
Lysophosphatidic Acid Receptor 5 (LPA5) Knockout Ameliorates the Neuroinflammatory Response In Vivo and Modifies the Inflammatory and Metabolic Landscape of Primary Microglia In Vitro

 , ,
, ,  , , ,
, , ,
Abstract
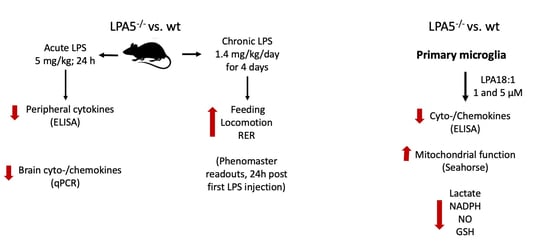
:
1. Introduction
2. Materials and Methods
2.1. Animals
- GFP Int Rev, 5-GTGGTGCAGATGAACTTCAGG- 3;
- 92GTFor, 5-CAGAGTCTGTATTGCCACCAG- 3; and
- 92GT Rev, 5-GTCCACGTTGATGAGCATCAG- 3.
2.2. Indirect Calorimetry (Metabolic Cage Monitoring)
2.3. RT-qPCR Analysis
2.4. Primary Microglia Cultures
2.5. LPA Treatment
2.6. Seahorse XF Analyzer Respiratory Assay
2.7. ELISA
2.8. Lactate Measurement
2.9. NADPH/NADP Assay
2.10. Glutathione Assay
2.11. Determination of Nitric Oxide (NO)
2.12. Statistical Analysis
3. Results
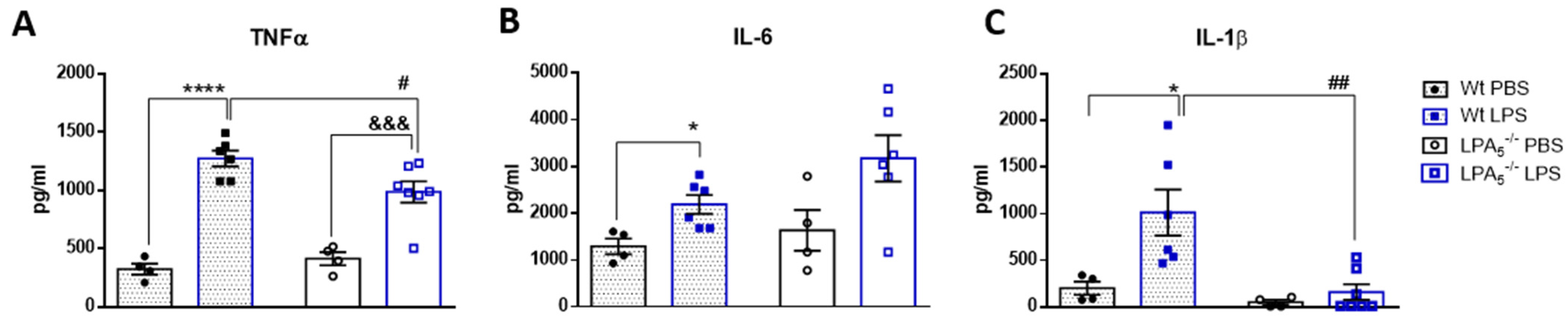
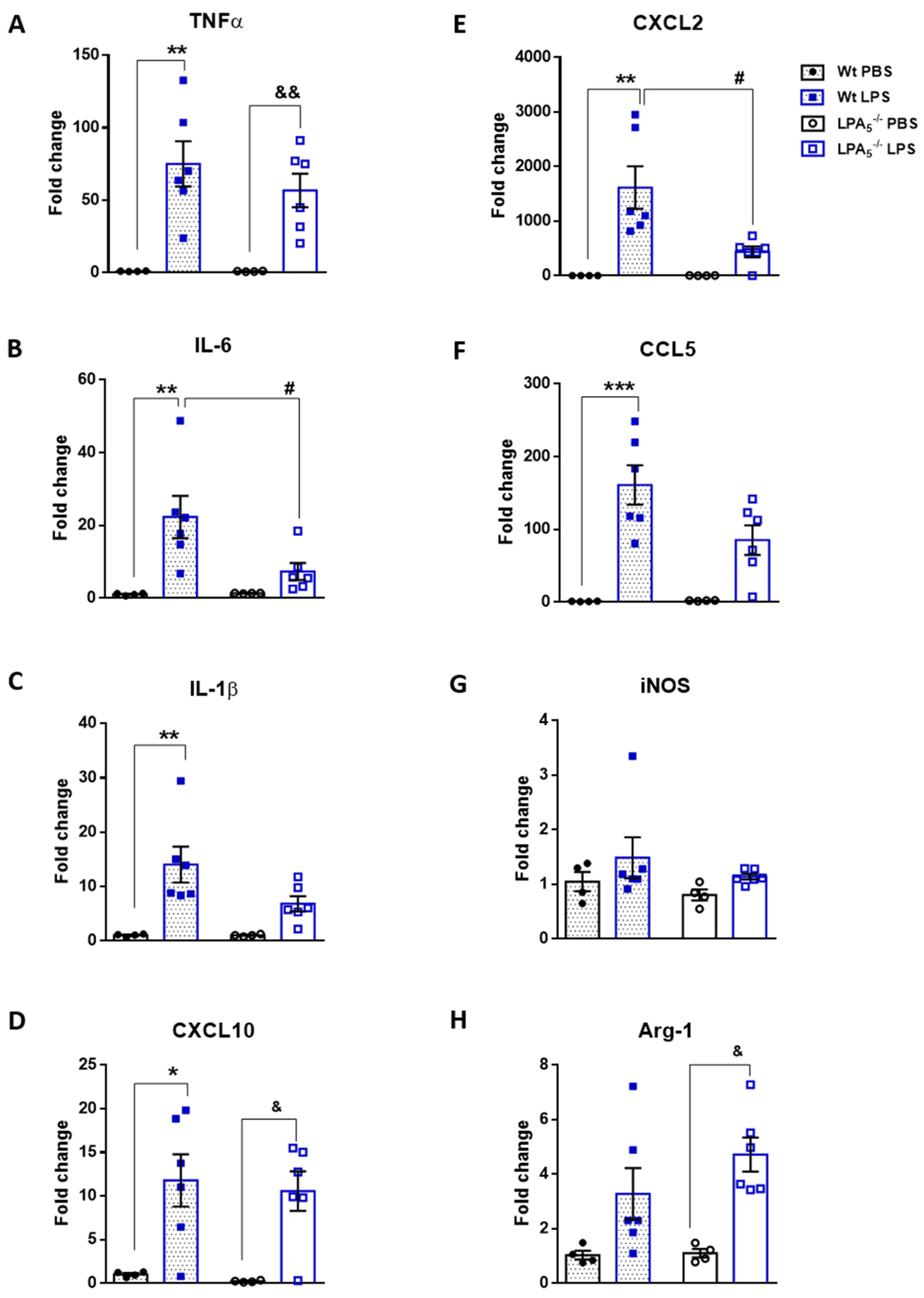
3.1. Endotoxemia Is Reduced in LPA5-/- Mice
3.2. Improved Metabolic Performance of LPA5-/- Mice after Short-Term, Low-Dose LPS Treatment
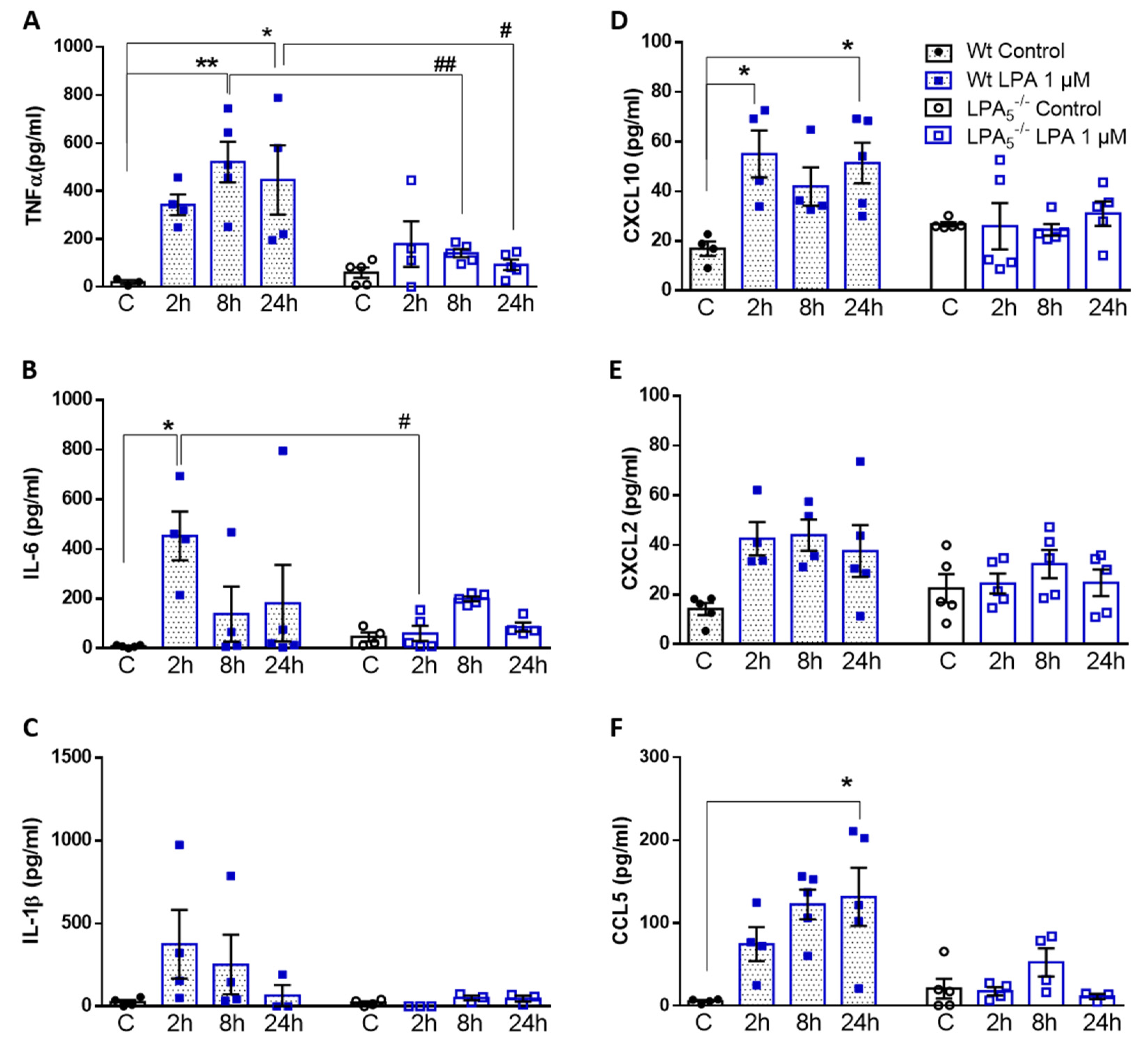
3.3. LPA5 Regulates LPA-Induced Secretion of Cyto-/Chemokines in Primary Microglia
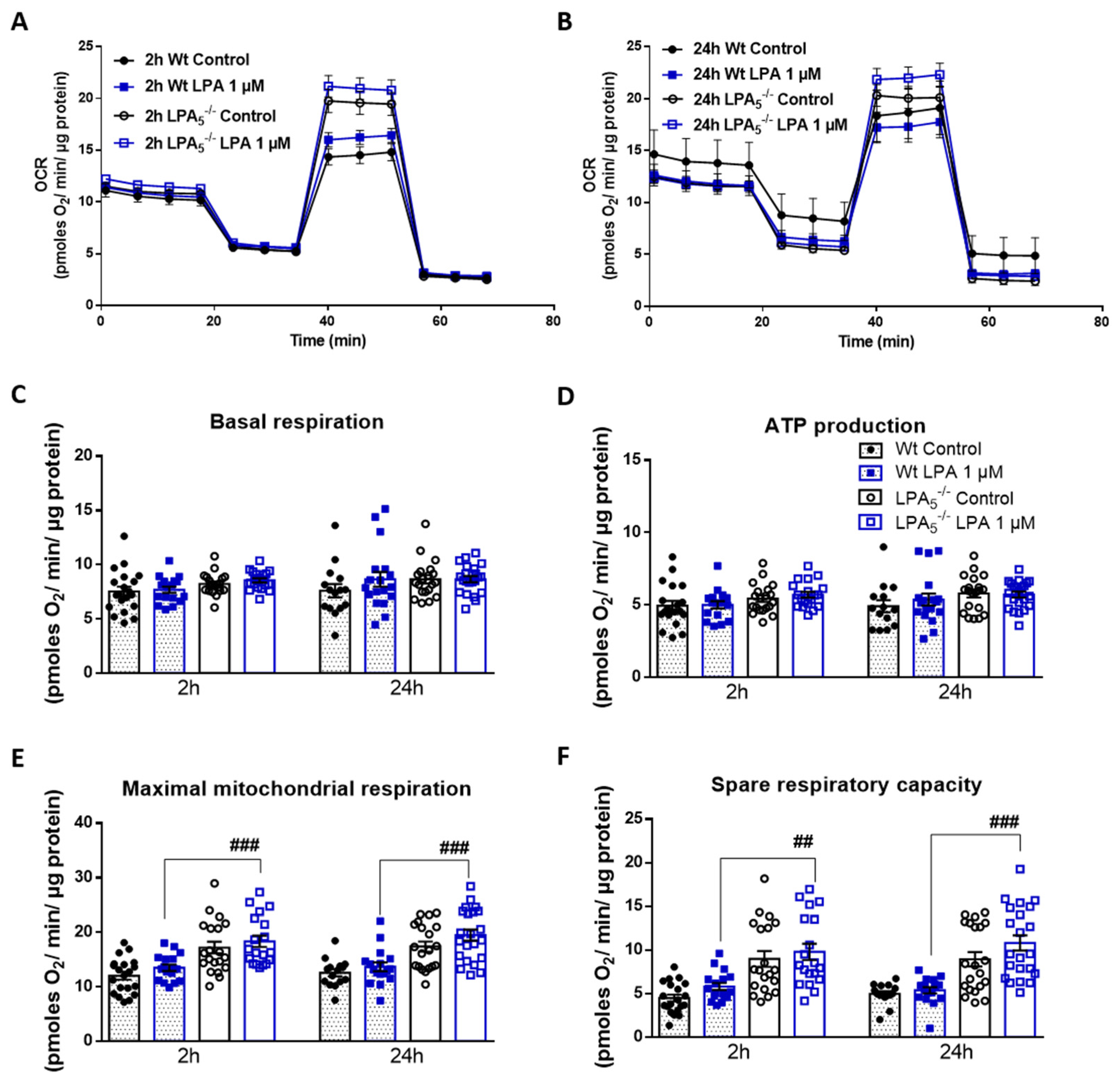
3.4. LPA5-/- Microglia Have Higher Mitochondrial Capacity as Compared to wt Cells
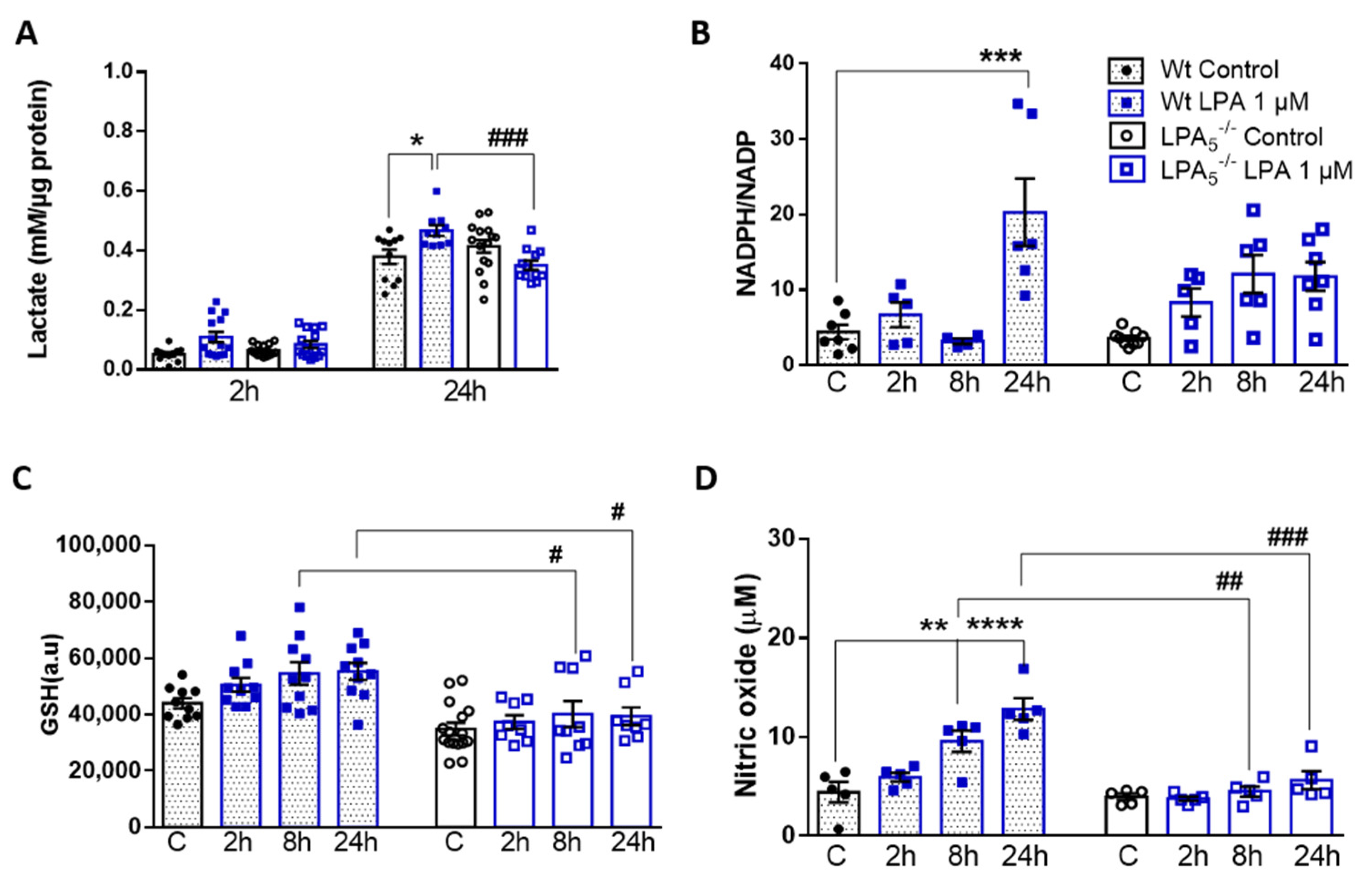
3.5. LPA5 Deletion Attenuates LPA-Induced Lactate, NADPH, GSH, and NO Synthesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATX | Autotaxin |
| BrP-LPA | Bromophosphonate lysophosphatidic acid |
| CNS | Central nervous system |
| DMSO | Dimethyl sulfoxide |
| GSH | Reduced glutathione |
| iNOS | Inducible nitric oxide synthase (NOS2) |
| LPA | Lysophosphatidic acid |
| LPA1-6 | Lysophosphatidic acid receptors 1-6 |
| LPS | Lipopolysaccharide |
| NADPH | Nicotinamide adenine dinucleotide phosphate, reduced |
| NO | Nitric oxide |
| OCR | Oxygen consumption rate |
| PBS | Phosphate buffered saline |
| PPP | Pentose phosphate pathway |
| TLR | Toll-like receptor |
| tMCAO | Transient middle cerebral artery occlusion |
| Wt | Wildtype |
References
