
Platelets, Bacterial Adhesins and the Pneumococcus



Abstract
1. Introduction
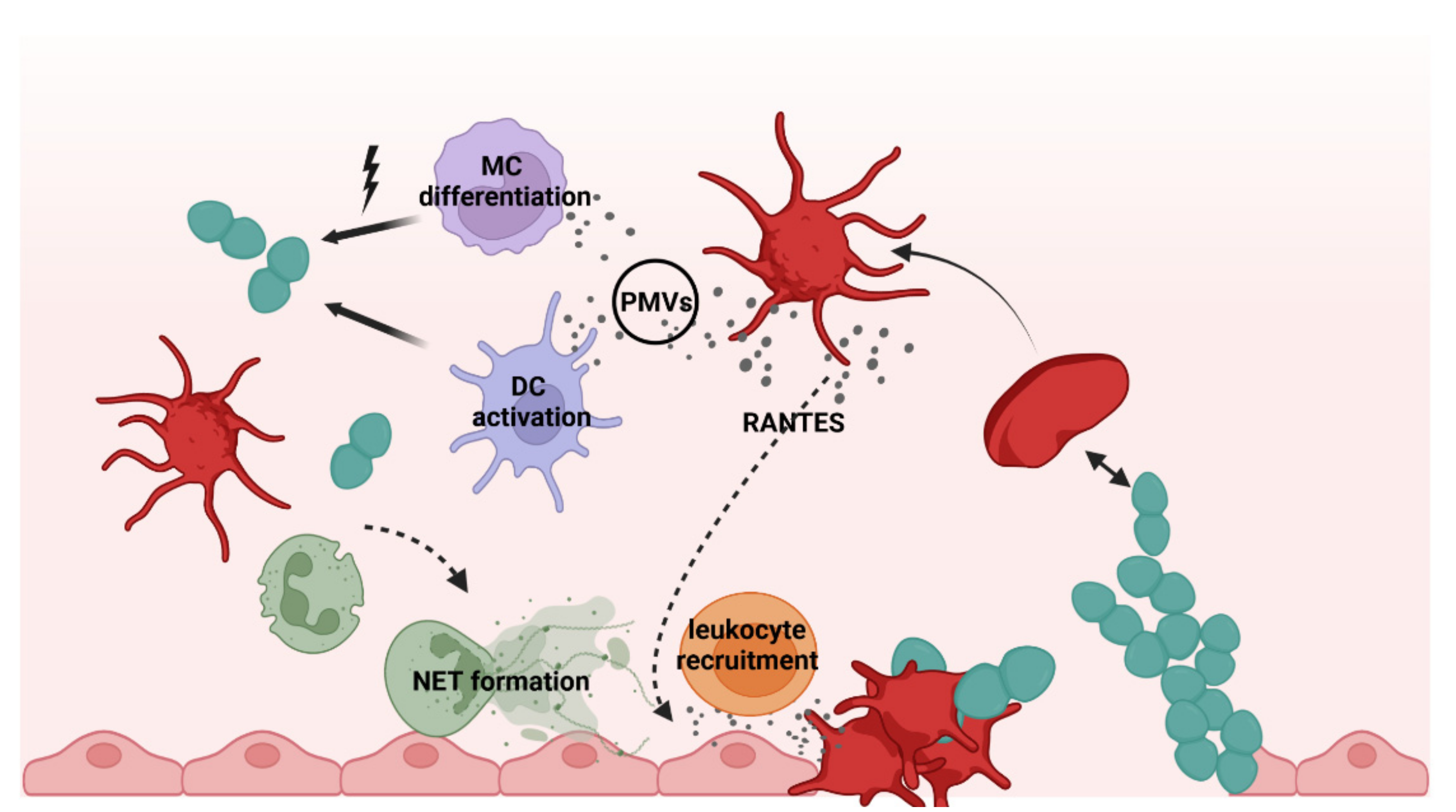
2. Platelets as Immune Cells in Infections
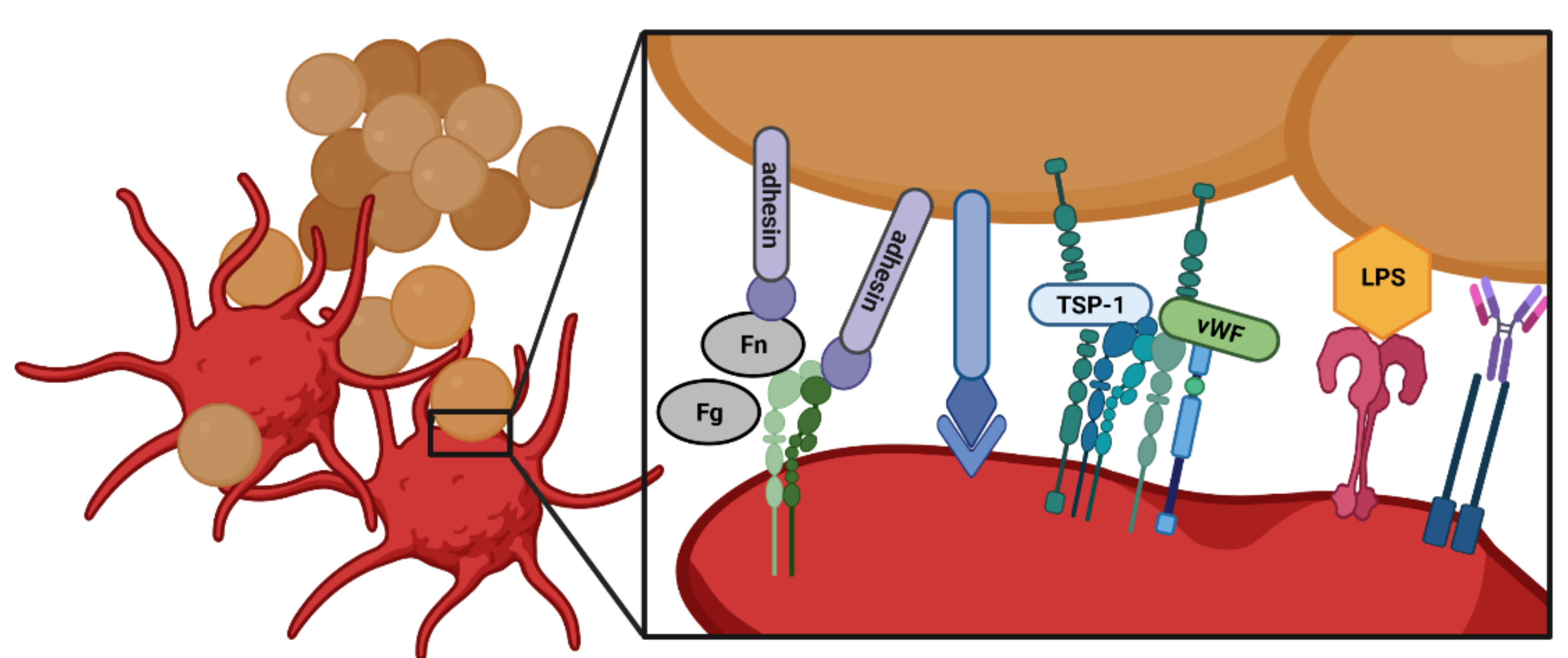
3. Interactions of Platelets with Bacteria
4. Platelet Receptors in Bacterial Infections and Bacterial Adhesins
5. Platelets in S. pneumoniae Infections
5.1. Community Acquired Pneumonia (CAP)
5.2. Sepsis
5.3. Infective Endocarditis
5.4. Pneumococcal Interactions with Platelets
6. Relevance of Findings for Disease
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robier, C. Platelet morphology. J. Lab. Med. 2020, 44, 231–239. [Google Scholar] [CrossRef]
- Lefrançais, E.; Looney, M.R. Platelet Biogenesis in the Lung Circulation. Physiology 2019, 34, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Machlus, K.R.; Italiano, J.E., Jr. The incredible journey: From megakaryocyte development to platelet formation. J. Cell Biol. 2013, 201, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, I.C.; Carr, P.; Gusnanto, A.; Ouwehand, W.H.; Fitzgerald, D.; Watkins, N.A. Platelet genomics and proteomics in human health and disease. J. Clin. Investig. 2005, 115, 3370–3377. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.L.; Mascelli, M.A.; Neblock, D.S.; Weisman, H.F.; Coller, B.S.; Jordan, R.E. Analysis of GPIIb/IIIa Receptor Number by Quantification of 7E3 Binding to Human Platelets. Blood 1996, 88, 907–914. [Google Scholar] [CrossRef]
- Bledzka, K.; Smyth, S.S.; Plow, E.F. Integrin αIIbβ3: From discovery to efficacious therapeutic target. Circ. Res. 2013, 112, 1189–1200. [Google Scholar] [CrossRef]
- Afshar-Kharghan, V.; Agah, R.; Andrews, R.K.; Aster, R.H.; Atkinson, B.; Awtry, E.H.; Bahou, W.F.; Barnard, M.R.; Bavry, A.A.; Bayer, A.S.; et al. Contributors. In Platelets, 2nd ed.; Michelson, A.D., Ed.; Academic Press: Cambridge, MA, USA, 2007. [Google Scholar] [CrossRef]
- Klinger, M.H.; Jelkmann, W. Role of blood platelets in infection and inflammation. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2002, 22, 913–922. [Google Scholar] [CrossRef]
- Portier, I.; Campbell, R.A. Role of Platelets in Detection and Regulation of Infection. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 70–78. [Google Scholar] [CrossRef]
- Assinger, A. Platelets and infection-an emerging role of platelets in viral infection. Front. Immunol. 2014, 5, 649. [Google Scholar] [CrossRef]
- Burkard, P.; Vögtle, T.; Nieswandt, B. Platelets in Thrombo-Inflammation: Concepts, Mechanisms, and Therapeutic Strategies for Ischemic Stroke. Hamostaseologie 2020, 40, 153–164. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, X.M.; Fang, L.; Jennings, N.L.; Su, Y.; Xu, Q.; Samson, A.L.; Kiriazis, H.; Wang, X.F.; Shan, L.; et al. Novel role of platelets in mediating inflammatory responses and ventricular rupture or remodeling following myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Smyth, S.S.; McEver, R.P.; Weyrich, A.S.; Morrell, C.N.; Hoffman, M.R.; Arepally, G.M.; French, P.A.; Dauerman, H.L.; Becker, R.C. Platelet functions beyond hemostasis. J. Thromb. Haemost. JTH 2009, 7, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.; Cockburn, I.A.; Swaim, A.; Thompson, L.E.; Tripathi, A.; Fletcher, C.A.; Shirk, E.M.; Sun, H.; Kowalska, M.A.; Fox-Talbot, K.; et al. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell Host Microbe 2008, 4, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Coppinger, J.A.; Cagney, G.; Toomey, S.; Kislinger, T.; Belton, O.; McRedmond, J.P.; Cahill, D.J.; Emili, A.; Fitzgerald, D.J.; Maguire, P.B. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 2004, 103, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.; Flaumenhaft, R. Platelet alpha-granules: Basic biology and clinical correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef]
- Palabrica, T.; Lobb, R.; Furie, B.C.; Aronovitz, M.; Benjamin, C.; Hsu, Y.M.; Sajer, S.A.; Furie, B. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature 1992, 359, 848–851. [Google Scholar] [CrossRef]
- Del Conde, I.; Crúz, M.A.; Zhang, H.; López, J.A.; Afshar-Kharghan, V. Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 2005, 201, 871–879. [Google Scholar] [CrossRef]
- Katoh, N.; Soga, F.; Nara, T.; Tamagawa-Mineoka, R.; Nin, M.; Kotani, H.; Masuda, K.; Kishimoto, S. Effect of serotonin on the differentiation of human monocytes into dendritic cells. Clin. Exp. Immunol. 2006, 146, 354–361. [Google Scholar] [CrossRef]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef]
- Aggrey, A.A.; Srivastava, K.; Ture, S.; Field, D.J.; Morrell, C.N. Platelet Induction of the Acute-Phase Response Is Protective in Murine Experimental Cerebral Malaria. J. Immunol. 2013, 190, 4685–4691. [Google Scholar] [CrossRef]
- Lindemann, S.; Tolley, N.D.; Dixon, D.A.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A.; Weyrich, A.S. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J. Cell Biol. 2001, 154, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; McIntyre, T.M. Lipopolysaccharide signaling without a nucleus: Kinase cascades stimulate platelet shedding of proinflammatory IL-1β-rich microparticles. J. Immunol. 2011, 186, 5489–5496. [Google Scholar] [CrossRef] [PubMed]
- Binsker, U.; Palankar, R.; Wesche, J.; Kohler, T.P.; Prucha, J.; Burchhardt, G.; Rohde, M.; Schmidt, F.; Bröker, B.M.; Mamat, U.; et al. Secreted Immunomodulatory Proteins of Staphylococcus aureus Activate Platelets and Induce Platelet Aggregation. Thromb. Haemost. 2018, 118, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.A.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ. Res. 2009, 104, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, L.M.; Lin, E.; Morin, K.M.; Tanriverdi, K.; Freedman, J.E. Regulatory effects of TLR2 on megakaryocytic cell function. Blood 2011, 117, 5963–5974. [Google Scholar] [CrossRef] [PubMed]
- Green, S.A.; Smith, M.; Hasley, R.B.; Stephany, D.; Harned, A.; Nagashima, K.; Abdullah, S.; Pittaluga, S.; Imamichi, T.; Qin, J.; et al. Activated platelet-T-cell conjugates in peripheral blood of patients with HIV infection: Coupling coagulation/inflammation and T cells. AIDS 2015, 29, 1297–1308. [Google Scholar] [CrossRef]
- Hundelshausen, P.v.; Weber, K.S.C.; Huo, Y.; Proudfoot, A.E.I.; Nelson, P.J.; Ley, K.; Weber, C. RANTES Deposition by Platelets Triggers Monocyte Arrest on Inflamed and Atherosclerotic Endothelium. Circulation 2001, 103, 1772–1777. [Google Scholar] [CrossRef]
