H.; Stefanis, L.; Xilouri, M. Autophagy mediates the clearance of oligodendroglial SNCA/alpha-synuclein and TPPP/p25A in multiple system atrophy models. Autophagy 2022, 1–30. [Google Scholar] [CrossRef]
- Sauerbeck, A.D.; Goldstein, E.Z.; Alfredo, A.N.; Norenberg, M.; Marcillo, A.; McTigue, D.M. Alpha-synuclein increases in rodent and human spinal cord injury and promotes inflammation and tissue loss. Sci. Rep. 2021, 11, 11720. [Google Scholar] [CrossRef]
- Ohri, S.S.; Bankston, A.N.; Mullins, S.A.; Liu, Y.; Andres, K.R.; Beare, J.E.; Howard, R.M.; Burke, D.A.; Riegler, A.S.; Smith, A.E.; et al. Blocking Autophagy in Oligodendrocytes Limits Functional Recovery after Spinal Cord Injury. J. Neurosci. 2018, 38, 5900–5912. [Google Scholar] [CrossRef] [PubMed]
- Washington, P.M.; Villapol, S.; Burns, M.P. Polypathology and dementia after brain trauma: Does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp. Neurol. 2015, 275, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Griesbach, G.S.; Masel, B.E.; Helvie, R.E.; Ashley, M.J. The Impact of Traumatic Brain Injury on Later Life: Effects on Normal Aging and Neurodegenerative Diseases. J. Neurotrauma 2018, 35, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Kawai, N.; Kawanishi, M.; Kudomi, N.; Maeda, Y.; Yamamoto, Y.; Nishiyama, Y.; Tamiya, T. Detection of brain amyloid β deposition in patients with neuropsychological impairment after traumatic brain injury: PET evaluation using Pittsburgh Compound-B. Brain Inj. 2013, 27, 1026–1031. [Google Scholar] [CrossRef]
- Ubukata, S.; Oishi, N.; Higashi, T.; Kagawa, S.; Yamauchi, H.; Okuyama, C.; Watanabe, H.; Ono, M.; Saji, H.; Aso, T.; et al. Spatial Patterns of Amyloid Deposition in Patients with Chronic Focal or Diffuse Traumatic Brain Injury Using 18F-FPYBF-2 PET. Neuropsychiatr. Dis. Treat. 2020, 16, 2719–2732. [Google Scholar] [CrossRef]
- Albayram, O.; Kondo, A.; Mannix, R.; Smith, C.; Tsai, C.-Y.; Colin, S.; Herbert, M.K.; Qiu, J.; Monuteaux, M.; Driver, J.; et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat. Commun. 2017, 8, 1000. [Google Scholar] [CrossRef]
- Bourdenx, M.; Martín-Segura, A.; Scrivo, A.; Rodriguez-Navarro, J.A.; Kaushik, S.; Tasset, I.; Diaz, A.; Storm, N.J.; Xin, Q.; Juste, Y.R.; et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 2021, 184, 2696–2714.e25. [Google Scholar] [CrossRef]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef]
- Ren, X.; Chen, J.-F. Caffeine and Parkinson’s Disease: Multiple Benefits and Emerging Mechanisms. Front. Neurosci. 2020, 14, 602697. [Google Scholar] [CrossRef]
- Towers, C.G.; Thorburn, A. Therapeutic Targeting of Autophagy. eBioMedicine 2016, 14, 15–23. [Google Scholar] [CrossRef]
- Moors, T.E.; Hoozemans, J.J.M.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.-C.; Van De Berg, W.D.J. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, H.; Handa, K.; Murakami, T.; Aizawa, T.; Ozawa, H. Chaperone-Mediated Autophagy in Neurodegenerative Diseases and Acute Neurological Insults in the Central Nervous System. Cells 2022, 11, 1205. https://doi.org/10.3390/cells11071205
Kanno H, Handa K, Murakami T, Aizawa T, Ozawa H. Chaperone-Mediated Autophagy in Neurodegenerative Diseases and Acute Neurological Insults in the Central Nervous System. Cells. 2022; 11(7):1205. https://doi.org/10.3390/cells11071205
Chicago/Turabian StyleKanno, Haruo, Kyoichi Handa, Taishi Murakami, Toshimi Aizawa, and Hiroshi Ozawa. 2022. "Chaperone-Mediated Autophagy in Neurodegenerative Diseases and Acute Neurological Insults in the Central Nervous System" Cells 11, no. 7: 1205. https://doi.org/10.3390/cells11071205
APA StyleKanno, H., Handa, K., Murakami, T., Aizawa, T., & Ozawa, H. (2022). Chaperone-Mediated Autophagy in Neurodegenerative Diseases and Acute Neurological Insults in the Central Nervous System. Cells, 11(7), 1205. https://doi.org/10.3390/cells11071205