
Leaky Gum: The Revisited Origin of Systemic Diseases


Abstract
:1. Introduction
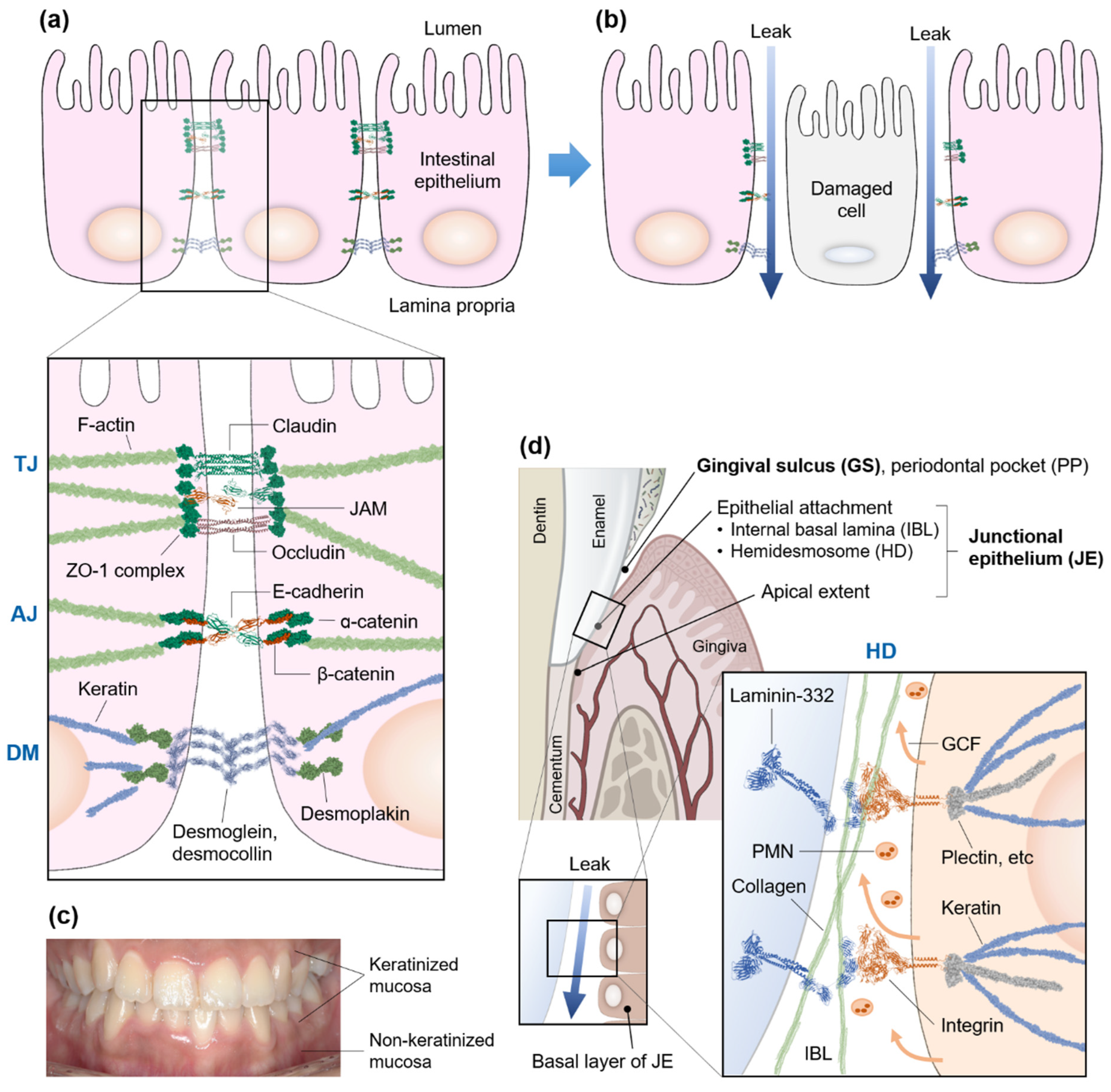
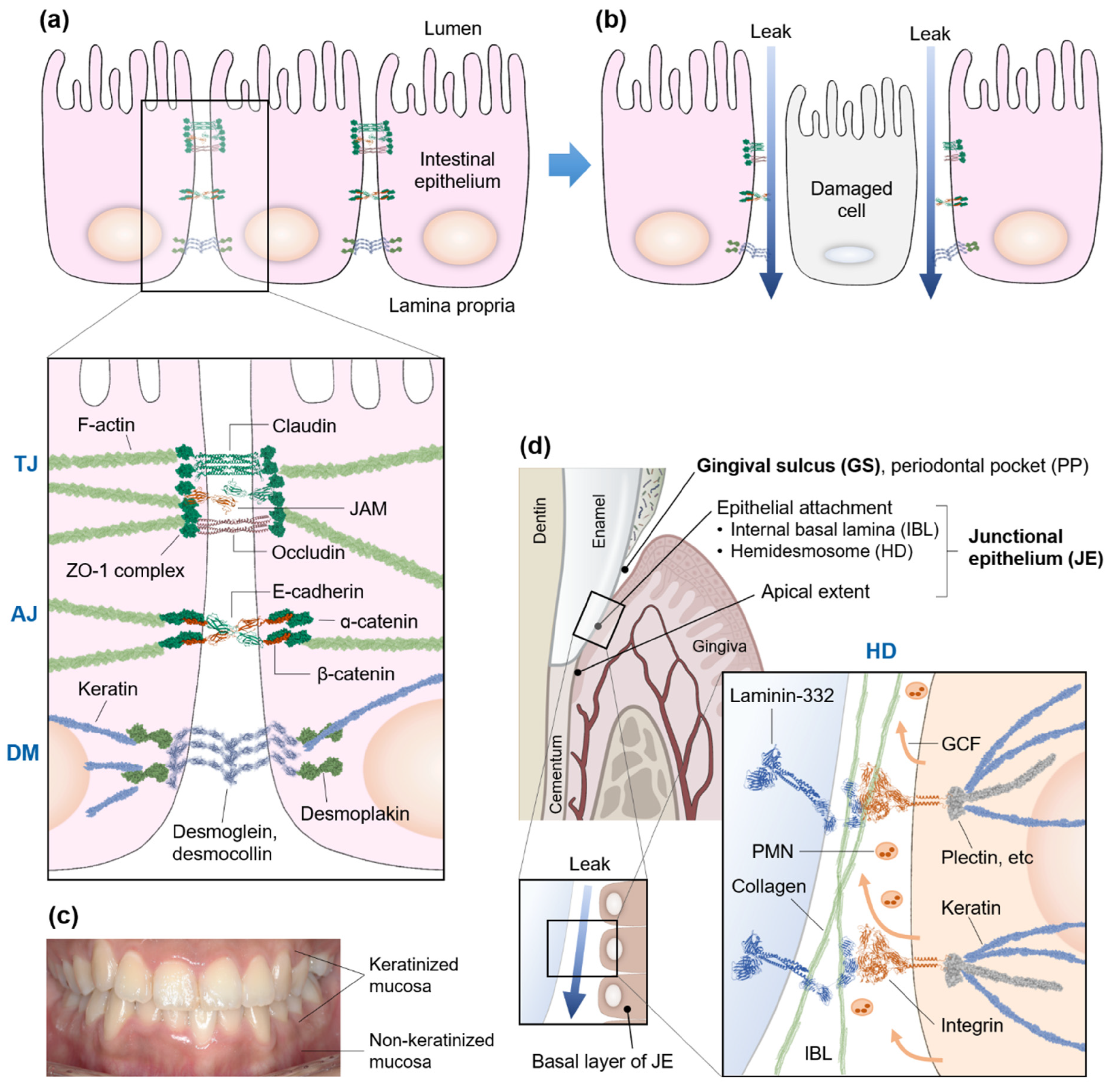
2. Hyperpermeable Intestine—Leaky Gut
3. Gum and Gut Mucosal Barriers
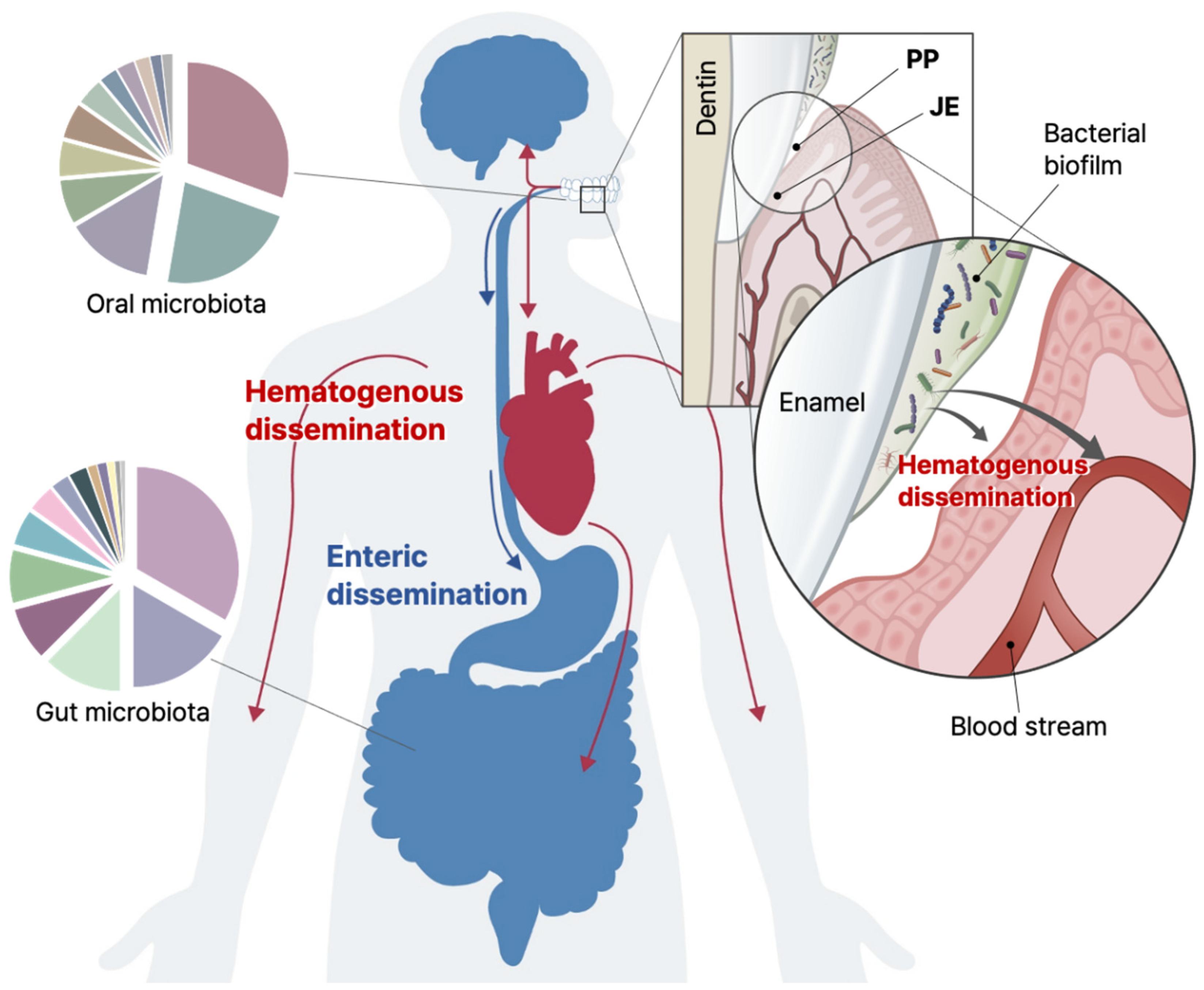
4. Gingival Sulcus and Junctional Epithelium
5. Focal Infection Theory and Leaky Gum
6. Oral Pathogens and Systemic Diseases
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6, 237ra265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Arango, L.F.; Barrett, H.L.; McIntyre, H.D.; Callaway, L.K.; Morrison, M.; Nitert, M.D. Contributions of the maternal oral and gut microbiome to placental microbial colonization in overweight and obese pregnant women. Sci. Rep. 2017, 7, 2860. [Google Scholar] [CrossRef] [PubMed]
- Goffau, M.C.d.; Lager, S.; Sovio, U.; Gaccioli, F.; Cook, E.; Peacock, S.J.; Parkhill, J.; Charnock-Jones, D.S.; Smith, G.C.S. Human placenta has no microbiome but can contain potential pathogens. Nature 2019, 572, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Kapila, Y. Oral microbiome shifts during pregnancy and adverse pregnancy outcomes: Hormonal and Immunologic changes at play. Periodontol. 2000 2021, 87, 276–281. [Google Scholar] [CrossRef]
- Whiteside, S.A.; McGinniss, J.E.; Collman, R.G. The lung microbiome: Progress and promise. J. Clin. Investig. 2021, 131, e150473. [Google Scholar] [CrossRef]
- Huffnagle, G.B.; Dickson, R.P.; Lukacs, N.W. The respiratory tract microbiome and lung inflammation: A two-way street. Mucosal Immunol. 2017, 10, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Dickson, R.P.; Erb-Downward, J.R.; Freeman, C.M.; McCloskey, L.; Falkowski, N.R.; Huffnagle, G.B.; Curtis, J.L.; Clemente, J.C.; Molyneaux, P.; Bogaert, D. Bacterial Topography of the Healthy Human Lower Respiratory Tract. MBio 2017, 8, e02287-16. [Google Scholar] [CrossRef]
- Mehta, R.S.; Nishihara, R.; Cao, Y.; Song, M.; Mima, K.; Qian, Z.R.; Nowak, J.A.; Kosumi, K.; Hamada, T.; Masugi, Y.; et al. Association of Dietary Patterns With Risk of Colorectal Cancer Subtypes Classified by Fusobacterium nucleatum in Tumor Tissue. JAMA Oncol. 2017, 3, 921–927. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Li, S.; Gan, R.-Y.; Zhao, C.-N.; Meng, X.; Li, H.-B. Targeting gut microbiota with dietary components on cancer: Effects and potential mechanisms of action. Crit. Rev. Food Sci. Nutr. 2020, 60, 1025–1037. [Google Scholar] [CrossRef]
- Agirman, G.; Yu, K.B.; Hsiao, E.Y. Signaling inflammation across the gut-brain axis. Science 2021, 374, 1087–1092. [Google Scholar] [CrossRef]
- Morais, L.H.; Schreiber, H.L.; Mazmanian, S.K. The gut microbiota–brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 2021, 19, 241–255. [Google Scholar] [CrossRef]
- Kitamoto, S.; Nagao-Kitamoto, H.; Jiao, Y.; Gillilland, M.G.; Hayashi, A.; Imai, J.; Sugihara, K.; Miyoshi, M.; Brazil, J.C.; Kuffa, P.; et al. The Intermucosal Connection between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell 2020, 182, 447–462.e14. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, S.; Nagao-Kitamoto, H.; Hein, R.; Schmidt, T.M.; Kamada, N. The Bacterial Connection between the Oral Cavity and the Gut Diseases. J. Dent. Res. 2020, 99, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Ray, K. The oral–gut axis in IBD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 532. [Google Scholar] [CrossRef] [PubMed]
- Narengaowa; Kong, W.; Lan, F.; Awan, U.F.; Qing, H.; Ni, J. The Oral–Gut–Brain AXIS: The Influence of Microbes in Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 113. [Google Scholar] [CrossRef]
- Byrd, K.M.; Gulati, A.S. The “Gum–Gut” Axis in Inflammatory Bowel Diseases: A Hypothesis-Driven Review of Associations and Advances. Front. Immunol. 2021, 12, 620124. [Google Scholar] [CrossRef]
- Hu, S.; Png, E.; Gowans, M.; Ong, D.E.H.; de Sessions, P.F.; Song, J.; Nagarajan, N. Ectopic gut colonization: A metagenomic study of the oral and gut microbiome in Crohn’s disease. Gut Pathog. 2021, 13, 13. [Google Scholar] [CrossRef]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Malard, F.; Dore, J.; Gaugler, B.; Mohty, M. Introduction to host microbiome symbiosis in health and disease. Mucosal Immunol. 2021, 14, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Relman, D.A. The human microbiome: Ecosystem resilience and health. Nutr. Rev. 2012, 70, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Bowman, A.A.; Young, V.B. Antibiotic-Induced Alterations of the Gut Microbiota Alter Secondary Bile Acid Production and Allow for Clostridium difficile Spore Germination and Outgrowth in the Large Intestine. mSphere 2016, 1, e00045-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessa, F.C.; Mu, Y.; Bamberg, W.M.; Beldavs, Z.G.; Dumyati, G.K.; Dunn, J.R.; Farley, M.M.; Holzbauer, S.M.; Meek, J.I.; Phipps, E.C.; et al. Burden of Clostridium difficile infection in the United States. N. Engl. J. Med. 2015, 372, 825–834. [Google Scholar] [CrossRef] [Green Version]