- Theilmeier, G.; Lenaerts, T.; Remacle, C.; Collen, D.; Vermylen, J.; Hoylaerts, M.F. Circulating Activated Platelets Assist THP-1 Monocytoid/Endothelial Cell Interaction Under Shear Stress. Blood 1999, 94, 2725–2734. [Google Scholar] [CrossRef]
- Weyrich, A.S.; McIntyre, T.M.; McEver, R.P.; Prescott, S.M.; Zimmerman, G.A. Monocyte tethering by P-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-alpha secretion. Signal integration and NF-kappa B translocation. J. Clin. Investig. 1995, 95, 2297–2303. [Google Scholar] [CrossRef]
- Weyrich, A.S.; Elstad, M.R.; McEver, R.P.; McIntyre, T.M.; Moore, K.L.; Morrissey, J.H.; Prescott, S.M.; Zimmerman, G.A. Activated platelets signal chemokine synthesis by human monocytes. J. Clin. Investig. 1996, 97, 1525–1534. [Google Scholar] [CrossRef]
- Cognasse, F.; Duchez, A.C.; Audoux, E.; Ebermeyer, T.; Arthaud, C.A.; Prier, A.; Eyraud, M.A.; Mismetti, P.; Garraud, O.; Bertoletti, L.; et al. Platelets as Key Factors in Inflammation: Focus on CD40L/CD40. Front. Immunol. 2022, 13, 825892. [Google Scholar] [CrossRef]
- Danese, S.; de la Motte, C.; Reyes, B.M.; Sans, M.; Levine, A.D.; Fiocchi, C. Cutting edge: T cells trigger CD40-dependent platelet activation and granular RANTES release: A novel pathway for immune response amplification. J. Immunol. 2004, 172, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Daub, K.; Braun, G.; Schönberger, T.; May, A.E.; Schaller, M.; Stein, G.M.; Stellos, K.; Bueltmann, A.; Siegel-Axel, D.; et al. Platelets recruit human dendritic cells via Mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Hagihara, M.; Higuchi, A.; Tamura, N.; Ueda, Y.; Hirabayashi, K.; Ikeda, Y.; Kato, S.; Sakamoto, S.; Hotta, T.; Handa, S.; et al. Platelets, after exposure to a high shear stress, induce IL-10-producing, mature dendritic cells in vitro. J. Immunol. 2004, 172, 5297–5303. [Google Scholar] [CrossRef]
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 2010, 327, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Mizrachi, L.; Jy, W.; Jimenez, J.J.; Pastor, J.; Mauro, L.M.; Horstman, L.L.; de Marchena, E.; Ahn, Y.S. High levels of circulating endothelial microparticles in patients with acute coronary syndromes. Am. Heart J. 2003, 145, 962–970. [Google Scholar] [CrossRef]
- Mallat, Z.; Benamer, H.; Hugel, B.; Benessiano, J.; Steg, P.G.; Freyssinet, J.M.; Tedgui, A. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation 2000, 101, 841–843. [Google Scholar] [CrossRef]
- Suades, R.; Padró, T.; Vilahur, G.; Badimon, L. Circulating and platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques. Thromb. Haemost. 2012, 108, 1208–1219. [Google Scholar] [CrossRef]
- Vajen, T.; Benedikter, B.J.; Heinzmann, A.C.A.; Vasina, E.M.; Henskens, Y.; Parsons, M.; Maguire, P.B.; Stassen, F.R.; Heemskerk, J.W.M.; Schurgers, L.J.; et al. Platelet extracellular vesicles induce a pro-inflammatory smooth muscle cell phenotype. J. Extracell. Vesicles 2017, 6, 1322454. [Google Scholar] [CrossRef]
- Badimon, L.; Suades, R.; Fuentes, E.; Palomo, I.; Padró, T. Role of Platelet-Derived Microvesicles As Crosstalk Mediators in Atherothrombosis and Future Pharmacology Targets: A Link between Inflammation, Atherosclerosis, and Thrombosis. Front. Pharmacol. 2016, 7, 293. [Google Scholar] [CrossRef]
- Laffont, B.; Corduan, A.; Rousseau, M.; Duchez, A.C.; Lee, C.H.; Boilard, E.; Provost, P. Platelet microparticles reprogram macrophage gene expression and function. Thromb. Haemost. 2016, 115, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Vasina, E.M.; Cauwenberghs, S.; Feijge, M.A.; Heemskerk, J.W.; Weber, C.; Koenen, R.R. Microparticles from apoptotic platelets promote resident macrophage differentiation. Cell Death Dis. 2011, 2, e211. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.A.; Wuescher, L.M.; Worth, R.G. Platelets: Essential components of the immune system. Curr. Trends Immunol. 2015, 16, 65–78. [Google Scholar] [PubMed]
- Kraemer, B.F.; Campbell, R.A.; Schwertz, H.; Cody, M.J.; Franks, Z.; Tolley, N.D.; Kahr, W.H.; Lindemann, S.; Seizer, P.; Yost, C.C.; et al. Novel anti-bacterial activities of β-defensin 1 in human platelets: Suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog. 2011, 7, e1002355. [Google Scholar] [CrossRef] [PubMed]
- Hamzeh-Cognasse, H.; Damien, P.; Chabert, A.; Pozzetto, B.; Cognasse, F.; Garraud, O. Platelets and infections-complex interactions with bacteria. Front. Immunol. 2015, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Jahn, K.; Handtke, S.; Palankar, R.; Weißmüller, S.; Nouailles, G.; Kohler, T.P.; Wesche, J.; Rohde, M.; Heinz, C.; Aschenbrenner, A.F.; et al. Pneumolysin induces platelet destruction, not platelet activation, which can be prevented by immunoglobulin preparations in vitro. Blood Adv. 2020, 4, 6315–6326. [Google Scholar] [CrossRef]
- Kerrigan, S.W.; Cox, D. Platelet-bacterial interactions. Cell. Mol. Life Sci. CMLS 2010, 67, 513–523. [Google Scholar] [CrossRef]
- Ford, I.; Douglas, C.W. The role of platelets in infective endocarditis. Platelets 1997, 8, 285–294. [Google Scholar] [CrossRef]
- Jung, C.-J.; Yeh, C.-Y.; Shun, C.-T.; Hsu, R.-B.; Cheng, H.-W.; Lin, C.-S.; Chia, J.-S. Platelets Enhance Biofilm Formation and Resistance of Endocarditis-Inducing Streptococci on the Injured Heart Valve. J. Infect. Dis. 2012, 205, 1066–1075. [Google Scholar] [CrossRef]
- Franchini, M.; Veneri, D. Helicobacter pylori-associated immune thrombocytopenia. Platelets 2006, 17, 71–77. [Google Scholar] [CrossRef]
- Karpman, D.; Manea, M.; Vaziri-Sani, F.; Ståhl, A.L.; Kristoffersson, A.C. Platelet activation in hemolytic uremic syndrome. Semin. Thromb. Hemost. 2006, 32, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.; Cointe, A.; Mariani Kurkdjian, P.; Rafat, C.; Hertig, A. Shiga Toxin-Associated Hemolytic Uremic Syndrome: A Narrative Review. Toxins 2020, 12, 67. [Google Scholar] [CrossRef] [PubMed]
- Assinger, A.; Schrottmaier, W.C.; Salzmann, M.; Rayes, J. Platelets in Sepsis: An Update on Experimental Models and Clinical Data. Front. Immunol. 2019, 10, 1687. [Google Scholar] [CrossRef] [PubMed]
- Clawson, C.C. Platelet interaction with bacteria. 3. Ultrastructure. Am. J. Pathol. 1973, 70, 449–471. [Google Scholar] [PubMed]