- Dawson, G. Measuring brain lipids. Biochim. Biophys. Acta 2015, 1851, 1026–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozek, K.; Wei, Y.; Yan, Z.; Liu, X.; Xiong, J.; Sugimoto, M.; Tomita, M.; Paabo, S.; Sherwood, C.C.; Hof, P.R.; et al. Organization and evolution of brain lipidome revealed by large-scale analysis of human, chimpanzee, macaque, and mouse tissues. Neuron 2015, 85, 695–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, J. Lysophospholipid receptors: Implications for neural signaling. Crit. Rev. Neurobiol. 1999, 13, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Ishii, I.; Fukushima, N.; Ye, X.; Chun, J. Lysophospholipid receptors: Signaling and biology. Annu. Rev. Biochem. 2004, 73, 321–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kihara, Y.; Maceyka, M.; Spiegel, S.; Chun, J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol. 2014, 171, 3575–3594. [Google Scholar] [CrossRef] [Green Version]
- Yung, Y.C.; Stoddard, N.C.; Mirendil, H.; Chun, J. Lysophosphatidic Acid signaling in the nervous system. Neuron 2015, 85, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef] [Green Version]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid. Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [Green Version]
- Czirr, E.; Wyss-Coray, T. The immunology of neurodegeneration. J. Clin. Investig. 2012, 122, 1156–1163. [Google Scholar] [CrossRef]
- Labzin, L.I.; Heneka, M.T.; Latz, E. Innate Immunity and Neurodegeneration. Annu. Rev. Med. 2018, 69, 437–449. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, T.; Contos, J.J.; Musante, D.B.; Chun, J.; Ransom, B.R. Expression and function of lysophosphatidic acid receptors in cultured rodent microglial cells. J. Biol. Chem. 2001, 276, 25946–25952. [Google Scholar] [CrossRef] [Green Version]
- Abels, E.R.; Nieland, L.; Hickman, S.; Broekman, M.L.D.; El Khoury, J.; Maas, S.L.N. Comparative Analysis Identifies Similarities between the Human and Murine Microglial Sensomes. Int. J. Mol. Sci. 2021, 22, 1495. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Rivera, R.; Gardell, S.; Dubin, A.E.; Chun, J. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J. Biol. Chem. 2006, 281, 23589–23597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasternack, S.M.; von Kugelgen, I.; Al Aboud, K.; Lee, Y.A.; Ruschendorf, F.; Voss, K.; Hillmer, A.M.; Molderings, G.J.; Franz, T.; Ramirez, A.; et al. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat. Genet. 2008, 40, 329–334. [Google Scholar] [CrossRef]