- CDC. Nearly Half a Million Americans Suffer from C. difficile Infections in Single Year. Available online: https://www.cdc.gov/media/releases/2015/p0225-clostridium-difficile.html (accessed on 16 February 2022).
- Zitvogel, L.; Galluzzi, L.; Viaud, S.; Vétizou, M.; Daillère, R.; Merad, M.; Kroemer, G. Cancer and the gut microbiota: An unexpected link. Sci. Transl. Med. 2015, 7, 271ps1. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yu, Y.-B. Intestinal microbiota and chronic constipation. Springerplus 2016, 5, 1130. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.Y.; Tsoi, H.; Wu, W.K.K.; et al. Gavage of Fecal Samples from Patients with Colorectal Cancer Promotes Intestinal Carcinogenesis in Germ-Free and Conventional Mice. Gastroenterology 2017, 153, 1621–1633. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Liu, X.; An, Y.; Zhou, G.; Liu, Y.; Xu, M.; Dong, W.; Wang, S.; Yan, F.; Jiang, K.; et al. Dysbiosis contributes to chronic constipation development via regulation of serotonin transporter in the intestine. Sci. Rep. 2017, 7, 10322. [Google Scholar] [CrossRef] [Green Version]
- Ohkusa, T.; Koido, S.; Nishikawa, Y.; Sato, N. Gut Microbiota and Chronic Constipation: A Review and Update. Front. Med. 2019, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Ding, R.X.; Goh, W.R.; Wu, R.N.; Yue, X.Q.; Luo, X.; Khine, W.W.T.; Wu, J.R.; Lee, Y.K. Revisit gut microbiota and its impact on human health and disease. J. Food Drug Anal. 2019, 27, 623–631. [Google Scholar] [CrossRef]
- Atreya, C.E.; Turnbaugh, P.J. Probing the tumor micro(b)environment. Science 2020, 368, 938–939. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, R.; Li, D.; Zhao, L.; Zhu, L. Role of gut microbiota in functional constipation. Gastroenterol. Rep. 2021, 9, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Hollander, D. Intestinal permeability, leaky gut, and intestinal disorders. Curr. Gastroenterol. Rep. 1999, 1, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.-D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability—A new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [Green Version]
- Fukui, H. Increased Intestinal Permeability and Decreased Barrier Function: Does It Really Influence the Risk of Inflammation? Inflamm. Intest. Dis. 2016, 1, 135–145. [Google Scholar] [CrossRef]
- Mu, Q.; Kirby, J.; Reilly, C.M.; Luo, X.M. Leaky gut as a danger signal for autoimmune diseases. Front. Immunol. 2017, 8, 598. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.R.; Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G.; Hyland, N.P. Breaking down the barriers: The gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front. Cell. Neurosci. 2015, 9, 392. [Google Scholar] [CrossRef] [Green Version]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef]
- Fukui, H. Role of Gut Dysbiosis in Liver Diseases: What Have We Learned So Far? Diseases 2019, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Harbison, J.E.; Roth-Schulze, A.J.; Giles, L.C.; Tran, C.D.; Ngui, K.M.; Penno, M.A.; Thomson, R.L.; Wentworth, J.M.; Colman, P.G.; Craig, M.E.; et al. Gut microbiome dysbiosis and increased intestinal permeability in children with islet autoimmunity and type 1 diabetes: A prospective cohort study. Pediatr. Diabetes 2019, 20, 574–583. [Google Scholar] [CrossRef]
- Schoultz, I.; Keita, Å.V. The Intestinal Barrier and Current Techniques for the Assessment of Gut Permeability. Cells 2020, 9, 1909. [Google Scholar] [CrossRef] [PubMed]
- Rogler, G.; Singh, A.; Kavanaugh, A.; Rubin, D.T. Extraintestinal Manifestations of Inflammatory Bowel Disease: Current Concepts, Treatment, and Implications for Disease Management. Gastroenterology 2021, 161, 1118–1132. [Google Scholar] [CrossRef] [PubMed]
- Vavricka, S.R.; Schoepfer, A.; Scharl, M.; Lakatos, P.L.; Navarini, A.; Rogler, G. Extraintestinal Manifestations of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1982–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravina, A.; Federico, A.; Ruocco, E.; Lo Schiavo, A.; Romano, F.; Miranda, A.; Sgambato, D.; Dallio, M.; Ruocco, V.; Loguercio, C.; et al. Crohn’s disease and skin. United Eur. Gastroenterol. J. 2016, 4, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argyriou, K.; Khan, M.; Samuel, S. Multiple Unusual Ulcerated Skin Lesions in a Crohn’s Disease Patient. Gastroenterology 2018, 155, e17–e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chebli, J.M.F.; de Oliveira Moreira, B.; da Rocha Ribeiro, T.C. An Unusual Cause of Skin Rash in Crohn’s Disease. Gastroenterology 2018, 155, 618–620. [Google Scholar] [CrossRef] [Green Version]