- Clawson, C.C.; Rao, G.H.; White, J.G. Platelet interaction with bacteria. IV. Stimulation of the release reaction. Am. J. Pathol. 1975, 81, 411–420. [Google Scholar]
- Li, X.; Iwai, T.; Nakamura, H.; Inoue, Y.; Chen, Y.; Umeda, M.; Suzuki, H. An ultrastructural study of Porphyromonas gingivalis-induced platelet aggregation. Thromb. Res. 2008, 122, 810–819. [Google Scholar] [CrossRef]
- Youssefian, T.; Drouin, A.; Massé, J.M.; Guichard, J.; Cramer, E.M. Host defense role of platelets: Engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood 2002, 99, 4021–4029. [Google Scholar] [CrossRef]
- Worth, R.G.; Chien, C.D.; Chien, P.; Reilly, M.P.; McKenzie, S.E.; Schreiber, A.D. Platelet FcgammaRIIA binds and internalizes IgG-containing complexes. Exp. Hematol. 2006, 34, 1490–1495. [Google Scholar] [CrossRef]
- Antczak, A.J.; Vieth, J.A.; Singh, N.; Worth, R.G. Internalization of IgG-coated targets results in activation and secretion of soluble CD40 ligand and RANTES by human platelets. Clin. Vaccine Immunol. 2011, 18, 210–216. [Google Scholar] [CrossRef]
- Gaertner, F.; Ahmad, Z.; Rosenberger, G.; Fan, S.; Nicolai, L.; Busch, B.; Yavuz, G.; Luckner, M.; Ishikawa-Ankerhold, H.; Hennel, R.; et al. Migrating Platelets Are Mechano-scavengers that Collect and Bundle Bacteria. Cell 2017, 171, 1368–1382. [Google Scholar] [CrossRef]
- Verschoor, A.; Neuenhahn, M.; Navarini, A.A.; Graef, P.; Plaumann, A.; Seidlmeier, A.; Nieswandt, B.; Massberg, S.; Zinkernagel, R.M.; Hengartner, H.; et al. A platelet-mediated system for shuttling blood-borne bacteria to CD8α+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat. Immunol. 2011, 12, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.; Lozano, M.L.; Navarro-Núñez, L.; Vicente, V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica 2009, 94, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Capel, P.J.; van de Winkel, J.G.; van den Herik-Oudijk, I.E.; Verbeek, J.S. Heterogeneity of human IgG Fc receptors. ImmunoMethods 1994, 4, 25–34. [Google Scholar] [CrossRef] [PubMed]
- McCrae, K.R.; Shattil, S.J.; Cines, D.B. Platelet activation induces increased Fc gamma receptor expression. J. Immunol. 1990, 144, 3920–3927. [Google Scholar] [PubMed]
- Moriarty, R.D.; Cox, A.; McCall, M.; Smith, S.G.; Cox, D. Escherichia coli induces platelet aggregation in an FcγRIIa-dependent manner. J. Thromb. Haemost. JTH 2016, 14, 797–806. [Google Scholar] [CrossRef]
- Fitzgerald, J.R.; Loughman, A.; Keane, F.; Brennan, M.; Knobel, M.; Higgins, J.; Visai, L.; Speziale, P.; Cox, D.; Foster, T.J. Fibronectin-binding proteins of Staphylococcus aureus mediate activation of human platelets via fibrinogen and fibronectin bridges to integrin GPIIb/IIIa and IgG binding to the FcgammaRIIa receptor. Mol. Microbiol. 2006, 59, 212–230. [Google Scholar] [CrossRef]
- Byrne, M.F.; Kerrigan, S.W.; Corcoran, P.A.; Atherton, J.C.; Murray, F.E.; Fitzgerald, D.J.; Cox, D.M. Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology 2003, 124, 1846–1854. [Google Scholar] [CrossRef]
- Josefsson, E.; McCrea, K.W.; Eidhin, D.N.; O’Connell, D.; Cox, J.; Hook, M.; Foster, T.J. Three new members of the serine-aspartate repeat protein multigene family of Staphylococcus aureus. Microbiology 1998, 144 Pt 12, 3387–3395. [Google Scholar] [CrossRef]
- Ní Eidhin, D.; Perkins, S.; Francois, P.; Vaudaux, P.; Höök, M.; Foster, T.J. Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol. Microbiol. 1998, 30, 245–257. [Google Scholar] [CrossRef]
- Heilmann, C.; Niemann, S.; Sinha, B.; Herrmann, M.; Kehrel, B.E.; Peters, G. Staphylococcus aureus fibronectin-binding protein (FnBP)-mediated adherence to platelets, and aggregation of platelets induced by FnBPA but not by FnBPB. J. Infect. Dis. 2004, 190, 321–329. [Google Scholar] [CrossRef]
- Flock, J.I.; Fröman, G.; Jönsson, K.; Guss, B.; Signäs, C.; Nilsson, B.; Raucci, G.; Höök, M.; Wadström, T.; Lindberg, M. Cloning and expression of the gene for a fibronectin-binding protein from Staphylococcus aureus. EMBO J. 1987, 6, 2351–2357. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.; Kerrigan, S.W.; Kaw, G.; Hogan, M.; Penades, J.; Litt, D.; Fitzgerald, D.J.; Foster, T.J.; Cox, D. Multiple mechanisms for the activation of human platelet aggregation by Staphylococcus aureus: Roles for the clumping factors ClfA and ClfB, the serine-aspartate repeat protein SdrE and protein A. Mol. Microbiol. 2002, 44, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Miajlovic, H.; Loughman, A.; Brennan, M.; Cox, D.; Foster, T.J. Both complement-and fibrinogen-dependent mechanisms contribute to platelet aggregation mediated by Staphylococcus aureus clumping factor B. Infect. Immun. 2007, 75, 3335–3343. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miajlovic, H.; Zapotoczna, M.; Geoghegan, J.A.; Kerrigan, S.W.; Speziale, P.; Foster, T.J. Direct interaction of iron-regulated surface determinant IsdB of Staphylococcus aureus with the GPIIb/IIIa receptor on platelets. Microbiology 2010, 156, 920–928. [Google Scholar] [CrossRef]
- Brennan, M.P.; Loughman, A.; Devocelle, M.; Arasu, S.; Chubb, A.J.; Foster, T.J.; Cox, D. Elucidating the role of Staphylococcus epidermidis serine–aspartate repeat protein G in platelet activation. J. Thromb. Haemost. 2009, 7, 1364–1372. [Google Scholar] [CrossRef]
- Bertling, A.; Niemann, S.; Hussain, M.; Holbrook, L.; Stanley, R.G.; Brodde, M.F.; Pohl, S.; Schifferdecker, T.; Roth, J.; Jurk, K.; et al. Staphylococcal extracellular adherence protein induces platelet activation by stimulation of thiol isomerases. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1979–1990. [Google Scholar] [CrossRef]
- Wallis, S.; Wolska, N.; Englert, H.; Posner, M.; Upadhyay, A.; Renné, T.; Eggleston, I.; Bagby, S.; Pula, G. A peptide from the staphylococcal protein Efb binds P-selectin and inhibits the interaction of platelets with leukocytes. J. Thromb. Haemost. 2021, 20, 729–741. [Google Scholar] [CrossRef]
- Posner, M.G.; Upadhyay, A.; Abubaker, A.A.; Fortunato, T.M.; Vara, D.; Canobbio, I.; Bagby, S.; Pula, G. Extracellular Fibrinogen-binding Protein (Efb) from Staphylococcus aureus Inhibits the Formation of Platelet-Leukocyte Complexes. J. Biol. Chem. 2016, 291, 2764–2776. [Google Scholar] [CrossRef]
- Li, R.; Emsley, J. The organizing principle of the platelet glycoprotein Ib–IX–V complex. J. Thromb. Haemost. 2013, 11, 605–614. [Google Scholar] [CrossRef]
