
The Effect of Ex Vivo Human Serum from Liver Disease Patients on Cellular Protein Synthesis and Growth

 , , , , ,
, , , , ,
Abstract
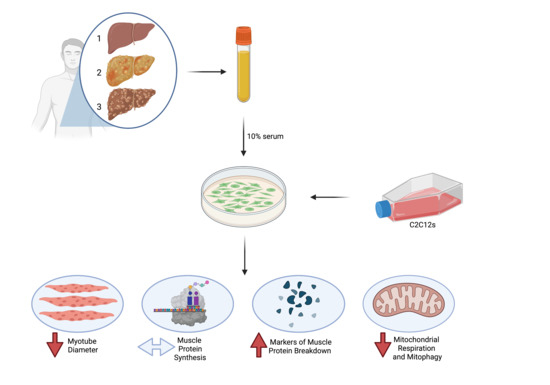
1. Introduction
2. Materials and Methods
2.1. Participant Characteristics and Ethical Approval
2.2. Study Design
2.3. Experimental Procedures
Blood Analyses
2.4. Cell Culture
2.5. Myotube Diameter
2.6. Muscle Protein Synthesis
2.7. Immunoblotting
2.8. Mitochondrial Respirometry
2.9. Mitophagy Assay
2.10. Statistical Analysis
3. Results
3.1. Body Composition and Strength
3.2. Blood Analysis
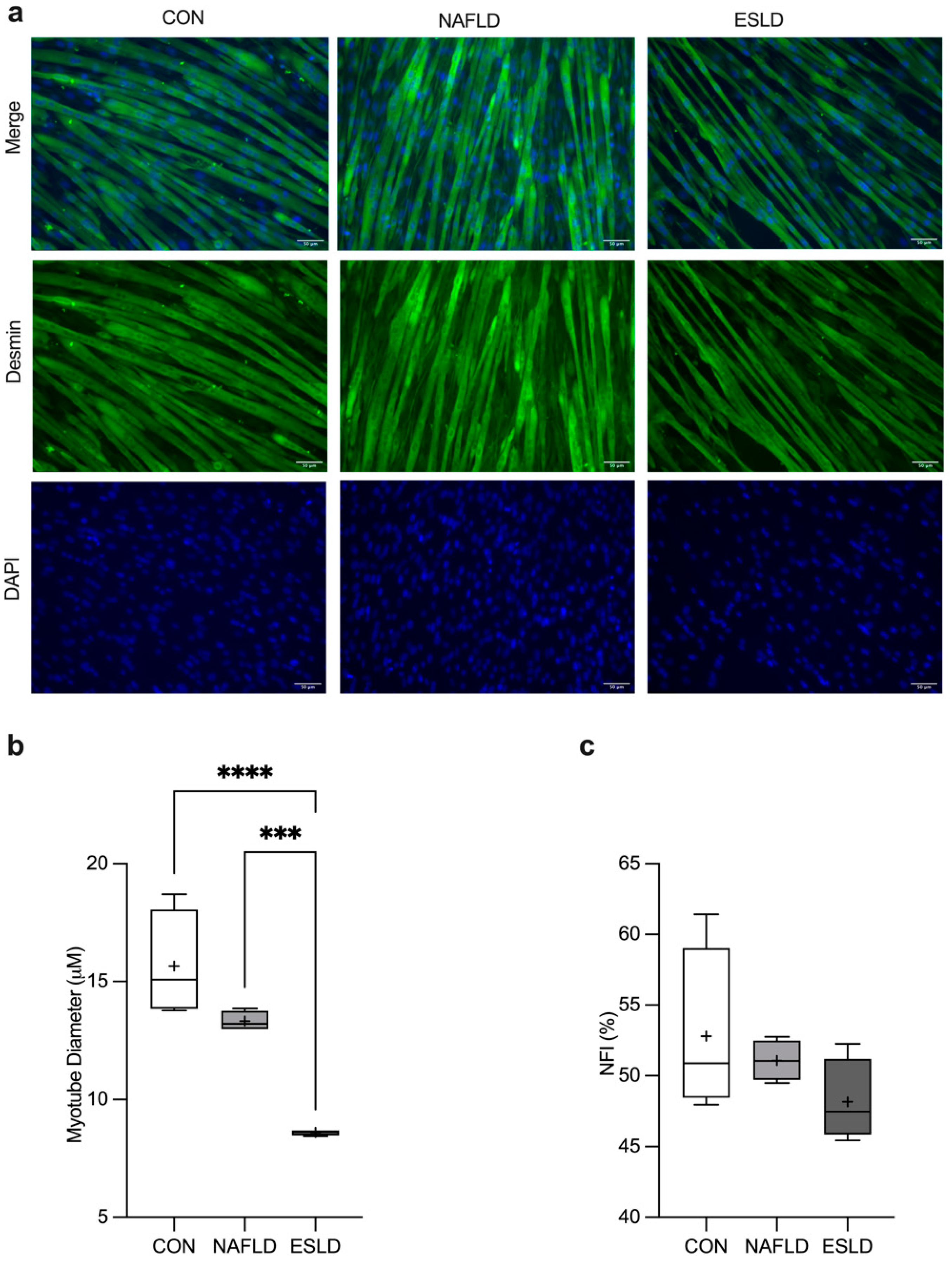
3.3. Myotube Diameter and Nuclear Fusion Index
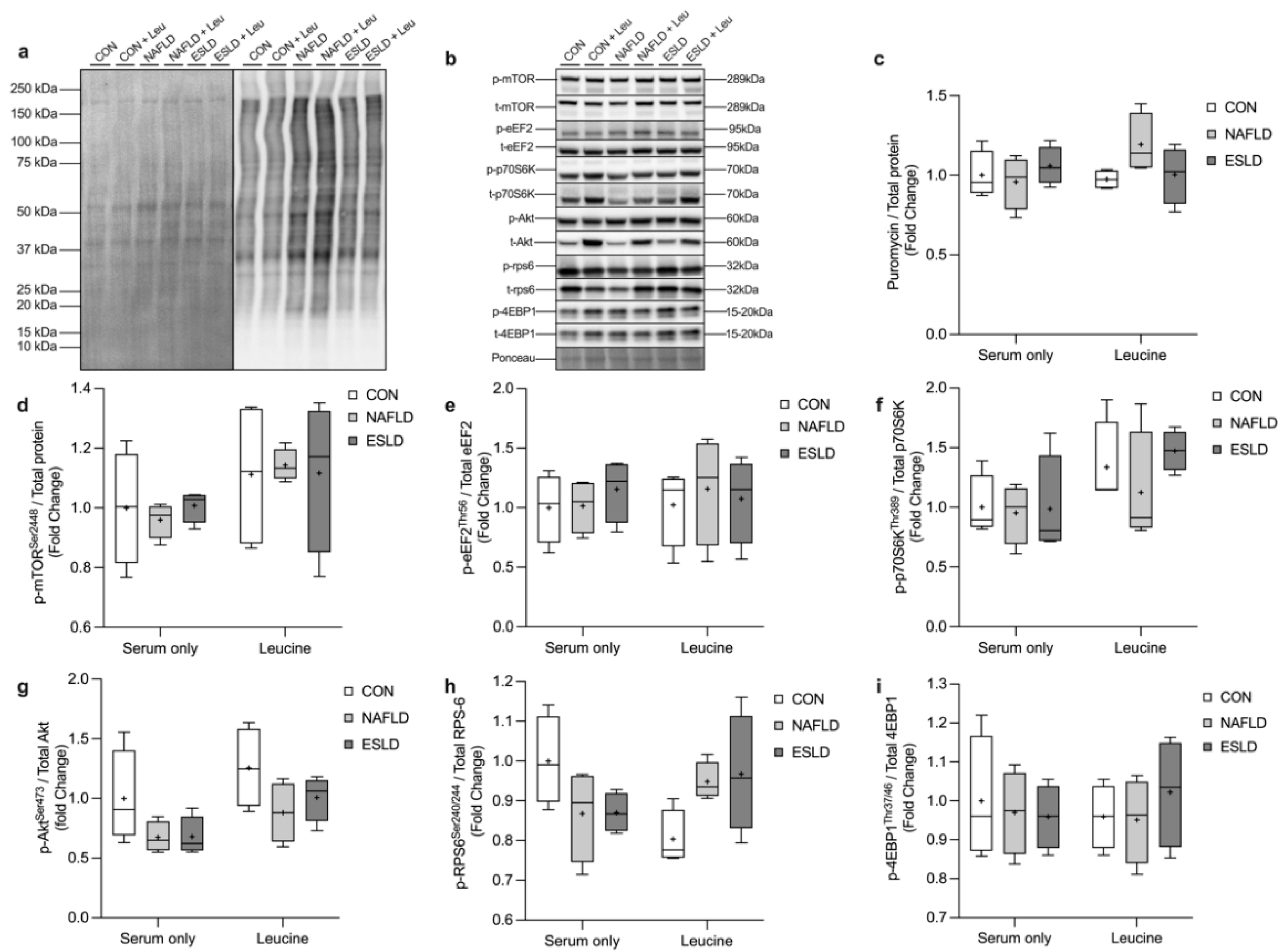
3.4. Muscle Protein Synthesis and Anabolic Signaling
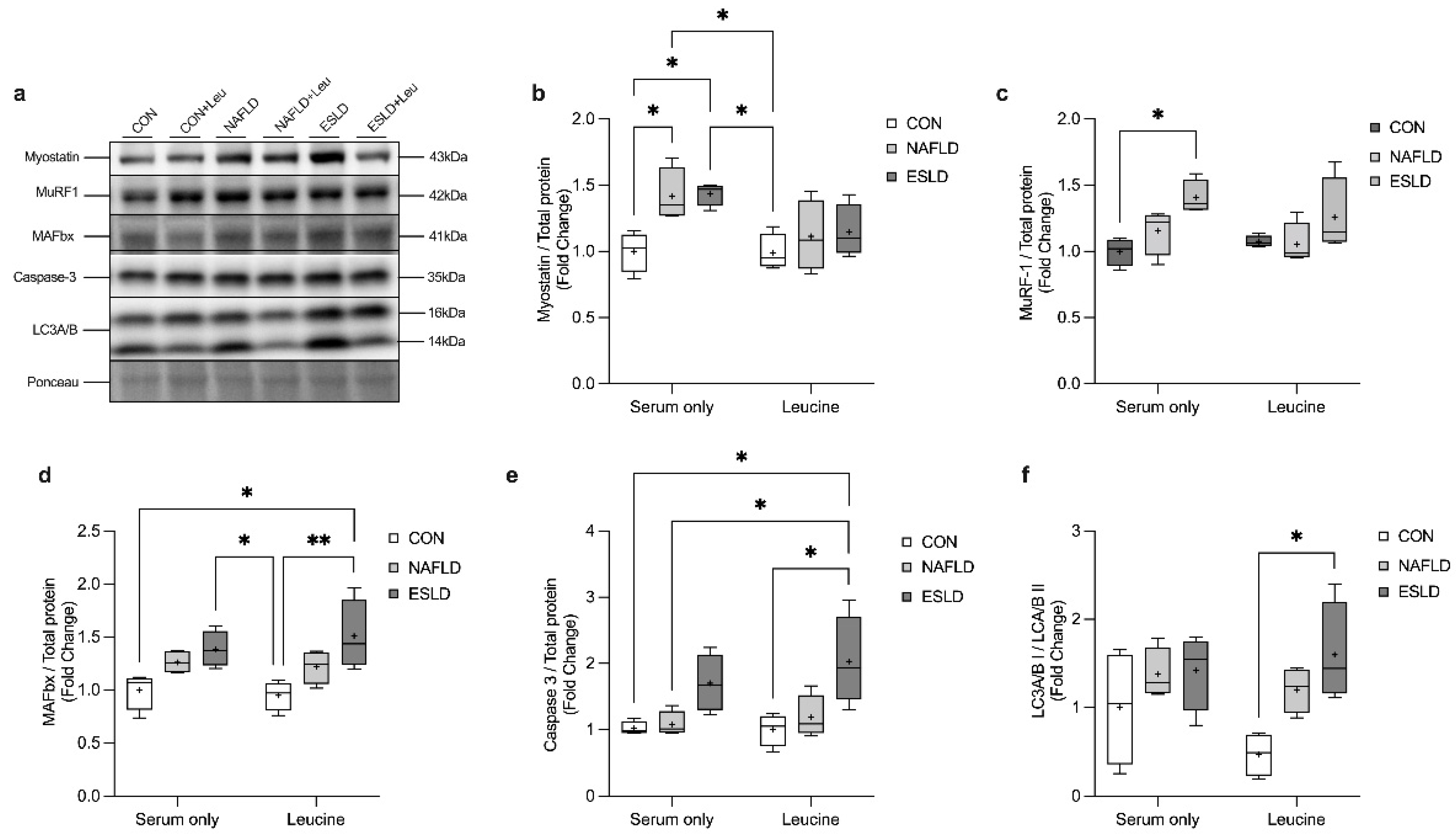
3.5. Proteolytic Signaling
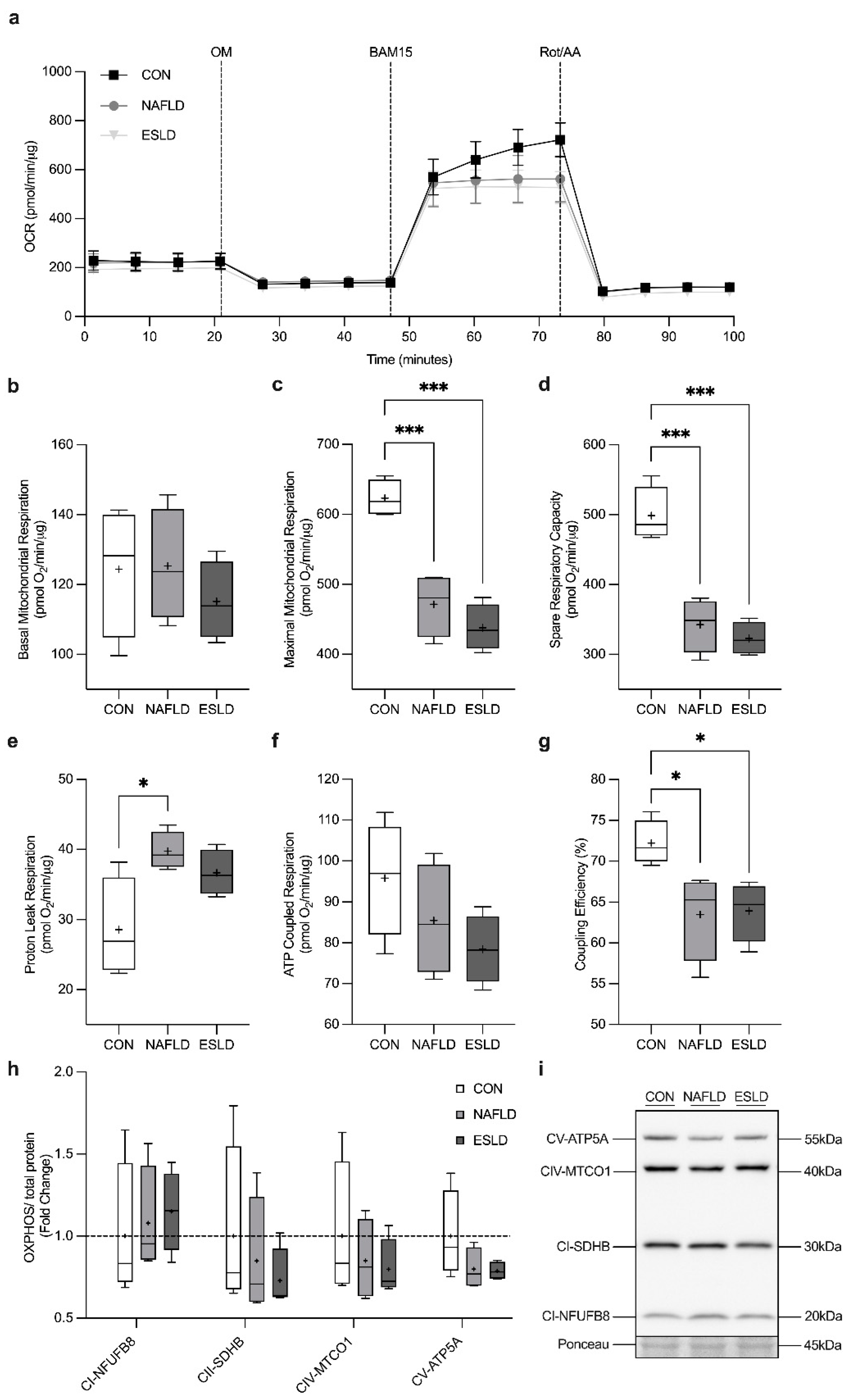
3.6. Mitochondrial Respiration
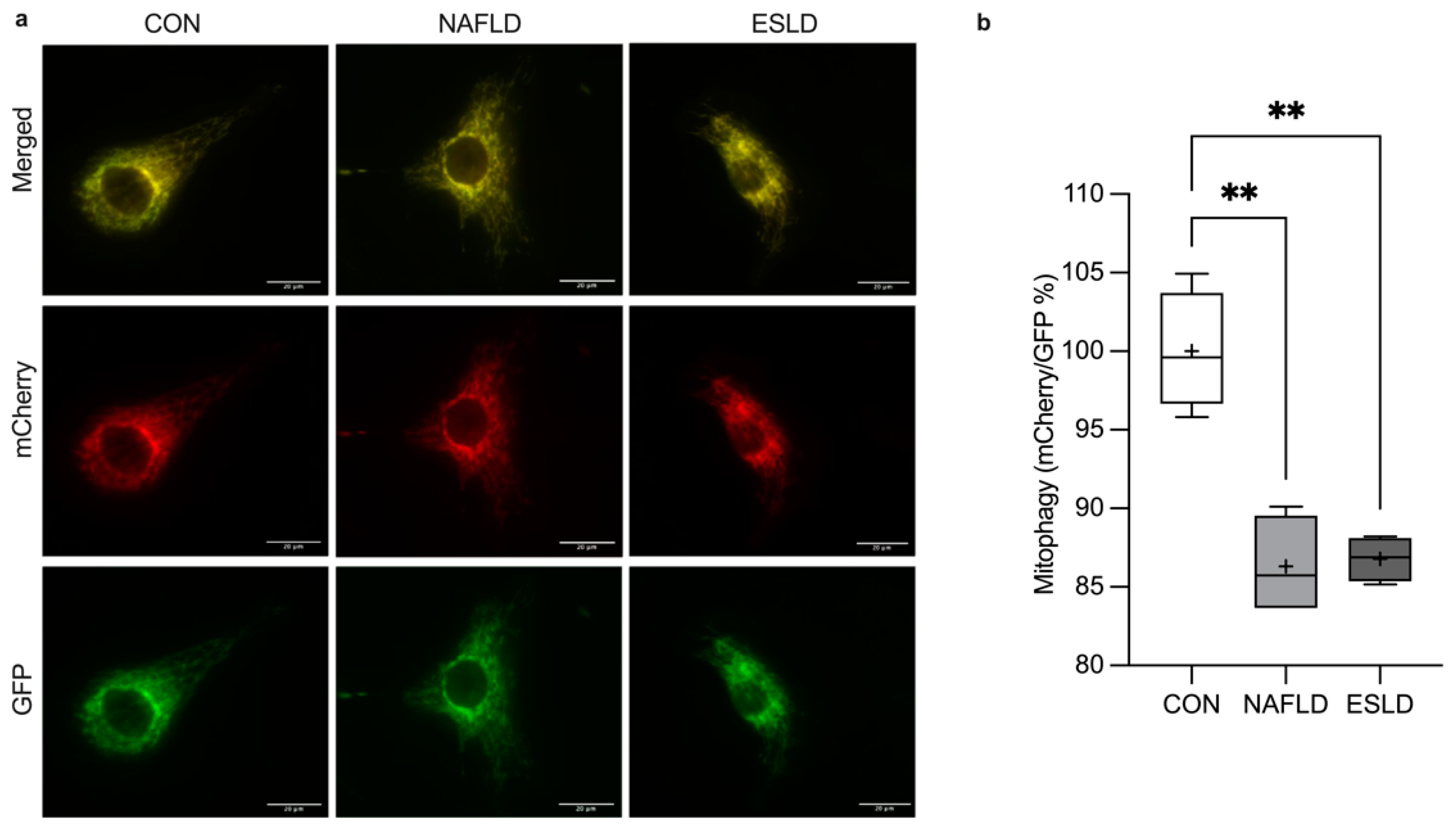
3.7. Mitophagy
3.8. Mitochondrial Content
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, G.; Kang, S.H.; Kim, M.Y.; Baik, S.K. Prognostic value of sarcopenia in patients with liver cirrhosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0186990. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Dasarathy, S. Consilience in sarcopenia of cirrhosis. J. Cachex-Sarcopenia Muscle 2012, 3, 225–237. [Google Scholar] [CrossRef]
- Van Vugt, J.L.A.; Levolger, S.; De Bruin, R.W.F.; van Rosmalen, J.; Metselaar, H.J.; Ijzermans, J.N.M. Systematic Review and Meta-Analysis of the Impact of Computed Tomography-Assessed Skeletal Muscle Mass on Outcome in Patients Awaiting or Undergoing Liver Transplantation. Am. J. Transplant. 2016, 16, 2277–2292. [Google Scholar] [CrossRef]
- Montano–Loza, A.J.; Meza–Junco, J.; Prado, C.M.; Lieffers, J.R.; Baracos, V.E.; Bain, V.G.; Sawyer, M.B. Muscle Wasting Is Associated With Mortality in Patients With Cirrhosis. Clin. Gastroenterol. Hepatol. 2012, 10, 166–173.e1. [Google Scholar] [CrossRef]