- Chung-Yee Hui, R.; Kuo, K.L.; Yang, T.W. A Rare Skin Complication Associated With Crohn’s Disease. Gastroenterology 2021, 160, 669–670. [Google Scholar] [CrossRef]
- Rabiei, S.; Mohebbi, S.Z.; Patja, K.; Virtanen, J.I. Physicians’ knowledge of and adherence to improving oral health. BMC Public Health 2012, 12, 855. [Google Scholar] [CrossRef] [Green Version]
- Elad, S.; Zadik, Y.; Caton, J.G.; Epstein, J.B. Oral mucosal changes associated with primary diseases in other body systems. Periodontol. 2000 2019, 80, 28–48. [Google Scholar] [CrossRef]
- Del Monte, U. Does the cell number 109 still really fit one gram of tumor tissue? Cell Cycle 2009, 8, 505–506. [Google Scholar] [CrossRef] [Green Version]
- Vighi, G.; Marcucci, F.; Sensi, L.; Di Cara, G.; Frati, F. Allergy and the gastrointestinal system. Clin. Exp. Immunol. 2008, 153, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Antoni, L.; Nuding, S.; Weller, D.; Gersemann, M.; Ott, G.; Wehkamp, J.; Stange, E.F. Human colonic mucus is a reservoir for antimicrobial peptides. J. Crohns Colitis 2013, 7, e652–e664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila, T.; Rizk, A.M.; Sultan, A.S.; Jabra-Rizk, M.A. The power of saliva: Antimicrobial and beyond. PLoS Pathog. 2019, 15, e1008058. [Google Scholar] [CrossRef] [PubMed]
- Wickett, R.R.; Visscher, M.O. Structure and function of the epidermal barrier. Am. J. Infect. Control 2006, 34, S98–S110. [Google Scholar] [CrossRef]
- Wells, J.M.; Brummer, R.J.; Derrien, M.; MacDonald, T.T.; Troost, F.; Cani, P.D.; Theodorou, V.; Dekker, J.; Méheust, A.; De Vos, W.M. Homeostasis of the gut barrier and potential biomarkers. Am. J. Physiol. Gastrointest. Liver. Physiol. 2017, 312, G171–G193. [Google Scholar] [CrossRef]
- Cornick, S.; Tawiah, A.; Chadee, K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers 2015, 3, e982426. [Google Scholar] [CrossRef] [Green Version]
- Iweala, O.I.; Nagler, C.R. The Microbiome and Food Allergy. Annu. Rev. Immunol. 2019, 37, 377–403. [Google Scholar] [CrossRef]
- Zenobia, C.; Herpoldt, K.-L.; Freire, M. Is the oral microbiome a source to enhance mucosal immunity against infectious diseases? NPJ Vaccines 2021, 6, 80. [Google Scholar] [CrossRef]
- Lin, D.; Yang, L.; Wen, L.; Lu, H.; Chen, Q.; Wang, Z. Crosstalk between the oral microbiota, mucosal immunity, and the epithelial barrier regulates oral mucosal disease pathogenesis. Mucosal Immunol. 2021, 14, 1247–1258. [Google Scholar] [CrossRef]
- Adams, D. Keratinization of the oral epithelium. Ann. R. Coll. Surg. Engl. 1976, 58, 351–358. [Google Scholar]
- Walmsley, A.D.; Walsh, T.F.; Lumley, P.J.; Burke, F.J.T.; Shortall, A.C.C.; Hayes-Hall, R.; Pretty, I.A. Chapter 2—The healthy mouth. In Restorative Dentistry, 2nd ed.; Walmsley, A.D., Walsh, T.F., Lumley, P.J., Burke, F.J.T., Shortall, A.C.C., Hayes-Hall, R., Pretty, I.A., Eds.; Churchill Livingstone: Edinburgh, UK, 2007; pp. 3–11. [Google Scholar]
- Brodala, N.; Merricks, E.P.; Bellinger, D.A.; Damrongsri, D.; Offenbacher, S.; Beck, J.; Madianos, P.; Sotres, D.; Chang, Y.-L.; Koch, G.; et al. Porphyromonas gingivalis Bacteremia Induces Coronary and Aortic Atherosclerosis in Normocholesterolemic and Hypercholesterolemic Pigs. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1446–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forner, L.; Larsen, T.; Kilian, M.; Holmstrup, P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J. Clin. Periodontol. 2006, 33, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, P.B.; Brennan, M.T.; Sasser, H.C.; Fox, P.C.; Paster, B.J.; Bahrani-Mougeot, F.K. Bacteremia associated with toothbrushing and dental extraction. Circulation 2008, 117, 3118–3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosales, C.; Uribe-Querol, E. Neutrophil Role in Periodontal Disease; Khajah, M.A., Ed.; IntechOpen: London, UK, 2017; p. 67. [Google Scholar]
- Groeger, S.; Meyle, J. Oral Mucosal Epithelial Cells. Front. Immunol. 2019, 10, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosshardt, D.D.; Lang, N.P. The junctional epithelium: From health to disease. J. Dent. Res. 2005, 84, 9–20. [Google Scholar] [CrossRef]
- Lasserre, J.F.; Brecx, M.C.; Toma, S. Oral Microbes, Biofilms and Their Role in Periodontal and Peri-Implant Diseases. Materials 2018, 11, 1802. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.P.; Mullany, P. Oral biofilms: A reservoir of transferable, bacterial, antimicrobial resistance. Expert Rev. Anti-Infect. Ther. 2010, 8, 1441–1450. [Google Scholar] [CrossRef]