- Bennett, J.S.; Berger, B.W.; Billings, P.C. The structure and function of platelet integrins. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 200–205. [Google Scholar] [CrossRef]
- Bensing, B.A.; López, J.A.; Sullam, P.M. The Streptococcus gordonii surface proteins GspB and Hsa mediate binding to sialylated carbohydrate epitopes on the platelet membrane glycoprotein Ibalpha. Infect. Immun. 2004, 72, 6528–6537. [Google Scholar] [CrossRef] [PubMed]
- Plummer, C.; Wu, H.; Kerrigan, S.W.; Meade, G.; Cox, D.; Ian Douglas, C.W. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br. J. Haematol. 2005, 129, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Claes, J.; Vanassche, T.; Peetermans, M.; Liesenborghs, L.; Vandenbriele, C.; Vanhoorelbeke, K.; Missiakas, D.; Schneewind, O.; Hoylaerts, M.F.; Heying, R.; et al. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor-binding protein. Blood 2014, 124, 1669–1676. [Google Scholar] [CrossRef] [PubMed]
- Botos, I.; Segal, D.M.; Davies, D.R. The Structural Biology of Toll-like Receptors. Structure 2011, 19, 447–459. [Google Scholar] [CrossRef]
- Cognasse, F.; Nguyen, K.A.; Damien, P.; McNicol, A.; Pozzetto, B.; Hamzeh-Cognasse, H.; Garraud, O. The Inflammatory Role of Platelets via Their TLRs and Siglec Receptors. Front. Immunol. 2015, 6, 83. [Google Scholar] [CrossRef]
- Cognasse, F.; Hamzeh, H.; Chavarin, P.; Acquart, S.; Genin, C.; Garraud, O. Evidence of Toll-like receptor molecules on human platelets. Immunol. Cell Biol. 2005, 83, 196–198. [Google Scholar] [CrossRef]
- Gautam, J.K.; Comeau, L.D.; Krueger, J.K.; Smith, M.F., Jr. Structural and functional evidence for the role of the TLR2 DD loop in TLR1/TLR2 heterodimerization and signaling. J. Biol. Chem. 2006, 281, 30132–30142. [Google Scholar] [CrossRef]
- Zähringer, U.; Lindner, B.; Inamura, S.; Heine, H.; Alexander, C. TLR2–promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 2008, 213, 205–224. [Google Scholar] [CrossRef]
- Keane, C.; Tilley, D.; Cunningham, A.; Smolenski, A.; Kadioglu, A.; Cox, D.; Jenkinson, H.F.; Kerrigan, S.W. Invasive Streptococcus pneumoniae trigger platelet activation via Toll-like receptor 2. J. Thromb. Haemost. 2010, 8, 2757–2765. [Google Scholar] [CrossRef]
- Liu, X.; Liu, H.; Luo, X.; Zhang, P.; Gao, Y.; Xie, S.; Xu, K.; Chang, J.; Ma, L. Strains of Group B streptococci from septic patients induce platelet activation via Toll-like Receptor 2. Clin. Exp. Pharmacol. Physiol. 2017, 44, 335–343. [Google Scholar] [CrossRef]
- El Kebir, D.; Damlaj, A.; Makhezer, N.; Filep, J.G. Toll-like receptor 9 signaling regulates tissue factor and tissue factor pathway inhibitor expression in human endothelial cells and coagulation in mice. Crit. Care Med. 2015, 43, e179–e189. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Powers, M.E.; Becker, R.E.; Sailer, A.; Turner, J.R.; Bubeck Wardenburg, J. Synergistic Action of Staphylococcus aureus α-Toxin on Platelets and Myeloid Lineage Cells Contributes to Lethal Sepsis. Cell Host Microbe 2015, 17, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Berube, B.J.; Bubeck Wardenburg, J. Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [PubMed]
- Bhakdi, S.; Tranum-Jensen, J. Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.; Diep, B.A.; Mai, T.T.; Vo, N.H.; Warrener, P.; Suzich, J.; Stover, C.K.; Sellman, B.R. Differential expression and roles of Staphylococcus aureus virulence determinants during colonization and disease. MBio 2015, 6, e02272-14. [Google Scholar] [CrossRef] [PubMed]
- Kebaier, C.; Chamberland, R.R.; Allen, I.C.; Gao, X.; Broglie, P.M.; Hall, J.D.; Jania, C.; Doerschuk, C.M.; Tilley, S.L.; Duncan, J.A. Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J. Infect. Dis. 2012, 205, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Bhakdi, S.; Muhly, M.; Mannhardt, U.; Hugo, F.; Klapettek, K.; Mueller-Eckhardt, C.; Roka, L. Staphylococcal alpha toxin promotes blood coagulation via attack on human platelets. J. Exp. Med. 1988, 168, 527–542. [Google Scholar] [CrossRef]
- Schubert, S.; Schwertz, H.; Weyrich, A.S.; Franks, Z.G.; Lindemann, S.; Otto, M.; Behr, H.; Loppnow, H.; Schlitt, A.; Russ, M.; et al. Staphylococcus aureus α-Toxin Triggers the Synthesis of B-Cell Lymphoma 3 by Human Platelets. Toxins 2011, 3, 120. [Google Scholar] [CrossRef]
- Jahn, K.; Handtke, S.; Palankar, R.; Kohler, T.P.; Wesche, J.; Wolff, M.; Bayer, J.; Wolz, C.; Greinacher, A.; Hammerschmidt, S. α-hemolysin of Staphylococcus aureus impairs thrombus formation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol Rev 2008, 32, 149–167. [Google Scholar] [CrossRef]
- Weidenmaier, C.; Peschel, A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 2008, 6, 276–287. [Google Scholar] [CrossRef]
- Lopez, R. Streptococcus pneumoniae and its bacteriophages: One long argument. Int. Microbiol. 2004, 7, 163–171. [Google Scholar] [PubMed]
- Gisch, N.; Kohler, T.; Ulmer, A.J.; Muthing, J.; Pribyl, T.; Fischer, K.; Lindner, B.; Hammerschmidt, S.; Zahringer, U. Structural reevaluation of Streptococcus pneumoniae Lipoteichoic acid and new insights into its immunostimulatory potency. J. Biol. Chem. 2013, 288, 15654–15667. [Google Scholar] [CrossRef] [PubMed]
- Bentley, S.D.; Aanensen, D.M.; Mavroidi, A.; Saunders, D.; Rabbinowitsch, E.; Collins, M.; Donohoe, K.; Harris, D.; Murphy, L.; Quail, M.A.; et al. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet. 2006, 2, e0020031. [Google Scholar] [CrossRef] [PubMed]
- Hyams, C.; Camberlein, E.; Cohen, J.M.; Bax, K.; Brown, J.S. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect. Immun. 2010, 78, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Pribyl, T.; Moche, M.; Dreisbach, A.; Bijlsma, J.J.E.; Saleh, M.; Abdullah, M.R.; Hecker, M.; van Dijl, J.M.; Becher, D.; Hammerschmidt, S. Influence of Impaired Lipoprotein Biogenesis on Surface and Exoproteome of Streptococcus pneumoniae. J. Proteome Res. 2014, 13, 650–667. [Google Scholar] [CrossRef] [PubMed]
- Kohler, S.; Voß, F.; Gómez Mejia, A.; Brown, J.S.; Hammerschmidt, S. Pneumococcal lipoproteins involved in bacterial fitness, virulence, and immune evasion. FEBS Lett. 2016, 590, 3820–3839. [Google Scholar] [CrossRef]
- Bergmann, S.; Hammerschmidt, S. Versatility of pneumococcal surface proteins. Microbiology 2006, 152, 295–303. [Google Scholar] [CrossRef]