- Yadav, A.; Chang, Y.-H.; Carpenter, S.; Silva, A.C.; Rakela, J.; Aqel, B.A.; Byrne, T.J.; Douglas, D.D.; Vargas, H.E.; Carey, E.J. Relationship between sarcopenia, six-minute walk distance and health-related quality of life in liver transplant candidates. Clin. Transplant. 2015, 29, 134–141. [Google Scholar] [CrossRef]
- Dasarathy, S. Etiology and Management of Muscle Wasting in Chronic Liver Disease. Curr. Opin. Gastroenterol. 2016, 32, 159–165. [Google Scholar]
- Qiu, J.; Thapaliya, S.; Runkana, A.; Yang, Y.; Tsien, C.; Mohan, M.L.; Narayanan, A.; Eghtesad, B.; Mozdziak, P.E.; McDonald, C.; et al. Hyperammonemia in cirrhosis induces transcriptional regulation of myostatin by an NF-kappaB-mediated mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 18162–18167. [Google Scholar]
- Allen, S.L.; Quinlan, J.I.; Dhaliwal, A.; Armstrong, M.J.; Elsharkawy, A.M.; Greig, C.A.; Lord, J.M.; Lavery, G.G.; Breen, L. Sarcopenia in chronic liver disease: Mechanisms and countermeasures. Am. J. Physiol. Liver Physiol. 2021, 320, G241–G257. [Google Scholar] [CrossRef]
- Singh, S.S.; Kumar, A.; Welch, N.; Sekar, J.; Mishra, S.; Bellar, A.; Gangadhariah, M.; Attaway, A.; Al Khafaji, H.; Wu, X.; et al. Multiomics-Identified Intervention to Restore Ethanol-Induced Dysregulated Proteostasis and Secondary Sarcopenia in Alcoholic Liver Disease. Cell. Physiol. Biochem. 2021, 55, 91–116. [Google Scholar] [CrossRef]
- Ohashi, K.; Ishikawa, T.; Hoshi, A.; Suzuki, M.; Mitobe, Y.; Yamada, E.; Abeywickrama, H.; Seki, N.; Koyama, C.; Aoki, H.; et al. Relationship between Sarcopenia and Both Physical Activity and Lifestyle in Patients with Chronic Liver Disease. J. Clin. Med. Res. 2018, 10, 920–927. [Google Scholar] [CrossRef] [PubMed]
- El Sherif, O.; Dhaliwal, A.; Newsome, P.; Armstrong, M.J. Sarcopenia in nonalcoholic fatty liver disease: New challenges for clinical practice. Expert Rev. Gastroenterol. Hepatol. 2020, 23, 197–205. [Google Scholar] [CrossRef]