- Sharma, N.; Bhatia, S.; Sodhi, A.S.; Batra, N. Oral microbiome and health. AIMS Microbiol. 2018, 4, 42–66. [Google Scholar] [CrossRef]
- Yoneyama, T.; Yoshida, M.; Ohrui, T.; Mukaiyama, H.; Okamoto, H.; Hoshiba, K.; Ihara, S.; Yanagisawa, S.; Ariumi, S.; Morita, T. Oral care reduces pneumonia in older patients in nursing homes. J. Am. Geriatr. Soc. 2002, 50, 430–433. [Google Scholar] [CrossRef]
- Paju, S.; Scannapieco, F.A. Oral biofilms, periodontitis, and pulmonary infections. Oral Dis. 2007, 13, 508–512. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.D. The Micro-Organisms of the Human Mouth; S.S. White Dental Manufacturing Co.: Philadelphia, PA, USA, 1890. [Google Scholar]
- He, X.S.; Shi, W.Y. Oral Microbiology: Past, Present and Future. Int. J. Oral Sci. 2009, 1, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.D. The human mouth as a focus of infection. Lancet 1891, 138, 340–342. [Google Scholar] [CrossRef] [Green Version]
- Hunter, W. Oral Sepsis as a Cause of Disease. Br. Med. J. 1900, 2, 215–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billings, F. Chronic focal infections and their etiologic relations to arthritis and nephritis. JAMA Intern. Med. 1912, 9, 484–498. [Google Scholar] [CrossRef] [Green Version]
- Wessely, S. Surgery for the treatment of psychiatric illness: The need to test untested theories. J. R. Soc. Med. 2009, 102, 445–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimann, H.A.; Havens, W.P. Focal infection and systemic disease: A critical appraisal: The case against indiscriminate removal of teeth and tonsils clinical lecture at St. Louis session. J. Am. Med. Assoc. 1940, 114, 1–6. [Google Scholar] [CrossRef]
- Gutmann, J.L. Focal Infection Revisited—The Swinging of the Pendulum. Dent. Hist. 2017, 62, 81–92. [Google Scholar]
- Potgieter, M.; Bester, J.; Kell, D.B.; Pretorius, E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol. Rev. 2015, 39, 567–591. [Google Scholar] [CrossRef] [Green Version]
- Païssé, S.; Valle, C.; Servant, F.; Courtney, M.; Burcelin, R.; Amar, J.; Lelouvier, B.J.T. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 2016, 56, 1138–1147. [Google Scholar] [CrossRef]
- Whittle, E.; Leonard, M.O.; Harrison, R.; Gant, T.W.; Tonge, D.P. Multi-Method Characterization of the Human Circulating Microbiome. Front. Microbiol. 2019, 9, 3266. [Google Scholar] [CrossRef] [Green Version]
- Dickson, R.P.; Huffnagle, G.B. The lung microbiome: New principles for respiratory bacteriology in health and disease. PLoS Pathog. 2015, 11, e1004923. [Google Scholar] [CrossRef] [PubMed]
- Man, W.H.; de Steenhuijsen Piters, W.A.A.; Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell 2019, 176, 998–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elwishahy, A.; Antia, K.; Bhusari, S.; Ilechukwu, N.C.; Horstick, O.; Winkler, V. Porphyromonas Gingivalis as a Risk Factor to Alzheimer’s Disease: A Systematic Review. J. Alzheimers Dis. Rep. 2021, 5, 721–732. [Google Scholar] [CrossRef]
- Nara, P.L.; Sindelar, D.; Penn, M.S.; Potempa, J.; Griffin, W.S.T. Porphyromonas gingivalis Outer Membrane Vesicles as the Major Driver of and Explanation for Neuropathogenesis, the Cholinergic Hypothesis, Iron Dyshomeostasis, and Salivary Lactoferrin in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 82, 1417–1450. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, D.; Liu, S.; Zhang, S.; Pan, Y. The Role of Porphyromonas gingivalis Outer Membrane Vesicles in Periodontal Disease and Related Systemic Diseases. Front. Cell. Infect. Microbiol. 2021, 10, 585917. [Google Scholar] [CrossRef]
- Patel, S.; Howard, D.; Chowdhury, N.; Derieux, C.; Wellslager, B.; Yilmaz, Ö.; French, L. Characterization of Human Genes Modulated by Porphyromonas gingivalis Highlights the Ribosome, Hypothalamus, and Cholinergic Neurons. Front. Immunol. 2021, 12, 2165. [Google Scholar] [CrossRef]
- Fardini, Y.; Chung, P.; Dumm, R.; Joshi, N.; Han, Y.W. Transmission of diverse oral bacteria to murine placenta: Evidence for the oral microbiome as a potential source of intrauterine infection. Infect. Immun. 2010, 78, 1789–1796. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.S. From focal sepsis to periodontal medicine: A century of exploring the role of the oral microbiome in systemic disease. J. Physiol. 2017, 595, 465–476. [Google Scholar] [CrossRef]
- Kleinstein, S.; Nelson, K.; Freire, M. Inflammatory networks linking oral microbiome with systemic health and disease. J. Dent. Res. 2020, 99, 1131–1139. [Google Scholar] [CrossRef]