- Yanagida, K.; Masago, K.; Nakanishi, H.; Kihara, Y.; Hamano, F.; Tajima, Y.; Taguchi, R.; Shimizu, T.; Ishii, S. Identification and characterization of a novel lysophosphatidic acid receptor, p2y5/LPA6. J. Biol. Chem. 2009, 284, 17731–17741. [Google Scholar] [CrossRef] [Green Version]
- Plastira, I.; Bernhart, E.; Goeritzer, M.; Reicher, H.; Kumble, V.B.; Kogelnik, N.; Wintersperger, A.; Hammer, A.; Schlager, S.; Jandl, K.; et al. 1-Oleyl-lysophosphatidic acid (LPA) promotes polarization of BV-2 and primary murine microglia towards an M1-like phenotype. J. Neuroinflamm. 2016, 13, 205. [Google Scholar] [CrossRef] [Green Version]
- Kozian, D.H.; von Haeften, E.; Joho, S.; Czechtizky, W.; Anumala, U.R.; Roux, P.; Dudda, A.; Evers, A.; Nazare, M. Modulation of Hexadecyl-LPA-Mediated Activation of Mast Cells and Microglia by a Chemical Probe for LPA5. Chembiochem. A Eur. J. Chem. Biol. 2016, 17, 861–865. [Google Scholar] [CrossRef]
- Suckau, O.; Gross, I.; Schrotter, S.; Yang, F.; Luo, J.; Wree, A.; Chun, J.; Baska, D.; Baumgart, J.; Kano, K.; et al. LPA1, LPA2, LPA4, and LPA6 receptor expression during mouse brain development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2019, 248, 375–395. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.H.; Gaire, B.P.; Park, S.J.; Shin, D.Y.; Choi, J.W. Identifying lysophosphatidic acid receptor subtype 1 (LPA1) as a novel factor to modulate microglial activation and their TNF-alpha production by activating ERK1/2. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2018, 1863, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, A.; Lee, C.H.; Park, S.J.; Choi, J.W. Lysophosphatidic Acid Receptor 5 Plays a Pathogenic Role in Brain Damage after Focal Cerebral Ischemia by Modulating Neuroinflammatory Responses. Cells 2020, 9, 1446. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, A.; Park, S.J.; Choi, J.W. Inhibition of LPA5 Activity Provides Long-Term Neuroprotection in Mice with Brain Ischemic Stroke. Biomol. Ther. 2020, 28, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, A.; Park, S.J.; Choi, J.W. Receptor for Advanced Glycation End Products Is Involved in LPA5-Mediated Brain Damage after a Transient Ischemic Stroke. Life 2021, 11, 80. [Google Scholar] [CrossRef]
- Lin, M.E.; Rivera, R.R.; Chun, J. Targeted deletion of LPA5 identifies novel roles for lysophosphatidic acid signaling in development of neuropathic pain. J. Biol. Chem. 2012, 287, 17608–17617. [Google Scholar] [CrossRef] [Green Version]