- Manzano, C.; Contreras-Martel, C.; El Mortaji, L.; Izore, T.; Fenel, D.; Vernet, T.; Schoehn, G.; Di Guilmi, A.M.; Dessen, A. Sortase-mediated pilus fiber biogenesis in Streptococcus pneumoniae. Structure 2008, 16, 1838–1848. [Google Scholar] [CrossRef]
- Abdullah, M.R.; Gutierrez-Fernandez, J.; Pribyl, T.; Gisch, N.; Saleh, M.; Rohde, M.; Petruschka, L.; Burchhardt, G.; Schwudke, D.; Hermoso, J.A.; et al. Structure of the pneumococcal l,d-carboxypeptidase DacB and pathophysiological effects of disabled cell wall hydrolases DacA and DacB. Mol. Microbiol. 2014, 93, 1183–1206. [Google Scholar] [CrossRef]
- Saleh, M.; Bartual, S.G.; Abdullah, M.R.; Jensch, I.; Asmat, T.M.; Petruschka, L.; Pribyl, T.; Gellert, M.; Lillig, C.H.; Antelmann, H.; et al. Molecular architecture of Streptococcus pneumoniae surface thioredoxin-fold lipoproteins crucial for extracellular oxidative stress resistance and maintenance of virulence. EMBO Mol. Med. 2013, 5, 1852–1870. [Google Scholar] [CrossRef] [PubMed]
- Hermans, P.W.; Adrian, P.V.; Albert, C.; Estevao, S.; Hoogenboezem, T.; Luijendijk, I.H.; Kamphausen, T.; Hammerschmidt, S. The streptococcal lipoprotein rotamase A (SlrA) is a functional peptidyl-prolyl isomerase involved in pneumococcal colonization. J. Biol. Chem. 2006, 281, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Maestro, B.; Sanz, J.M. Choline Binding Proteins from Streptococcus pneumoniae: A Dual Role as Enzybiotics and Targets for the Design of New Antimicrobials. Antibiotics 2016, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Perez-Dorado, I.; Galan-Bartual, S.; Hermoso, J.A. Pneumococcal surface proteins: When the whole is greater than the sum of its parts. Mol. Oral Microbiol. 2012, 27, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Garcia, E. Recent trends on the molecular biology of pneumococcal capsules, lytic enzymes, and bacteriophage. FEMS Microbiol. Rev. 2004, 28, 553–580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.R.; Mostov, K.E.; Lamm, M.E.; Nanno, M.; Shimida, S.; Ohwaki, M.; Tuomanen, E. The polymeric immunoglobulin receptor translocates pneumococci across human nasopharyngeal epithelial cells. Cell 2000, 102, 827–837. [Google Scholar] [CrossRef]
- Elm, C.; Braathen, R.; Bergmann, S.; Frank, R.; Vaerman, J.P.; Kaetzel, C.S.; Chhatwal, G.S.; Johansen, F.E.; Hammerschmidt, S. Ectodomains 3 and 4 of human polymeric Immunoglobulin receptor (hpIgR) mediate invasion of Streptococcus pneumoniae into the epithelium. J. Biol. Chem. 2004, 279, 6296–6304. [Google Scholar] [CrossRef]
- Hammerschmidt, S. Adherence molecules of pathogenic pneumococci. Curr. Opin. Microbiol. 2006, 9, 12–20. [Google Scholar] [CrossRef]
- Hoskins, J.; Alborn, W.E., Jr.; Arnold, J.; Blaszczak, L.C.; Burgett, S.; DeHoff, B.S.; Estrem, S.T.; Fritz, L.; Fu, D.J.; Fuller, W.; et al. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 2001, 183, 5709–5717. [Google Scholar] [CrossRef]
- Tettelin, H.; Nelson, K.E.; Paulsen, I.T.; Eisen, J.A.; Read, T.D.; Peterson, S.; Heidelberg, J.; DeBoy, R.T.; Haft, D.H.; Dodson, R.J.; et al. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 2001, 293, 498–506. [Google Scholar] [CrossRef]
- Perry, A.M.; Ton-That, H.; Mazmanian, S.K.; Schneewind, O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. III. Lipid II is an in vivo peptidoglycan substrate for sortase-catalyzed surface protein anchoring. J. Biol. Chem. 2002, 277, 16241–16248. [Google Scholar] [CrossRef] [PubMed]
- Strominger, J.L.; Izaki, K.; Matsuhashi, M.; Tipper, D.J. Peptidoglycan transpeptidase and D-alanine carboxypeptidase: Penicillin-sensitive enzymatic reactions. Fed. Proc. 1967, 26, 9–22. [Google Scholar] [PubMed]
- Henderson, B.; Martin, A. Bacterial virulence in the moonlight: Multitasking bacterial moonlighting proteins are virulence determinants in infectious disease. Infect. Immun. 2011, 79, 3476–3491. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, S.; Schoenen, H.; Hammerschmidt, S. The interaction between bacterial enolase and plasminogen promotes adherence of Streptococcus pneumoniae to epithelial and endothelial cells. Int. J. Med. Microbiol. 2013, 303, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Hammerschmidt, S.; Malm, S.; Bergmann, S.; Riesbeck, K.; Blom, A.M. Enolase of Streptococcus pneumoniae binds human complement inhibitor C4b-binding protein and contributes to complement evasion. J. Immunol. 2012, 189, 3575–3584. [Google Scholar] [CrossRef]
- Fine, M.J.; Smith, M.A.; Carson, C.A.; Mutha, S.S.; Sankey, S.S.; Weissfeld, L.A.; Kapoor, W.N. Prognosis and outcomes of patients with community-acquired pneumonia. A meta-analysis. JAMA 1996, 275, 134–141. [Google Scholar] [CrossRef]
- Bartlett, J.G.; Dowell, S.F.; Mandell, L.A.; File, T.M., Jr.; Musher, D.M.; Fine, M.J. Practice guidelines for the management of community-acquired pneumonia in adults. Infectious Diseases Society of America. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2000, 31, 347–382. [Google Scholar] [CrossRef]
- Tunjungputri, R.N.; Mobegi, F.M.; Cremers, A.J.; van der Gaast-de Jongh, C.E.; Ferwerda, G.; Meis, J.F.; Roeleveld, N.; Bentley, S.D.; Pastura, A.S.; van Hijum, S.A.; et al. Phage-Derived Protein Induces Increased Platelet Activation and Is Associated with Mortality in Patients with Invasive Pneumococcal Disease. MBio 2017, 8, e01984-16. [Google Scholar] [CrossRef]
- Luquero-Bueno, S.; Galván-Román, J.M.; Curbelo, J.; Lancho-Sánchez, A.; Roy-Vallejo, E.; Ortega-Gómez, M.; Gómez, M.; Mateo-Jimenez, G.; Manzaneque-Pradales, M.; Aspa Marco, J. Platelet count as an evolution marker of late mortality and cardiovascular events after an episode of community-acquired pneumonia. Eur. Respir. J. 2019, 54, PA4532. [Google Scholar] [CrossRef]
- Feldman, C.; Kallenbach, J.M.; Levy, H.; Reinach, S.G.; Hurwitz, M.D.; Thorburn, J.R.; Koornhof, H.J. Community-acquired pneumonia of diverse aetiology: Prognostic features in patients admitted to an intensive care unit and a “severity of illness” core. Intensive Care Med. 1989, 15, 302–307. [Google Scholar] [CrossRef]