- Tsien, C.D.; McCullough, A.J.; Dasarathy, S. Late evening snack: Exploiting a period of anabolic opportunity in cirrhosis. J. Gastroenterol. Hepatol. 2012, 27, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Tsien, C.; Davuluri, G.; Singh, D.; Allawy, A.; Have, G.A.T.; Thapaliya, S.; Schulze, J.M.; Barnes, D.; McCullough, A.J.; Engelen, M.P.; et al. Metabolic and molecular responses to leucine-enriched branched chain amino acid supplementation in the skeletal muscle of alcoholic cirrhosis. Hepatology 2015, 61, 2018–2029. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Tsien, C.; Thapalaya, S.; Narayanan, A.; Weihl, C.C.; Ching, J.K.; Eghtesad, B.; Singh, K.; Fu, X.; Dubyak, G.; et al. Hyperammonemia-mediated autophagy in skeletal muscle contributes to sarcopenia of cirrhosis. Am. J. Physiol. Metab. 2012, 303, E983–E993. [Google Scholar] [CrossRef]
- Kumar, A.; Davuluri, G.; DeSilva, R.N.; Engelen, M.P.; Have, G.A.T.; Prayson, R.; Deutz, N.E.; Dasarathy, S. Ammonia lowering reverses sarcopenia of cirrhosis by restoring skeletal muscle proteostasis. Hepatology 2017, 65, 2045–2058. [Google Scholar] [CrossRef]
- Gao, B.; Xu, M.-J.; Bertola, A.; Wang, H.; Zhou, Z.; Liangpunsakul, S. Animal Models of Alcoholic Liver Disease: Pathogenesis and Clinical Relevance. Gene Expr. 2017, 17, 173–186. [Google Scholar] [CrossRef]
- Allen, S.L.; Marshall, R.N.; Edwards, S.J.; Lord, J.M.; Lavery, G.G.; Breen, L. The effect of young and old ex vivo human serum on cellular protein synthesis and growth in an in vitro model of aging. Am. J. Physiol. Physiol. 2021, 321, C26–C37. [Google Scholar] [CrossRef]
- Cuthbertson, D.; Smith, K.; Babraj, J.; Leese, G.; Waddell, T.; Atherton, P.; Wackerhage, H.; Taylor, P.M.; Rennie, M.J. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J. 2005, 19, 422–424. [Google Scholar] [CrossRef]
- Davuluri, G.; Allawy, A.; Thapaliya, S.; Rennison, J.H.; Singh, D.; Kumar, A.; Sandlers, Y.; Van Wagoner, D.R.; Flask, C.A.; Hoppel, C.; et al. Hyperammonaemia-induced skeletal muscle mitochondrial dysfunction results in cataplerosis and oxidative stress. J. Physiol. 2016, 594, 7341–7360. [Google Scholar] [CrossRef]