- Callaerts-Vegh, Z.; Leo, S.; Vermaercke, B.; Meert, T.; D’Hooge, R. LPA5 receptor plays a role in pain sensitivity, emotional exploration and reversal learning. Genes Brain Behav. 2012, 11, 1009–1019. [Google Scholar] [CrossRef]
- Murai, N.; Hiyama, H.; Kiso, T.; Sekizawa, T.; Watabiki, T.; Oka, H.; Aoki, T. Analgesic effects of novel lysophosphatidic acid receptor 5 antagonist AS2717638 in rodents. Neuropharmacology 2017, 126, 97–107. [Google Scholar] [CrossRef]
- Kozian, D.H.; Evers, A.; Florian, P.; Wonerow, P.; Joho, S.; Nazare, M. Selective non-lipid modulator of LPA5 activity in human platelets. Bioorg. Med. Chem. Lett. 2012, 22, 5239–5243. [Google Scholar] [CrossRef]
- Plastira, I.; Joshi, L.; Bernhart, E.; Schoene, J.; Specker, E.; Nazare, M.; Sattler, W. Small-Molecule Lysophosphatidic Acid Receptor 5 (LPAR5) Antagonists: Versatile Pharmacological Tools to Regulate Inflammatory Signaling in BV-2 Microglia Cells. Front. Cell. Neurosci. 2019, 13, 531. [Google Scholar] [CrossRef]
- Awada, R.; Saulnier-Blache, J.S.; Gres, S.; Bourdon, E.; Rondeau, P.; Parimisetty, A.; Orihuela, R.; Harry, G.J.; d’Hellencourt, C.L. Autotaxin downregulates LPS-induced microglia activation and pro-inflammatory cytokines production. J. Cell Biochem. 2014, 115, 2123–2132. [Google Scholar] [CrossRef] [Green Version]
- Plastira, I.; Bernhart, E.; Joshi, L.; Koyani, C.N.; Strohmaier, H.; Reicher, H.; Malle, E.; Sattler, W. MAPK signaling determines lysophosphatidic acid (LPA)-induced inflammation in microglia. J. Neuroinflamm. 2020, 17, 127. [Google Scholar] [CrossRef] [PubMed]
- Joshi, L.; Plastira, I.; Bernhart, E.; Reicher, H.; Koyani, C.N.; Madl, T.; Madreiter-Sokolowski, C.; Koshenov, Z.; Graier, W.F.; Hallstrom, S.; et al. Lysophosphatidic Acid Induces Aerobic Glycolysis, Lipogenesis, and Increased Amino Acid Uptake in BV-2 Microglia. Int. J. Mol. Sci. 2021, 22, 1968. [Google Scholar] [CrossRef]
- Xu, F.; Deng, C.; Ren, Z.; Sun, L.; Meng, Y.; Liu, W.; Wan, J.; Chen, G. Lysophosphatidic acid shifts metabolic and transcriptional landscapes to induce a distinct cellular state in human pluripotent stem cells. Cell Rep. 2021, 37, 110063. [Google Scholar] [CrossRef] [PubMed]