- Kartal, E.; Schmidt, T.S.B.; Molina-Montes, E.; Rodríguez-Perales, S.; Wirbel, J.; Maistrenko, O.M.; Akanni, W.A.; Alashkar Alhamwe, B.; Alves, R.J.; Carrato, A.; et al. A faecal microbiota signature with high specificity for pancreatic cancer. Gut 2022. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, Q.; Fu, X. Fusobacterium nucleatum Contributes to the Carcinogenesis of Colorectal Cancer by Inducing Inflammation and Suppressing Host Immunity. Transl. Oncol. 2019, 12, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Casasanta, M.A.; Yoo, C.C.; Udayasuryan, B.; Sanders, B.E.; Umaña, A.; Zhang, Y.; Peng, H.; Duncan, A.J.; Wang, Y.; Li, L.; et al. Fusobacterium nucleatum host-cell binding and invasion induces IL-8 and CXCL1 secretion that drives colorectal cancer cell migration. Sci. Signal. 2020, 13, eaba9157. [Google Scholar] [CrossRef] [PubMed]
- Loftus, M.; Hassouneh, S.A.-D.; Yooseph, S. Bacterial community structure alterations within the colorectal cancer gut microbiome. BMC Microbiol. 2021, 21, 98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, C.; Zhang, Z.; Li, Y.; Li, Q.; Geng, F.; Liu, J.; Pan, Y. Analysis of differentially expressed genes in oral epithelial cells infected with Fusobacterium nucleatum for revealing genes associated with oral cancer. J. Cell. Mol. Med. 2021, 25, 892–904. [Google Scholar] [CrossRef]
- Han, Y.W.; Redline, R.W.; Li, M.; Yin, L.; Hill, G.B.; McCormick, T.S. Fusobacterium nucleatum induces premature and term stillbirths in pregnant mice: Implication of oral bacteria in preterm birth. Infect. Immun. 2004, 72, 2272–2279. [Google Scholar] [CrossRef] [Green Version]
- Vander Haar, E.L.; So, J.; Gyamfi-Bannerman, C.; Han, Y.W. Fusobacterium nucleatum and adverse pregnancy outcomes: Epidemiological and mechanistic evidence. Anaerobe 2018, 50, 55–59. [Google Scholar] [CrossRef]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2018, 67, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Okumura, S.; Konishi, Y.; Narukawa, M.; Sugiura, Y.; Yoshimoto, S.; Arai, Y.; Sato, S.; Yoshida, Y.; Tsuji, S.; Uemura, K.; et al. Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat. Commun. 2021, 12, 5674. [Google Scholar] [CrossRef]
- Wang, X.; Jia, Y.; Wen, L.; Mu, W.; Wu, X.; Liu, T.; Liu, X.; Fang, J.; Luan, Y.; Chen, P.; et al. Porphyromonas gingivalis Promotes Colorectal Carcinoma by Activating the Hematopoietic NLRP3 Inflammasome. Cancer Res. 2021, 81, 2745–2759. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, Z.; Tang, Z.; Huang, Y.; Huang, M.; Liu, H.; Ziebolz, D.; Schmalz, G.; Jia, B.; Zhao, J. More Than Just a Periodontal Pathogen–the Research Progress on Fusobacterium nucleatum. Front. Cell. Infect. Microbiol. 2022, 12, 64. [Google Scholar] [CrossRef]
- Mohammed, H.; Varoni, E.; Cochis, A.; Cordaro, M.; Gallenzi, P.; Patini, R.; Staderini, E.; Lajolo, C.; Rimondini, L.; Rocchetti, V. Oral Dysbiosis in Pancreatic Cancer and Liver Cirrhosis: A Review of the Literature. Biomedicines 2018, 6, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flak, M.B.; Colas, R.A.; Muñoz-Atienza, E.; Curtis, M.A.; Dalli, J.; Pitzalis, C. Inflammatory arthritis disrupts gut resolution mechanisms, promoting barrier breakdown by Porphyromonas gingivalis. JCI Insight 2019, 4, e125191. [Google Scholar] [CrossRef]
- Reyes, L. Porphyromonas gingivalis. Trends Microbiol. 2021, 29, 376–377. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qi, J.; Zhao, H.; He, S.; Zhang, Y.; Wei, S.; Zhao, F. Metagenomic sequencing reveals microbiota and its functional potential associated with periodontal disease. Sci. Rep. 2013, 3, 1843. [Google Scholar] [CrossRef]
- Padmalatha, G.; Bavle, R.; Satyakiran, G.; Paremala, K.; Sudhakara, M.; Makarla, S. Quantification of Porphyromonas gingivalis in chronic periodontitis patients associated with diabetes mellitus using real-time polymerase chain reaction. J. Oral Maxillofac. Pathol. 2016, 20, 413–418. [Google Scholar] [CrossRef]
- Matsha, T.E.; Prince, Y.; Davids, S.; Chikte, U.; Erasmus, R.T.; Kengne, A.P.; Davison, G.M. Oral Microbiome Signatures in Diabetes Mellitus and Periodontal Disease. J. Dent. Res. 2020, 99, 658–665. [Google Scholar] [CrossRef]
- Wu, J.S.; Zheng, M.; Zhang, M.; Pang, X.; Li, L.; Wang, S.S.; Yang, X.; Wu, J.B.; Tang, Y.J.; Tang, Y.L.; et al. Porphyromonas gingivalis Promotes 4-Nitroquinoline-1-Oxide-Induced Oral Carcinogenesis With an Alteration of Fatty Acid Metabolism. Front. Microbiol. 2018, 9, 2081. [Google Scholar] [CrossRef]
- Wen, L.; Mu, W.; Lu, H.; Wang, X.; Fang, J.; Jia, Y.; Li, Q.; Wang, D.; Wen, S.; Guo, J.; et al. Porphyromonas gingivalis Promotes Oral Squamous Cell Carcinoma Progression in an Immune Microenvironment. J. Dent. Res. 2020, 99, 666–675. [Google Scholar] [CrossRef]