- Dasarathy, S.; Merli, M. Sarcopenia from mechanism to diagnosis and treatment in liver disease. J. Hepatol. 2016, 65, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.A.; Wiggs, M.P.; et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachex-Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef]
- Foucher, J.; Chanteloup, E.; Vergniol, J.; Castera, L.; Le Bail, B.; Adhoute, X.; Bertet, J.; Couzigou, P.; De Lédinghen, V. Diagnosis of cirrhosis by transient elastography (FibroScan): A prospective study. Gut 2006, 55, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Carson, B.P.; Patel, B.; Amigo-Benavent, M.; Pauk, M.; Kumar Gujulla, S.; Murphy, S.M.; Kiely, P.A.; Jakeman, P.M. Regulation of muscle protein synthesis in an in vitro cell model using ex vivo human serum. Exp. Physiol. 2018, 103, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Agley, C.C.; Velloso, C.P.; Lazarus, N.R.; Harridge, S.D.R. An Image Analysis Method for the Precise Selection and Quantification of Fluorescently Labeled Cellular Constituents: Application to the Measurement of Human Muscle Cells in Culture. J. Histochem. Cytochem. 2012, 60, 428–438. [Google Scholar]
- Goodman, C.A.; Mabrey, D.M.; Frey, J.W.; Miu, M.H.; Schmidt, E.K.; Pierre, P.; Hornberger, T.A. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactivein vivotechnique. FASEB J. 2011, 25, 1028–1039. [Google Scholar] [CrossRef]
- Affourtit, C.; Brand, M.D. Measuring mitochondrial bioengertics in INS-1E insulinoma cells. Methods Enzym. 2009, 457, 405–424. [Google Scholar]
- Seabright, A.P.; Fine, N.H.F.; Barlow, J.P.; Lord, S.O.; Musa, I.; Gray, A.; Bryant, J.A.; Banzhaf, M.; Lavery, G.G.; Hardie, D.G.; et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. 2020, 34, 6284–6301. [Google Scholar] [CrossRef]
- Allen, G.F.G.; Toth, R.; James, J.; Ganley, I.G. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013, 14, 1127–1135. [Google Scholar]
- Dasarathy, S.; Dodig, M.; Muc, S.M.; Kalhan, S.C.; McCullough, A.J. Skeletal muscle atrophy is associated with an increased expression of myostatin and impaired satellite cell function in the portacaval anastamosis rat. Am. J. Physiol. Liver Physiol. 2004, 287, G1124–G1130. [Google Scholar] [CrossRef]