- Goeritzer, M.; Bernhart, E.; Plastira, I.; Reicher, H.; Leopold, C.; Eichmann, T.O.; Rechberger, G.; Madreiter-Sokolowski, C.T.; Prasch, J.; Eller, P.; et al. Myeloperoxidase and Septic Conditions Disrupt Sphingolipid Homeostasis in Murine Brain Capillaries In Vivo and Immortalized Human Brain Endothelial Cells In Vitro. Int. J. Mol. Sci. 2020, 21, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Jalabi, W.; Shpargel, K.B.; Farabaugh, K.T.; Dutta, R.; Yin, X.; Kidd, G.J.; Bergmann, C.C.; Stohlman, S.A.; Trapp, B.D. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J. Neurosci. 2012, 32, 11706–11715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, L.; Plastira, I.; Bernhart, E.; Reicher, H.; Triebl, A.; Kofeler, H.C.; Sattler, W. Inhibition of Autotaxin and Lysophosphatidic Acid Receptor 5 Attenuates Neuroinflammation in LPS-Activated BV-2 Microglia and a Mouse Endotoxemia Model. Int. J. Mol. Sci. 2021, 22, 8519. [Google Scholar] [CrossRef]
- Lummis, N.C.; Sánchez-Pavón, P.; Kennedy, G.; Frantz, A.J.; Kihara, Y.; Blaho, V.A.; Chun, J. LPA(1/3) overactivation induces neonatal posthemorrhagic hydrocephalus through ependymal loss and ciliary dysfunction. Sci. Adv. 2019, 5, eaax2011. [Google Scholar] [CrossRef] [Green Version]
- Bernier, L.P.; York, E.M.; MacVicar, B.A. Immunometabolism in the Brain: How Metabolism Shapes Microglial Function. Trends Neurosci. 2020, 43, 854–869. [Google Scholar] [CrossRef]
- Lynch, M.A. Can the emerging field of immunometabolism provide insights into neuroinflammation? Prog. Neurobiol. 2020, 184, 101719. [Google Scholar] [CrossRef]
- Hu, Y.; Mai, W.; Chen, L.; Cao, K.; Zhang, B.; Zhang, Z.; Liu, Y.; Lou, H.; Duan, S.; Gao, Z. mTOR-mediated metabolic reprogramming shapes distinct microglia functions in response to lipopolysaccharide and ATP. Glia 2020, 68, 1031–1045. [Google Scholar] [CrossRef]
- Banks, W.A.; Robinson, S.M. Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav. Immun. 2010, 24, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas-Caraveo, A.; Sayd, A.; Maus, S.R.; Caso, J.R.; Madrigal, J.L.M.; Garcia-Bueno, B.; Leza, J.C. Lipopolysaccharide enters the rat brain by a lipoprotein-mediated transport mechanism in physiological conditions. Sci. Rep. 2017, 7, 13113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.; Hernandes, M.S. Sepsis-Associated Encephalopathy and Blood-Brain Barrier Dysfunction. Inflammation 2021, 44, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. The endotoxin hypothesis of neurodegeneration. J. Neuroinflamm. 