- Wolff, M.; Handtke, S.; Palankar, R.; Wesche, J.; Kohler, T.P.; Kohler, C.; Gruel, Y.; Hammerschmidt, S.; Greinacher, A. Activated platelets kill Staphylococcus aureus, but not Streptococcus pneumoniae-The role of FcγRIIa and platelet factor 4/heparinantibodies. J. Thromb. Haemost. 2020, 18, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Vardon-Bounes, F.; Ruiz, S.; Gratacap, M.P.; Garcia, C.; Payrastre, B.; Minville, V. Platelets Are Critical Key Players in Sepsis. Int. J. Mol. Sci. 2019, 20, 3494. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Wartha, F.; Beiter, K.; Albiger, B.; Fernebro, J.; Zychlinsky, A.; Normark, S.; Henriques-Normark, B. Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell. Microbiol. 2007, 9, 1162–1171. [Google Scholar] [CrossRef]
- Martinez, P.J.; Farhan, A.; Mustafa, M.; Javaid, N.; Darkoh, C.; Garrido-Sanabria, E.; Fisher-Hoch, S.P.; Briles, D.E.; Kantarci, A.; Mirza, S. PspA facilitates evasion of pneumococci from bactericidal activity of neutrophil extracellular traps (NETs). Microb. Pathog. 2019, 136, 103653. [Google Scholar] [CrossRef]
- Beiter, K.; Wartha, F.; Albiger, B.; Normark, S.; Zychlinsky, A.; Henriques-Normark, B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 2006, 16, 401–407. [Google Scholar] [CrossRef]
- Levi, M. The coagulant response in sepsis and inflammation. Hamostaseologie 2010, 30, 14–16. [Google Scholar] [CrossRef]
- Hoshino, K.; Kitamura, T.; Nakamura, Y.; Irie, Y.; Matsumoto, N.; Kawano, Y.; Ishikura, H. Usefulness of plasminogen activator inhibitor-1 as a predictive marker of mortality in sepsis. J. Intensive Care 2017, 5, 42. [Google Scholar] [CrossRef]
- Tipoe, T.L.; Wu, W.K.K.; Chung, L.; Gong, M.; Dong, M.; Liu, T.; Roever, L.; Ho, J.; Wong, M.C.S.; Chan, M.T.V.; et al. Plasminogen Activator Inhibitor 1 for Predicting Sepsis Severity and Mortality Outcomes: A Systematic Review and Meta-Analysis. Front. Immunol. 2018, 9, 1218. [Google Scholar] [CrossRef] [PubMed]
- Iskander, K.N.; Osuchowski, M.F.; Stearns-Kurosawa, D.J.; Kurosawa, S.; Stepien, D.; Valentine, C.; Remick, D.G. Sepsis: Multiple abnormalities, heterogeneous responses, and evolving understanding. Physiol. Rev. 2013, 93, 1247–1288. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. The immunopathogenesis of sepsis. Nature 2002, 420, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Waller, A.K.; Sage, T.; Kumar, C.; Carr, T.; Gibbins, J.M.; Clarke, S.R. Staphylococcus aureus lipoteichoic acid inhibits platelet activation and thrombus formation via the Paf receptor. J. Infect. Dis. 2013, 208, 2046–2057. [Google Scholar] [CrossRef]
- Daix, T.; Guerin, E.; Tavernier, E.; Mercier, E.; Gissot, V.; Hérault, O.; Mira, J.P.; Dumas, F.; Chapuis, N.; Guitton, C.; et al. Multicentric Standardized Flow Cytometry Routine Assessment of Patients With Sepsis to Predict Clinical Worsening. Chest 2018, 154, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Camicia, G.; Pozner, R.; de Larrañaga, G. Neutrophil extracellular traps in sepsis. Shock 2014, 42, 286–294. [Google Scholar] [CrossRef]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1α and Cathepsin G. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef]
- Tyml, K. Critical role for oxidative stress, platelets, and coagulation in capillary blood flow impairment in sepsis. Microcirculation 2011, 18, 152–162. [Google Scholar] [CrossRef]
- Lorente, L.; Martín, M.M.; Varo, N.; Borreguero-León, J.M.; Solé-Violán, J.; Blanquer, J.; Labarta, L.; Díaz, C.; Jiménez, A.; Pastor, E.; et al. Association between serum soluble CD40 ligand levels and mortality in patients with severe sepsis. Crit. Care 2011, 15, R97. [Google Scholar] [CrossRef]
- Zhang, S.; Rahman, M.; Zhang, S.; Qi, Z.; Thorlacius, H. Simvastatin antagonizes CD40L secretion, CXC chemokine formation, and pulmonary infiltration of neutrophils in abdominal sepsis. J. Leukoc. Biol. 2011, 89, 735–742. [Google Scholar] [CrossRef]
- Chew, M.; Rahman, M.; Ihrman, L.; Erson, A.; Zhang, S.; Thorlacius, H. Soluble CD40L (CD154) is increased in patients with shock. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2010, 59, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Zhang, S.; Chew, M.; Ersson, A.; Jeppsson, B.; Thorlacius, H. Platelet-derived CD40L (CD154) mediates neutrophil upregulation of Mac-1 and recruitment in septic lung injury. Ann. Surg. 2009, 250, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yu, S.; Song, Z.; Zhu, X.; Wang, C.; Yan, J.; Wu, F.; Nanda, A.; Granger, D.N.; Li, G. Soluble CD40 ligand stimulates CD40-dependent activation of the β2 integrin Mac-1 and protein kinase C zeda (PKCζ) in neutrophils: Implications for neutrophil-platelet interactions and neutrophil oxidative burst. PLoS ONE 2013, 8, e0064631. [Google Scholar] [CrossRef] [PubMed]
- Yost, C.C.; Weyrich, A.S.; Zimmerman, G.A. The platelet activating factor (PAF) signaling cascade in systemic inflammatory responses. Biochimie 2010, 92, 692–697. [Google Scholar] [CrossRef]
- Martínez, E.; Miró, J.M.; Almirante, B.; Aguado, J.M.; Fernandez-Viladrich, P.; Fernandez-Guerrero, M.L.; Villanueva, J.L.; Dronda, F.; Moreno-Torrico, A.; Montejo, M.; et al. Effect of penicillin resistance of Streptococcus pneumoniae on the presentation, prognosis, and treatment of pneumococcal endocarditis in adults. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2002, 35, 130–139. [Google Scholar] [CrossRef][Green Version]
- Kan, B.; Ries, J.; Normark, B.H.; Chang, F.Y.; Feldman, C.; Ko, W.C.; Rello, J.; Snydman, D.R.; Yu, V.L.; Ortqvist, A. Endocarditis and pericarditis complicating pneumococcal bacteraemia, with special reference to the adhesive abilities of pneumococci: Results from a prospective study. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2006, 12, 338–344. [Google Scholar] [CrossRef] [PubMed]
- de Egea, V.; Muñoz, P.; Valerio, M.; de Alarcón, A.; Lepe, J.A.; Miró, J.M.; Gálvez-Acebal, J.; García-Pavía, P.; Navas, E.; Goenaga, M.A.; et al. Characteristics and Outcome of Streptococcus pneumoniae Endocarditis in the XXI Century: A Systematic Review of 111 Cases (2000-2013). Medicine 2015, 94, e1562. [Google Scholar] [CrossRef] [PubMed]
- Liesenborghs, L.; Meyers, S.; Vanassche, T.; Verhamme, P. Coagulation: At the heart of infective endocarditis. J. Thromb. Haemost. 2020, 18, 995–1008. [Google Scholar] [CrossRef]
- Durack, D.T.; Beeson, P.B. Experimental bacterial endocarditis. I. Colonization of a sterile vegetation. Br. J. Exp. Pathol. 1972, 53, 44–49. [Google Scholar]
- Angrist, A.A.; Oka, M. Pathogenesis of bacterial endocarditis. JAMA 1963, 183, 249–252. [Google Scholar] [CrossRef]