- Smeuninx, B.; McKendry, J.; Wilson, D.; Martin, U.; Breen, L. Age-Related Anabolic Resistance of Myofibrillar Protein Synthesis Is Exacerbated in Obese Inactive Individuals. J. Clin. Endocrinol. Metab. 2017, 102, 3535–3545. [Google Scholar] [CrossRef] [PubMed]
- Wåhlin-Larsson, B.; Wilkinson, D.J.; Strandberg, E.; Hosford-Donovan, A.; Atherton, P.J.; Kadi, F. Mechanistic Links Underlying the Impact of C-Reactive Protein on Muscle Mass in Elderly. Cell. Physiol. Biochem. 2017, 44, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Katsanos, C.S.; Kobayashi, H.; Sheffield-Moore, M.; Aarsland, A.; Wolfe, R.R. A high proportion of leucine is required for optimal stimulation of the rate of muscle protein synthesis by essential amino acids in the elderly. Am. J. Physiol. Metab. 2006, 291, E381–E387. [Google Scholar] [CrossRef]
- Kant, S.; Davuluri, G.; Alchirazi, K.A.; Welch, N.; Heit, C.; Kumar, A.; Gangadhariah, M.; Kim, A.; McMullen, M.R.; Willard, B.; et al. Ethanol sensitizes skeletal muscle to ammonia-induced molecular perturbations. J. Biol. Chem. 2019, 294, 7231–7244. [Google Scholar] [CrossRef]
- Trendelenburg, A.U.; Meyer, A.; Rohner, D.; Boyle, J.; Hatakeyama, S.; Glass, D.J. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am. J. Physiol. Physiol. 2009, 296, C1258–C1270. [Google Scholar] [CrossRef]
- Lokireddy, S.; Mouly, V.; Butler-Browne, G.; Gluckman, P.D.; Sharma, M.; Kambadur, R.; McFarlane, C. Myostatin promotes the wasting of human myoblast cultures through promoting ubiquitin-proteasome pathway-mediated loss of sarcomeric proteins. Am. J. Physiol. Physiol. 2011, 301, C1316–C1324. [Google Scholar] [CrossRef]
- Porter, C.; Hurren, N.M.; Cotter, M.V.; Bhattarai, N.; Reidy, P.T.; Dillon, E.L.; Durham, W.J.; Tuvdendorj, D.; Sheffield-Moore, M.; Volpi, E.; et al. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am. J. Physiol. Metab. 2015, 309, E224–E232. [Google Scholar] [CrossRef]