- Riviere, G.R.; Riviere, K.; Smith, K. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol. Immunol. 2002, 17, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Immunomicrobial pathogenesis of periodontitis: Keystones, pathobionts, and host response. Trends Immunol. 2014, 35, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Lamont, R.J. Dancing with the stars: How choreographed bacterial interactions dictate nososymbiocity and give rise to keystone pathogens, accessory pathogens, and pathobionts. Trends Microbiol. 2016, 24, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.J.F.; Araújo, I.D.T.d.; Alves, L.d.R.; Silva, R.L.d.; Calderon, P.d.S.; Borges, B.C.D.; Martins, A.R.L.d.A.; Gurgel, B.C.d.V.; Lins, R.D.A.U. Relationship of Porphyromonas gingivalis and Alzheimer’s disease: A systematic review of pre-clinical studies. Clin. Oral Investig. 2021, 25, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Mougeot, J.L.C.; Stevens, C.B.; Paster, B.J.; Brennan, M.T.; Lockhart, P.B.; Mougeot, F.K.B. Porphyromonas gingivalis is the most abundant species detected in coronary and femoral arteries. J. Oral Microbiol. 2017, 9, 1281562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slocum, C.; Kramer, C.; Genco, C. Immune dysregulation mediated by the oral microbiome: Potential link to chronic inflammation and atherosclerosis. J. Intern. Med. 2016, 280, 114–128. [Google Scholar] [CrossRef]
- Farrugia, C.; Stafford, G.P.; Potempa, J.; Wilkinson, R.N.; Chen, Y.; Murdoch, C.; Widziolek, M. Mechanisms of vascular damage by systemic dissemination of the oral pathogen Porphyromonas gingivalis. FEBS J. 2021, 288, 1479–1495. [Google Scholar] [CrossRef]
- Lee, K.; Roberts, J.S.; Choi, C.H.; Atanasova, K.R.; Yilmaz, Ö. Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence 2018, 9, 845–859. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Chowdhury, N.; Roberts, J.S.; Yilmaz, Ö. Host surface ectonucleotidase-CD73 and the opportunistic pathogen, Porphyromonas gingivalis, cross-modulation underlies a new homeostatic mechanism for chronic bacterial survival in human epithelial cells. Virulence 2020, 11, 414–429. [Google Scholar] [CrossRef]
- Lee, J.S.; Spooner, R.; Chowdhury, N.; Pandey, V.; Wellslager, B.; Atanasova, K.R.; Evans, Z.; Yilmaz, Ö. In Situ Intraepithelial Localizations of Opportunistic Pathogens, Porphyromonas gingivalis and Filifactor alocis, in Human Gingiva. Curr. Res. Microb. Sci. 2020, 1, 7–17. [Google Scholar] [CrossRef]
- Dong, J.; Li, Y.; Xiao, H.; Zhang, S.; Wang, B.; Wang, H.; Li, Y.; Fan, S.; Cui, M. Oral microbiota affects the efficacy and prognosis of radiotherapy for colorectal cancer in mouse models. Cell Rep. 2021, 37, 109866. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, G.; Guiglia, R.; Russo, L.L.; Campisi, G. Dentistry and internal medicine: From the focal infection theory to the periodontal medicine concept. Eur. J. Intern. Med. 2010, 21, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.D.; Papapanou, P.N.; Philips, K.H.; Offenbacher, S. Periodontal Medicine: 100 Years of Progress. J. Dent. Res. 2019, 98, 1053–1062. [Google Scholar] [CrossRef]
- Chou, H.H.; Yumoto, H.; Davey, M.; Takahashi, Y.; Miyamoto, T.; Gibson, F.C., 3rd; Genco, C.A. Porphyromonas gingivalis fimbria-dependent activation of inflammatory genes in human aortic endothelial cells. Infect. Immun. 2005, 73, 5367–5378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalla, E.; Lamster, I.B.; Hofmann, M.A.; Bucciarelli, L.; Jerud, A.P.; Tucker, S.; Lu, Y.; Papapanou, P.N.; Schmidt, A.M. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Yoshida, M.; Umeda, M.; Huang, Y.; Kitajima, S.; Inoue, Y.; Ishikawa, I.; Iwai, T. Extended exposure of lipopolysaccharide fraction from Porphyromonas gingivalis facilitates mononuclear cell adhesion to vascular endothelium via Toll-like receptor-2 dependent mechanism. Atherosclerosis 2008, 196, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kurita-Ochiai, T.; Hashizume, T.; Du, Y.; Oguchi, S.; Yamamoto, M. Aggregatibacter actinomycetemcomitans accelerates atherosclerosis with an increase in atherogenic factors in spontaneously hyperlipidemic mice. FEMS Immunol. Med. Microbiol. 2010, 59, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Tuomainen, A.M.; Jauhiainen, M.; Kovanen, P.T.; Metso, J.; Paju, S.; Pussinen, P.J. Aggregatibacter actinomycetemcomitans induces MMP-9 expression and proatherogenic lipoprotein profile in apoE-deficient mice. Microb. Pathog. 2008, 44, 111–117. [Google Scholar] [CrossRef]