2019, 16, 180. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Hopkins, A.M.; Hou, J. The development of modulators for lysophosphatidic acid receptors: A comprehensive review. Bioorg. Chem. 2021, 117, 105386. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Banks, W.A. From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Stamova, B.; Jin, L.W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.J.; Jacob, A.; Cunningham, P.; Hensley, L.; Quigg, R.J. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem. Int. 2008, 52, 447–456. [Google Scholar] [CrossRef] [Green Version]
- Nagyoszi, P.; Wilhelm, I.; Farkas, A.E.; Fazakas, C.; Dung, N.T.; Hasko, J.; Krizbai, I.A. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem. Int. 2010, 57, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.C.D.; Rustenhoven, J.; Park, T.I.; Schweder, P.; Jansson, D.; Heppner, P.A.; O’Carroll, S.J.; Mee, E.W.; Faull, R.L.M.; Curtis, M.; et al. Unique and shared inflammatory profiles of human brain endothelia and pericytes. J. Neuroinflamm. 2018, 15, 138. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Hao, F.; Zhang, F.; Kong, W.; Chun, J.; Xu, X.; Cui, M.Z. CD14 is a key mediator of both lysophosphatidic acid and lipopolysaccharide induction of foam cell formation. J. Biol. Chem. 2017, 292, 14391–14400. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, J.; Wang, Y.; Yang, L.; Han, B.; Zhang, Y.; Bai, Y.; Shen, L.; Li, M.; Jiang, T.; et al. PARP14 inhibits microglial activation via LPAR5 to promote post-stroke functional recovery. Autophagy 2021, 17, 2905–2922. [Google Scholar] [CrossRef]
- Kawamoto, Y.; Seo, R.; Murai, N.; Hiyama, H.; Oka, H. Identification of potent lysophosphatidic acid receptor 5 (LPA5) antagonists as potential analgesic agents. Bioorg. Med. Chem. 2018, 26, 257–265. [Google Scholar] [CrossRef]
- Kittaka, H.; Uchida, K.; Fukuta, N.; Tominaga, M. Lysophosphatidic acid-induced itch is mediated by signalling of LPA(5) receptor, phospholipase D and TRPA1/TRPV1. J. Physiol. 2017, 595, 2681–2698. [Google Scholar] [CrossRef] [Green Version]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Lopez-Collazo, E. Endotoxin tolerance: New mechanisms, molecules and clinical significance. Trends Immunol. 2009, 30, 475–487. [Google Scholar] [CrossRef]