- Herzberg, M.C.; Gong, K.; MacFarlane, G.D.; Erickson, P.R.; Soberay, A.H.; Krebsbach, P.H.; Manjula, G.; Schilling, K.; Bowen, W.H. Phenotypic characterization of Streptococcus sanguis virulence factors associated with bacterial endocarditis. Infect. Immun. 1990, 58, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, C.; Vaia, E.; Liccardo, D.; Marzano, F.; Valletta, A.; Spagnuolo, G.; Ferrara, N.; Rengo, C.; Cannavo, A.; Rengo, G. Infective Endocarditis: A Focus on Oral Microbiota. Microorganisms 2021, 9, 1218. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.O. Infective endocarditis and dental procedures: Evidence, pathogenesis, and prevention. J. Med. Investig. 2006, 53, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.-Q.; Van Wamel, W.; Nast, C.C.; Yeaman, M.R.; Cheung, A.L.; Bayer, A.S. Activation and Transcriptional Interaction between agr RNAII and RNAIII in Staphylococcus aureus In Vitro and in an Experimental Endocarditis Model. J. Infect. Dis. 2002, 186, 668–677. [Google Scholar] [CrossRef]
- Guckian, J.C. Effect of pneumococci on blood clotting, platelets, and polymorphonuclear leukocytes. Infect. Immun. 1975, 12, 910–918. [Google Scholar] [CrossRef]
- Clawson, C.C.; White, J.G. Platelet interaction with bacteria. I. Reaction phases and effects of inhibitors. Am. J. Pathol. 1971, 65, 367–380. [Google Scholar]
- de Stoppelaar, S.F.; Claushuis, T.A.M.; Schaap, M.C.L.; Hou, B.; van der Poll, T.; Nieuwland, R.; van ‘t Veer, C. Toll-Like Receptor Signalling Is Not Involved in Platelet Response to Streptococcus pneumoniae In Vitro or In Vivo. PLoS ONE 2016, 11, e0156977. [Google Scholar] [CrossRef]
- Niemann, S.; Kehrel, B.E.; Heilmann, C.; Rennemeier, C.; Peters, G.; Hammerschmidt, S. Pneumococcal association to platelets is mediated by soluble fibrin and supported by thrombospondin-1. Thromb. Haemost. 2009, 102, 735–742. [Google Scholar] [CrossRef]
- Binsker, U.; Kohler, T.P.; Krauel, K.; Kohler, S.; Schwertz, H.; Hammerschmidt, S. Pneumococcal Adhesins PavB and PspC Are Important for the Interplay with Human Thrombospondin-1*. J. Biol. Chem. 2015, 290, 14542–14555. [Google Scholar] [CrossRef]
- Binsker, U.; Kohler, T.P.; Krauel, K.; Kohler, S.; Habermeyer, J.; Schwertz, H.; Hammerschmidt, S. Serotype 3 pneumococci sequester platelet-derived human thrombospondin-1 via the adhesin and immune evasion protein Hic. J. Biol. Chem. 2017, 292, 5770–5783. [Google Scholar] [CrossRef]
- Marshall, J.E.; Faraj, B.H.A.; Gingras, A.R.; Lonnen, R.; Sheikh, M.A.; El-Mezgueldi, M.; Moody, P.C.E.; Andrew, P.W.; Wallis, R. The Crystal Structure of Pneumolysin at 2.0 Å Resolution Reveals the Molecular Packing of the Pre-pore Complex. Sci. Rep. 2015, 5, 13293. [Google Scholar] [CrossRef] [PubMed]
- Ohkuni, H.; Nagamune, H.; Ozaki, N.; Tabata, A.; Todome, Y.; Watanabe, Y.; Takahashi, H.; Ohkura, K.; Kourai, H.; Ohtsuka, H.; et al. Characterization of recombinant Streptococcus mitis-derived human platelet aggregation factor. Acta Pathol. Microbiol. Immunol. Scand. 2012, 120, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Nel, J.G.; Durandt, C.; Mitchell, T.J.; Feldman, C.; Anderson, R.; Tintinger, G.R. Pneumolysin Mediates Platelet Activation In Vitro. Lung 2016, 194, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Fernández, J.; Saleh, M.; Alcorlo, M.; Gómez-Mejía, A.; Pantoja-Uceda, D.; Treviño, M.A.; Voß, F.; Abdullah, M.R.; Galán-Bartual, S.; Seinen, J.; et al. Modular Architecture and Unique Teichoic Acid Recognition Features of Choline-Binding Protein L (CbpL) Contributing to Pneumococcal Pathogenesis. Sci. Rep. 2016, 6, 38094. [Google Scholar] [CrossRef][Green Version]
- Schmidt, F.; Kakar, N.; Meyer, T.C.; Depke, M.; Masouris, I.; Burchhardt, G.; Gómez-Mejia, A.; Dhople, V.; Håvarstein, L.S.; Sun, Z.; et al. In vivo proteomics identifies the competence regulon and AliB oligopeptide transporter as pathogenic factors in pneumococcal meningitis. PLOS Pathog. 2019, 15, e1007987. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Terao, Y.; Mori, Y.; Hamada, S.; Kawabata, S. PfbA, a novel plasmin- and fibronectin-binding protein of Streptococcus pneumoniae, contributes to fibronectin-dependent adhesion and antiphagocytosis. J. Biol. Chem. 2008, 283, 36272–36279. [Google Scholar] [CrossRef]
- Tomlinson, G.; Chimalapati, S.; Pollard, T.; Lapp, T.; Cohen, J.; Camberlein, E.; Stafford, S.; Periselneris, J.; Aldridge, C.; Vollmer, W.; et al. TLR-mediated inflammatory responses to Streptococcus pneumoniae are highly dependent on surface expression of bacterial lipoproteins. J. Immunol. 2014, 193, 3736–3745. [Google Scholar] [CrossRef]
- Voß, F.; van Beek, L.F.; Schwudke, D.; Ederveen, T.H.A.; van Opzeeland, F.J.; Thalheim, D.; Werner, S.; de Jonge, M.I.; Hammerschmidt, S. Lipidation of Pneumococcal Antigens Leads to Improved Immunogenicity and Protection. Vaccines 2020, 8, 310. [Google Scholar] [CrossRef]
- Aslam, R.; Speck, E.R.; Kim, M.; Crow, A.R.; Bang, K.W.; Nestel, F.P.; Ni, H.; Lazarus, A.H.; Freedman, J.; Semple, J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 2006, 107, 637–641. [Google Scholar] [CrossRef]
- Heß, N.; Waldow, F.; Kohler, T.P.; Rohde, M.; Kreikemeyer, B.; Gómez-Mejia, A.; Hain, T.; Schwudke, D.; Vollmer, W.; Hammerschmidt, S.; et al. Lipoteichoic acid deficiency permits normal growth but impairs virulence of Streptococcus pneumoniae. Nat. Commun. 2017, 8, 2093. [Google Scholar] [CrossRef]
- Tunjungputri, R.N.; de Jonge, M.I.; de Greeff, A.; van Selm, S.; Buys, H.; Harders-Westerveen, J.F.; Stockhofe-Zurwieden, N.; Urbanus, R.T.; de Groot, P.G.; Smith, H.E.; et al. Invasive pneumococcal disease leads to activation and hyperreactivity of platelets. Thromb. Res. 2016, 144, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Kullaya, V.; Jonge, M.I.d.; Langereis, J.D.; Jongh, C.E.v.d.G.-d.; Büll, C.; Adema, G.J.; Lefeber, D.; Cremers, A.J.; Mmbaga, B.T.; Groot, P.G.d.; et al. Desialylation of Platelets by Pneumococcal Neuraminidase A Induces ADP-Dependent Platelet Hyperreactivity. Infect. Immun. 2018, 86, e00213-18. [Google Scholar] [CrossRef] [PubMed]
- Wiebe, F.; Handtke, S.; Wesche, J.; Schnarre, A.; Palankar, R.; Wolff, M.; Jahn, K.; Voß, F.; Weißmüller, S.; Schüttrumpf, J.; et al. Polyvalent immunoglobulin preparations inhibit pneumolysin-induced platelet destruction. Thromb. Haemost. 2021, 1723–1880. [Google Scholar] [CrossRef] [PubMed]