- Julienne, C.M.; Dumas, J.-F.; Goupille, C.; Pinault, M.; Berri, C.; Collin, A.; Tesseraud, S.; Couet, C.; Servais, S. Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. J. Cachex-Sarcopenia Muscle 2012, 3, 265–275. [Google Scholar] [CrossRef]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Zhou, H.; Du, W.; Li, Y.; Shi, C.; Hu, N.; Ma, S.; Wang, W.; Ren, J. Effects of melatonin on fatty liver disease: The role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J. Pineal Res. 2017, 64, e12450. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective Mitochondrial Autophagy, or Mitophagy, as a Targeted Defense Against Oxidative Stress, Mitochondrial Dysfunction, and Aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Dornas, W.; Schuppan, D. Mitochondrial oxidative injury: A key player in nonalcoholic fatty liver disease. Am. J. Physiol. Liver Physiol. 2020, 319, G400–G411. [Google Scholar] [CrossRef]
- Sergi, D.; Naumovski, N.N.; Heilbronn, L.H.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N.L.-M. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef] [PubMed]
- Consitt, L.; Clark, B. The Vicious Cycle of Myostatin Signaling in Sarcopenic Obesity: Myostatin Role in Skeletal Muscle Growth, Insulin Signaling and Implications for Clinical Trials. J. Frailty Aging 2017, 7, 21–27. [Google Scholar] [CrossRef]
- Sinclair, M.; Grossmann, M.; Gow, P.J.; Angus, P.W. Testosterone in men with advanced liver disease: Abnormalities and implications. J. Gastroenterol. Hepatol. 2015, 30, 244–251. [Google Scholar] [CrossRef]
- Niu, L.; Geyer, P.E.; Albrechtsen, N.J.W.; Gluud, L.L.; Santos, A.; Doll, S.; Treit, P.V.; Holst, J.J.; Knop, F.K.; Vilsbøll, T.; et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 2019, 15, e8793. [Google Scholar] [CrossRef]
- Kumar, A.; Davuluri, G.; Welch, N.; Kim, A.; Gangadhariah, M.; Allawy, A.; Priyadarshini, A.; McMullen, M.R.; Sandlers, Y.; Willard, B.; et al. Oxidative stress mediates ethanol-induced skeletal muscle mitochondrial dysfunction and dysregulated protein synthesis and autophagy. Free Radic. Biol. Med. 2019, 145, 284–299. [Google Scholar] [CrossRef]