- Wendeln, A.C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Hasler, L.M.; Wild, K.; Skodras, A.; et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e613. [Google Scholar] [CrossRef]
- Bhattarai, S.; Sharma, S.; Ara, H.; Subedi, U.; Sun, G.; Li, C.; Bhuiyan, M.S.; Kevil, C.; Armstrong, W.P.; Minvielle, M.T.; et al. Disrupted Blood-Brain Barrier and Mitochondrial Impairment by Autotaxin-Lysophosphatidic Acid Axis in Postischemic Stroke. J. Am. Heart Assoc. 2021, 10, e021511. [Google Scholar] [CrossRef]
- D’Souza, K.; Nzirorera, C.; Cowie, A.M.; Varghese, G.P.; Trivedi, P.; Eichmann, T.O.; Biswas, D.; Touaibia, M.; Morris, A.J.; Aidinis, V.; et al. Autotaxin-LPA signaling contributes to obesity-induced insulin resistance in muscle and impairs mitochondrial metabolism. J. Lipid Res. 2018, 59, 1805–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federico, L.; Ren, H.; Mueller, P.A.; Wu, T.; Liu, S.; Popovic, J.; Blalock, E.M.; Sunkara, M.; Ovaa, H.; Albers, H.M.; et al. Autotaxin and its product lysophosphatidic acid suppress brown adipose differentiation and promote diet-induced obesity in mice. Mol. Endocrinol. 2012, 26, 786–797. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef]
- Gimeno-Bayon, J.; Lopez-Lopez, A.; Rodriguez, M.J.; Mahy, N. Glucose pathways adaptation supports acquisition of activated microglia phenotype. J. Neurosci. Res. 2014, 92, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Tu, D.; Gao, Y.; Yang, R.; Guan, T.; Hong, J.S.; Gao, H.M. The pentose phosphate pathway regulates chronic neuroinflammation and dopaminergic neurodegeneration. J. Neuroinflamm. 2019, 16, 255. [Google Scholar] [CrossRef] [Green Version]
- Castillo, E.; Mocanu, E.; Uruk, G.; Swanson, R.A. Glucose availability limits microglial nitric oxide production. J. Neurochem. 2021, 159, 1008–1015. [Google Scholar] [CrossRef]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef]
- Xiao, W.; Loscalzo, J. Metabolic Responses to Reductive Stress. Antioxid. Redox Signal. 2020, 32, 1330–1347. [Google Scholar] [CrossRef] [Green Version]
- Muri, J.; Kopf, M. Redox regulation of immunometabolism. Nat. Rev. Immunol. 2021, 21, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Company | Catalogue Number |