- Gough, P.J.; Gomez, I.G.; Wille, P.T.; Raines, E.W. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J. Clin. Investig. 2006, 116, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Arimatsu, K.; Yamada, H.; Miyazawa, H.; Minagawa, T.; Nakajima, M.; Ryder, M.I.; Gotoh, K.; Motooka, D.; Nakamura, S.; Iida, T.; et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci. Rep. 2014, 4, 4828. [Google Scholar] [CrossRef] [Green Version]
- Blasco-Baque, V.; Garidou, L.; Pomié, C.; Escoula, Q.; Loubieres, P.; Le Gall-David, S.; Lemaitre, M.; Nicolas, S.; Klopp, P.; Waget, A.; et al. Periodontitis induced by Porphyromonas gingivalis drives periodontal microbiota dysbiosis and insulin resistance via an impaired adaptive immune response. Gut 2017, 66, 872–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Liu, C.; Zheng, X.; Jia, X.; Peng, X.; Yang, R.; Zhou, X.; Xu, X. Porphyromonas gingivalis Induces Insulin Resistance by Increasing BCAA Levels in Mice. J. Dent. Res. 2020, 99, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, Z.; Nakanishi, Y.; Ni, J.; Hayashi, Y.; Takayama, F.; Zhou, Y.; Kadowaki, T.; Nakanishi, H. Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci. Rep. 2017, 7, 11759. [Google Scholar] [CrossRef] [PubMed]
- Memedovski, Z.; Czerwonka, E.; Han, J.; Mayer, J.; Luce, M.; Klemm, L.C.; Hall, M.L.; Mayer, A.M.S. Classical and Alternative Activation of Rat Microglia Treated with Ultrapure Porphyromonas gingivalis Lipopolysaccharide In Vitro. Toxins 2020, 12, 333. [Google Scholar] [CrossRef] [PubMed]
- Poole, S.; Singhrao, S.K.; Chukkapalli, S.; Rivera, M.; Velsko, I.; Kesavalu, L.; Crean, S. Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE-/- mice brains. J. Alzheimers Dis. 2015, 43, 67–80. [Google Scholar] [CrossRef] [Green Version]
- Ishida, N.; Ishihara, Y.; Ishida, K.; Tada, H.; Funaki-Kato, Y.; Hagiwara, M.; Ferdous, T.; Abdullah, M.; Mitani, A.; Michikawa, M.; et al. Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech. Dis. 2017, 3, 15. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Ni, J.; Liu, Y.; Teeling, J.L.; Takayama, F.; Collcutt, A.; Ibbett, P.; Nakanishi, H. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav. Immun. 2017, 65, 350–361. [Google Scholar] [CrossRef]
- Leira, Y.; Iglesias-Rey, R.; Gómez-Lado, N.; Aguiar, P.; Campos, F.; D’Aiuto, F.; Castillo, J.; Blanco, J.; Sobrino, T. Porphyromonas gingivalis lipopolysaccharide-induced periodontitis and serum amyloid-beta peptides. Arch. Oral Biol. 2019, 99, 120–125. [Google Scholar] [CrossRef]
- Tang, Z.; Liang, D.; Cheng, M.; Su, X.; Liu, R.; Zhang, Y.; Wu, H. Effects of Porphyromonas gingivalis and Its Underlying Mechanisms on Alzheimer-Like Tau Hyperphosphorylation in Sprague-Dawley Rats. J. Mol. Neurosci. 2021, 71, 89–100. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Metabolic Inflammation and Insulin Resistance in Obesity. Circ. Res. 2020, 126, 1549–1564. [Google Scholar] [CrossRef]
- Sharma, S.; Tripathi, P. Gut microbiome and type 2 diabetes: Where we are and where to go? J. Nutr. Biochem. 2019, 63, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Javed, F.; Warnakulasuriya, S. Is there a relationship between periodontal disease and oral cancer? A systematic review of currently available evidence. Crit. Rev. Oncol. Hematol. 2016, 97, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.Y.; Wong, T.Y.; Chiang, W.F.; Chen, Y.L. Fatty-acid-binding protein 5 promotes cell proliferation and invasion in oral squamous cell carcinoma. J. Oral Pathol. Med. 2010, 39, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Wada, K.; Taniguchi, Y.; Al-Shareef, H.; Masuda, T.; Usami, Y.; Aikawa, T.; Okura, M.; Kamisaki, Y.; Kogo, M. Expression of fatty acid binding protein 4 is involved in the cell growth of oral squamous cell carcinoma. Oncol. Rep. 2014, 31, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.Q.; Hu, X.W.; Liu, Y.L.; Ye, Z.J.; Gui, Y.H.; Zhou, D.L.; Qi, C.L.; He, X.D.; Wang, H.; Wang, L.J. CD11b deficiency suppresses intestinal tumor growth by reducing myeloid cell recruitment. Sci. Rep. 2015, 5, 15948. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Epithelium | Skin | Oral | Intestinal | |
|---|---|---|---|---|
| KERATINIZED TISSUE | Exist | Partially exist | Not exist | |
| EPITHELIAL LAYER | Multiple layers | Multiple layers | Single layer | |
| INTERCELLULAR JUNCTIONS | Tight junction | Exist | Exist | Exist |
| Adherence junction | Exist | Exist | Exist | |
| Desmosome | Exist | Exist | Exist | |
| Gap junction | Exist | Exist | Exist | |
| Hemidesomosome | Not exist | Exist | Not exist | |
| Oral Pathogens | Models | Infection Methods | Experimental Results | Year | Ref. | ||
|---|---|---|---|---|---|---|---|
| Atherosclerotic CVD | In vitro | P. gingivalis 381 | HAECs | 6 h infection | Increased ICAM-1, VCAM-1, and IL-6 expression | 2005 | [128] |