- Davuluri, G.; Giusto, M.; Chandel, R.; Welch, N.; Alsabbagh, K.; Kant, S.; Kumar, A.; Kim, A.; Gangadhariah, M.; Ghosh, P.K.; et al. Impaired Ribosomal Biogenesis by Noncanonical Degradation of β-Catenin during Hyperammonemia. Mol. Cell. Biol. 2019, 39, e00451-18. [Google Scholar] [CrossRef]
- Periyalwar, P.; Dasarathy, S. Malnutrition in Cirrhosis: Contribution and Consequences of Sarcopenia on Metabolic and Clinical Responses. Clin. Liver Dis. 2012, 16, 95–131. [Google Scholar] [CrossRef]
- Hallsworth, K.; Thoma, C.; Moore, S.; Ploetz, T.; Anstee, Q.M.; Taylor, R.; Day, C.P.; Trenell, M.I. Non-alcoholic fatty liver disease is associated with higher levels of objectively measured sedentary behaviour and lower levels of physical activity than matched healthy controls. Frontline Gastroenterol. 2015, 6, 44–51. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion Criteria | Exclusion Criteria | |
|---|---|---|
| ESLD | 1. Adults aged ≥ 18 years old 2. Male 3. Capable of providing written informed consent 4. Cirrhosis (defined by Child Pugh Scores A–C) 5. Radiological and serological evidence of cirrhosis +/− a liver biopsy 6. Listed on the UK transplant list | 1. Lack of capacity to provide informed consent. 2. Previously undergone liver transplant 3. Diagnosed with cancer 4. Receiving biliary intervention 5. Enrolled in an intervention trial for liver disease |
| NAFLD | 1. Adults aged ≥ 18 years old 2. Male 3. Capable of providing written informed consent 4. Significant fibrosis (F2–F4 Kleiner fibrosis stages) confirmed with liver biopsy 5. Fibroscan of < 12 kPa (to exclude cirrhosis) | 1. Lack of capacity to provide informed consent 2. Cirrhosis (defined by Child Pugh scores A–C) 3. Listed on the UK transplant list 4. Fibroscan > 15 kPa |
| CON | 1. Adults aged ≥ 18 years old 2. Male 3. Body mass index of 18.5–34.99 4. Capable of providing written informed consent Free from inflammatory chronic disease (i.e., good general health) Non-smoker Non-diabetic | 1. Lack of capacity to provide informed consent. 2. Diagnosed with diabetes 3. Diagnosed with any inflammatory disease condition Smoker |
| CON (n = 4) | NAFLD (n = 4) | ESLD (n = 4) | |