| iNOS | Qiagen | QT00100275 |
| HPRT | Qiagen | QT00166768 |
| Gene | Company | Forward/Reverse Primers |
| Arg-1 | Invitrogen | F: TGGCTTGCGAGACGTAGAC R: GCTCAGGTGAATCGGCCTTTT |
| TNFα | Invitrogen | F: ACTTCGGGGTGATCGGTCC R: GGCTACAGGCTTGTCACTCG |
| IL6 | Invitrogen | F: TGTTCTCTGGGAAATCGTGGA R: CAAGTGCATCATCGTTGTTCAT |
| IL1β | Invitrogen | F: CTCTCCACCTCAATGGACAGA R: CGTTGCTTGGTTCTCCTTGT |
| CXCL10 | Invitrogen | F: TTCTGCCTCATCCTGCTG R: AGACATCTCTGCTCATCATTC |
| CXCL2 | Invitrogen | F: AGTGAACTGCGCTGTCAATG R: GCCCTTGAGAGTGGCTATGA |
| CCL5 | Invitrogen | F: GCTGCTTTGCCTACCTCTCC R: TCGAGTGACAAACACGACTGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, L.; Plastira, I.; Bernhart, E.; Reicher, H.; Koshenov, Z.; Graier, W.F.; Vujic, N.; Kratky, D.; Rivera, R.; Chun, J.; et al. Lysophosphatidic Acid Receptor 5 (LPA5) Knockout Ameliorates the Neuroinflammatory Response In Vivo and Modifies the Inflammatory and Metabolic Landscape of Primary Microglia In Vitro. Cells 2022, 11, 1071. https://doi.org/10.3390/cells11071071
Joshi L, Plastira I, Bernhart E, Reicher H, Koshenov Z, Graier WF, Vujic N, Kratky D, Rivera R, Chun J, et al. Lysophosphatidic Acid Receptor 5 (LPA5) Knockout Ameliorates the Neuroinflammatory Response In Vivo and Modifies the Inflammatory and Metabolic Landscape of Primary Microglia In Vitro. Cells. 2022; 11(7):1071. https://doi.org/10.3390/cells11071071
Chicago/Turabian StyleJoshi, Lisha, Ioanna Plastira, Eva Bernhart, Helga Reicher, Zhanat Koshenov, Wolfgang F. Graier, Nemanja Vujic, Dagmar Kratky, Richard Rivera, Jerold Chun, and et al. 2022. "Lysophosphatidic Acid Receptor 5 (LPA5) Knockout Ameliorates the Neuroinflammatory Response In Vivo and Modifies the Inflammatory and Metabolic Landscape of Primary Microglia In Vitro" Cells 11, no. 7: 1071. https://doi.org/10.3390/cells11071071
APA StyleJoshi, L., Plastira, I., Bernhart, E., Reicher, H., Koshenov, Z., Graier, W. F., Vujic, N., Kratky, D., Rivera, R., Chun, J., & Sattler, W. (2022). Lysophosphatidic Acid Receptor 5 (LPA5) Knockout Ameliorates the Neuroinflammatory Response In Vivo and Modifies the Inflammatory and Metabolic Landscape of Primary Microglia In Vitro. Cells, 11(7), 1071. https://doi.org/10.3390/cells11071071