| P. gingivalis ATCC33277-driven PgLPS | HUVECs | 24 h infection | Increased adhesion of mononuclear cells to HUVECs via ICAM-1 and TLR-2 dependent mechanism. | 2008 | [130] | ||
| In vivo | P. gingivalis 381 | Apoe−/− mice
| Oral infection 5 times per week over 3 weeks | Increased aortic atherosclerosis. | 2003 | [129] | |
| P. gingivalis 381 | Apoe−/− mice
| Oral infection 5 times per week over 3 weeks | Increased aortic ICAM-1, VCAM-1 immunostaining. | 2005 | [128] | ||
| P. gingivalis 381 or A7436 | Pigs
| Subcutaneously infection 3 times per week for 5 months | Increased aortic and coronary arterial atherosclerosis. | 2005 | [63] | ||
| A. actinomycetemcomitans AT445b | Apoe−/− mice
| Intravenous infection once a week for 4, 6, or 8 weeks | Increased aortic MMP-9 expression and serum CRP. | 2008 | [132] | ||
| A. actinomycetemcomitans HK1651 | Apoeshl mice
| Intravenous infection 3 times per week over 3 weeks | Increased atherosclerotic plaque, serum C-reactive protein (CRP), IL-6, and aortic ICAM-1. | 2014 | [134] | ||
| T2DM | In vivo | P. gingivalis W83 | Mice | Oral infection twice per week for 5 weeks | Increased gut dysbiosis, gut barrier invasion, serum endotoxin, insulin resistance. | 2014 | [134] |
| P. gingivalis ATCC33277, F. nucleatum, P. intermedia | Mice
| Oral infection 4 times a week for 4 weeks, thereafter normal diet or HFD-fed for additional 3 months | Increased periodontal dysbiosis, insulin resistance in HFD-fed mice. | 2017 | [135] | ||
| P. gingivalis ATCC33277 (WT) or ∆bcat | Mice
| Oral infection twice per week for 4 weeks concomitantly HFD-fed | P. gingivalis (∆bcat) cannot induce insulin resistance in HFD-fed mice. | 2020 | [136] | ||
| OSCC | In vivo | P. gingivalis 381 | Mice
| 4NQO treatment for 8 weeks, thereafter oral infection with P. gingivalis for 8 weeks | Enhanced OSCC induction and dysregulated lipid metabolism in 4NQO-treated mice. | 2018 | [113] |
| P. gingivalis ATCC33277 | Mice
| 4NQO treatment for 16 weeks, thereafter oral infection with P. gingivalis for 10 weeks | Enhanced OSCC induction and increased infiltration of CD11b+ MDSCs in 4NQO-treated mice. | 2020 | [114] | ||
| AD | In vitro | P. gingivalis ATCC33277 | Immortalized mouse microglial cell line MG6 | 3, 6, or 12 h infection of P. gingivalis in the presence and absence of KYT1 (Rgp inhibitor) and KYT36 (Kgp inhibitor) | Increased expression levels of IL-6 and TNF-α, which was inhibited by KYT1 and KYT36 treatment. | 2017 | [137] |
| PgLPS | Rat brain neonatal microglia | 18 h infection | Activated microglial release of cytokine TNF-α, IL-6, and MMP-9. | 2020 | [138] | ||
| In vivo | P. gingivalis 381, Treponema denticola ATCC 35404, Tannerella forsythia ATCC 43037, and F. nucleatum ATCC 49256 | Apoe−/− mice
| Oral infection for 24 weeks | P. gingivalis genomic DNA was detected in mice brain (9 out of 12 at 24 weeks). | 2015 | [139] | |
| P. gingivalis ATCC33277 | APP transgenic mice
| Gingival infection | Exacerbated Aβ plaques and inflammatory cytokines in the brain of AD mouse model. | 2017 | [140] | ||
| PgLPS | Mice
| Intraperitoneal infection daily for 5 weeks | PgLPS induced learning and memory deficit in middle-aged WT mice, but not in young WT, young Catb−/−, and middle-aged Catb−/− mice. | 2017 | [141] | ||
| P. gingivalis ATCC33277, Kgp-deficient P. gingivalis KDP129 | Cx3cr1+/GFP mice | Injection of P. gingivalis into the somatosensory cortex of mice | GFP+ microglia accumulated around the injection site of P. gingivalis, but not of KDP129. | 2017 | [137] | ||
| PgLPS | Rats (n = 6) | Palatal gingival infection 3 times for 2 weeks | Induced alveolar bone loss and increased serum Aβ levels. | 2019 | [137,142] | ||
| P. gingivalis ATCC33277 | Rats
| Intravenous infection 3 times a week for 4 or 12 weeks | Induced tau hyperphosphorylation (pTau181 and pTau231) in the rat hippocampus. | 2021 | [143] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, D.-Y.; Park, J.Y.; Lee, D.; Hwang, I.; Kim, H.-S. Leaky Gum: The Revisited Origin of Systemic Diseases. Cells 2022, 11, 1079. https://doi.org/10.3390/cells11071079
Park D-Y, Park JY, Lee D, Hwang I, Kim H-S. Leaky Gum: The Revisited Origin of Systemic Diseases. Cells. 2022; 11(7):1079. https://doi.org/10.3390/cells11071079
Chicago/Turabian StylePark, Do-Young, Jin Young Park, Dahye Lee, Inseong Hwang, and Hye-Sung Kim. 2022. "Leaky Gum: The Revisited Origin of Systemic Diseases" Cells 11, no. 7: 1079. https://doi.org/10.3390/cells11071079
APA StylePark, D.-Y., Park, J. Y., Lee, D., Hwang, I., & Kim, H.-S. (2022). Leaky Gum: The Revisited Origin of Systemic Diseases. Cells, 11(7), 1079. https://doi.org/10.3390/cells11071079