- Ishikura, H.; Nishida, T.; Murai, A.; Nakamura, Y.; Irie, Y.; Tanaka, J.; Umemura, T. New diagnostic strategy for sepsis-induced disseminated intravascular coagulation: A prospective single-center observational study. Crit. Care 2014, 18, R19. [Google Scholar] [CrossRef]
- Tiwari, N.R.; Chaudhari, K.S.; Sharma, R.; Haas, K.P.; Sharma, V.R. Antiplatelet Agents in Sepsis-Putting it all together: A Call to Action. Indian J. Crit. Care Med. Peer-Rev. Off. Publ. Indian Soc. Crit. Care Med. 2020, 24, 483–484. [Google Scholar] [CrossRef]
- Chen, C.M.; Lu, H.C.; Tung, Y.T.; Chen, W. Antiplatelet Therapy for Acute Respiratory Distress Syndrome. Biomedicines 2020, 8, 230. [Google Scholar] [CrossRef]
- Wang, Y.; Ouyang, Y.; Liu, B.; Ma, X.; Ding, R. Platelet activation and antiplatelet therapy in sepsis: A narrative review. Thromb. Res. 2018, 166, 28–36. [Google Scholar] [CrossRef]
- O’Neal, H.R., Jr.; Koyama, T.; Koehler, E.A.; Siew, E.; Curtis, B.R.; Fremont, R.D.; May, A.K.; Bernard, G.R.; Ware, L.B. Prehospital statin and aspirin use and the prevalence of severe sepsis and acute lung injury/acute respiratory distress syndrome. Crit. Care Med. 2011, 39, 1343–1350. [Google Scholar] [CrossRef]
- Davì, G.; Santilli, F.; Vazzana, N. Thromboxane receptors antagonists and/or synthase inhibitors. Handb. Exp. Pharmacol. 2012, 261–286. [Google Scholar] [CrossRef]
- Tagami, T.; Matsui, H.; Horiguchi, H.; Fushimi, K.; Yasunaga, H. Antithrombin and mortality in severe pneumonia patients with sepsis-associated disseminated intravascular coagulation: An observational nationwide study. J. Thromb. Haemost. 2014, 12, 1470–1479. [Google Scholar] [CrossRef]
- Levi, M. Antithrombin in sepsis revisited. Crit. Care 2005, 9, 624–625. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
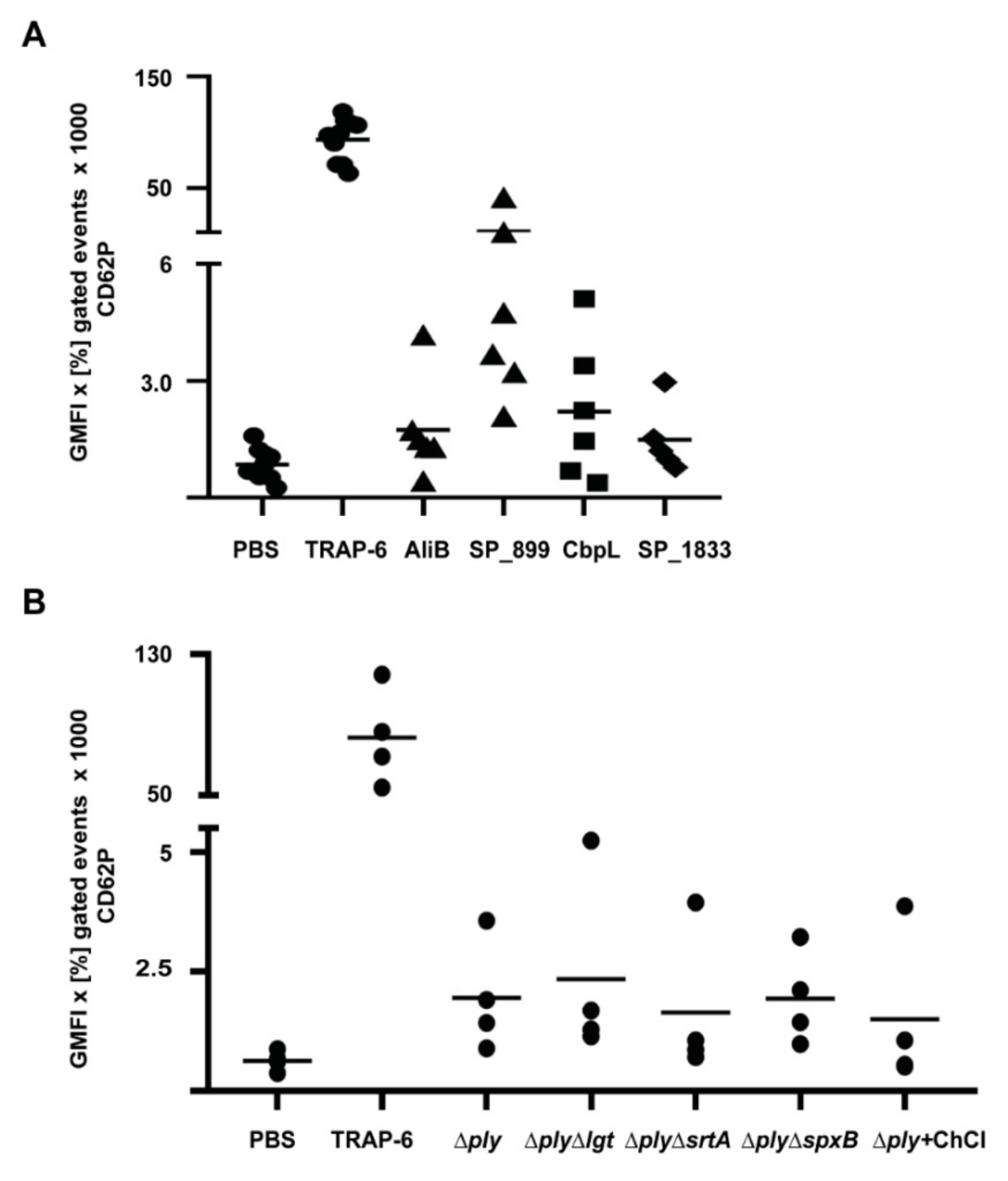
| Protein Class | No. | Protein Name SP Number | Function | Activation of Washed Platelets | Protein Concentration (µM) * |
|---|---|---|---|---|---|
| Lipoproteins | 1 | AdcAII (SP_1002) | substrate-binding protein of ABC transporter for zinc(II) ions | - | 4 |
| 2 | AliB (SP_1527) | substrate-binding protein of ABC transporter for oligopeptides | + | 2 | |
| 3 | AliC | substrate-binding protein of ABC transporter for oligopeptides | - | 4 | |
| 4 | AliD | substrate-binding protein of ABC transporter for oligopeptides | - | 4 | |
| 5 | AmiA (SP_1891) | substrate-binding protein of ABC transporter for oligopeptides | - | 4 | |
| 6 | DacB (SP_0629) | L,D-carboxypeptidase, peptidoglycan turnover | - | 4 | |
| 7 | Lipidated DacB | L,D-carboxypeptidase, peptidoglycan turnover | - | 4 | |
| 8 | Etrx1 (SP_0659) | extracellular thioredoxin protein 1 | - | 4 | |
| 9 | Etrx2 (SP_1000) | extracellular thioredoxin protein 2 | - | 4 | |
| 10 | MetQ (SP_0149) | substrate-binding protein of ABC transporter for methionine | - | 4 | |
| 11 | Lipidated MetQ | substrate-binding protein of ABC transporter for methionine | - | 4 | |
| 12 | PccL (SP_0198) | transport of small hydrophobic molecules such as siderophores | - | 4 | |
| 13 | PiaA (SP_1032) | substrate-binding protein of ABC transporter for iron | - | 4 | |
| 14 | PnrA (SP_0845) | substrate-binding protein of ABC transporter for nucleosides | - | 4 | |
| 15 | PpmA (SP_0981) | proteinase maturation protein, peptidyl-prolyl isomerase | - | 4 | |
| 16 | PsaA (SP_1650) | substrate-binding protein of ABC transporter for manganese | - | 4 | |
| 17 | SlrA (SP_0771) | streptococcal lipoprotein rotamase, peptidyl-prolyl isomerase | - | 4 | |
| 18 | GshT (SP_0148) | substrate-binding protein of ABC transporter for glutathione | - | 4 | |
| 19 | SP_0191 | unknown function | - | 4 | |
| 20 | SP_0899 | unknown function | +++ | 2/4 | |
| 21 | FusA (SP_1796) | substrate-binding protein of ABC transporter for fructo-oligosaccharides | - | 2 | |
| 22 | RafE (SP_1897) | substrate-binding protein of ABC transporter for multiple sugars | - | 4 | |
| 23 | PstS (SP_2084) | substrate-binding protein of ABC transporter for phosphate ions | - | - | |
| 24 | SP_1690 | substrate-binding protein of ABC transporter | - | 4 | |
| 25 | MalX (SP_2108) | substrate-binding protein of ABC transporter for maltose/maltodextrin | - | 4 | |
| 26 | SatA (SP_1683) | substrate-binding protein of ABC transporter for sialic acid | - | 4 | |
| CBPs | 27 | CbpC (SP_0377) | regulatory function for autolysis by inhibiting autolysin LytC | - | 4 |
| 28 | CbpF (SP_0391) | putative adhesin | - | 4 | |
| 29 | CbpL (SP_0667) | putative adhesin | ++ | 4 | |
| 30 | Chimeric (PspA+PspC) | fusion of N-terminal domains of PspA and PspC | - | 4 | |
| 31 | PcpA (SP_2136) | adhesin | - | 2 | |
| 32 | PspA_QP2 (SP_0117) | virulence factor, binds lactoferrinand inhibits complement activation | - | 4 | |
| 33 | PspC_SH2 (SP_2190) | adhesion, IgA inactivation, major factor H–binding protein | - | 4 | |
| Sortase– anchored proteins | 34 | PfbA (SP_1833) | plasmin- and fibronectin-binding protein | + | 2 |
| 35 | PitB (spt_1059) | pilin of pneumococcal pilus-2, adhesin | - | 2 | |
| 36 | PsrP (SP_1772) | adhesion, biofilm formation | - | 4 | |
| 37 | RrgB (SP_0463) | pilus-1 anchorage protein | - | 4 | |
| 38 | RrgC (SP_0464) | pilus-1 backbone protein, pilin | - | 4 | |
| 39 | SP_1992 | adhesin architecture, bind to collagen and lactoferrin in vitro | - | 4 | |
| Cell wall components | 40 | lipoteichoic acids | - | 4 | |
| 41 | wall teichoic acids | - | 40 µg/mL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahn, K.; Kohler, T.P.; Swiatek, L.-S.; Wiebe, S.; Hammerschmidt, S. Platelets, Bacterial Adhesins and the Pneumococcus. Cells 2022, 11, 1121. https://doi.org/10.3390/cells11071121
Jahn K, Kohler TP, Swiatek L-S, Wiebe S, Hammerschmidt S. Platelets, Bacterial Adhesins and the Pneumococcus. Cells. 2022; 11(7):1121. https://doi.org/10.3390/cells11071121
Chicago/Turabian StyleJahn, Kristin, Thomas P. Kohler, Lena-Sophie Swiatek, Sergej Wiebe, and Sven Hammerschmidt. 2022. "Platelets, Bacterial Adhesins and the Pneumococcus" Cells 11, no. 7: 1121. https://doi.org/10.3390/cells11071121
APA StyleJahn, K., Kohler, T. P., Swiatek, L.-S., Wiebe, S., & Hammerschmidt, S. (2022). Platelets, Bacterial Adhesins and the Pneumococcus. Cells, 11(7), 1121. https://doi.org/10.3390/cells11071121