|---|---|---|---|
| Anthropometrics | |||
| Age (years) | 10.0 | 7.6 | 1.7 |
| Body mass (kg) | 7.6 | 31.3 | 18.5 |
| Height (cm) | 7.4 | 3.6 | 4.3 |
| BMI (kg m−2) | 1.6 | 11.33 | 7.3 |
| FFM (kg) | 6.1 | 11.4 | 6.7 |
| WBFM (kg) | 2.2 | 20.1 | 15.5 |
| BF (%) | 1.9 | 7.3 * | 8.7 * |
| Strength | |||
| HGS (kg) | 9.8 | 7.1 | 8.2 |
| Blood Analyses | |||
| Fasting Serum Insulin (μIU/mL) | 4.1 | 3.5 | 10.1 * |
| Fasting Serum Glucose (mmol/L) | 0.1 | 0.1 | 0.1 |
| HOMA-IR | 0.7 | 0.6 | 2.0 * |
| Serum IL-6 (pg/mL) | 0.1 | 0.3 | 2.2 * |
| Serum CRP (ng/mL) | 0.7 | 0.5 | |
| Serum Ammonia | 0.09 | 0.1 | 0.04 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allen, S.L.; Seabright, A.P.; Quinlan, J.I.; Dhaliwal, A.; Williams, F.R.; Fine, N.H.F.; Hodson, D.J.; Armstrong, M.J.; Elsharkaway, A.M.; Greig, C.A.; et al. The Effect of Ex Vivo Human Serum from Liver Disease Patients on Cellular Protein Synthesis and Growth. Cells 2022, 11, 1098. https://doi.org/10.3390/cells11071098
Allen SL, Seabright AP, Quinlan JI, Dhaliwal A, Williams FR, Fine NHF, Hodson DJ, Armstrong MJ, Elsharkaway AM, Greig CA, et al. The Effect of Ex Vivo Human Serum from Liver Disease Patients on Cellular Protein Synthesis and Growth. Cells. 2022; 11(7):1098. https://doi.org/10.3390/cells11071098
Chicago/Turabian StyleAllen, Sophie L., Alex P. Seabright, Jonathan I. Quinlan, Amritpal Dhaliwal, Felicity R. Williams, Nicholas H. F. Fine, David J. Hodson, Matthew J. Armstrong, Ahmed M. Elsharkaway, Carolyn A. Greig, and et al. 2022. "The Effect of Ex Vivo Human Serum from Liver Disease Patients on Cellular Protein Synthesis and Growth" Cells 11, no. 7: 1098. https://doi.org/10.3390/cells11071098
APA StyleAllen, S. L., Seabright, A. P., Quinlan, J. I., Dhaliwal, A., Williams, F. R., Fine, N. H. F., Hodson, D. J., Armstrong, M. J., Elsharkaway, A. M., Greig, C. A., Lai, Y.-C., Lord, J. M., Lavery, G. G., & Breen, L. (2022). The Effect of Ex Vivo Human Serum from Liver Disease Patients on Cellular Protein Synthesis and Growth. Cells, 11(7), 1098. https://doi.org/10.3390/cells11071098