
Ex Vivo Generation of CAR Macrophages from Hematopoietic Stem and Progenitor Cells for Use in Cancer Therapy

 ,
,  ,
,
Abstract
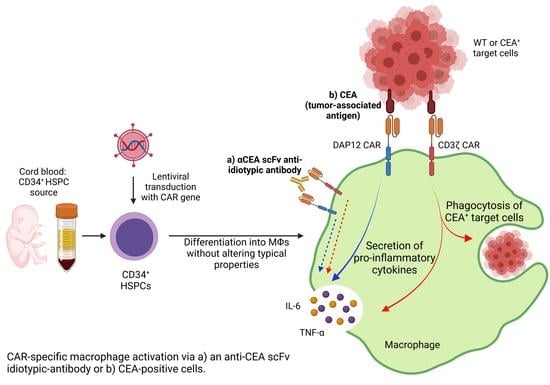
{kind=link}
{kind=link}
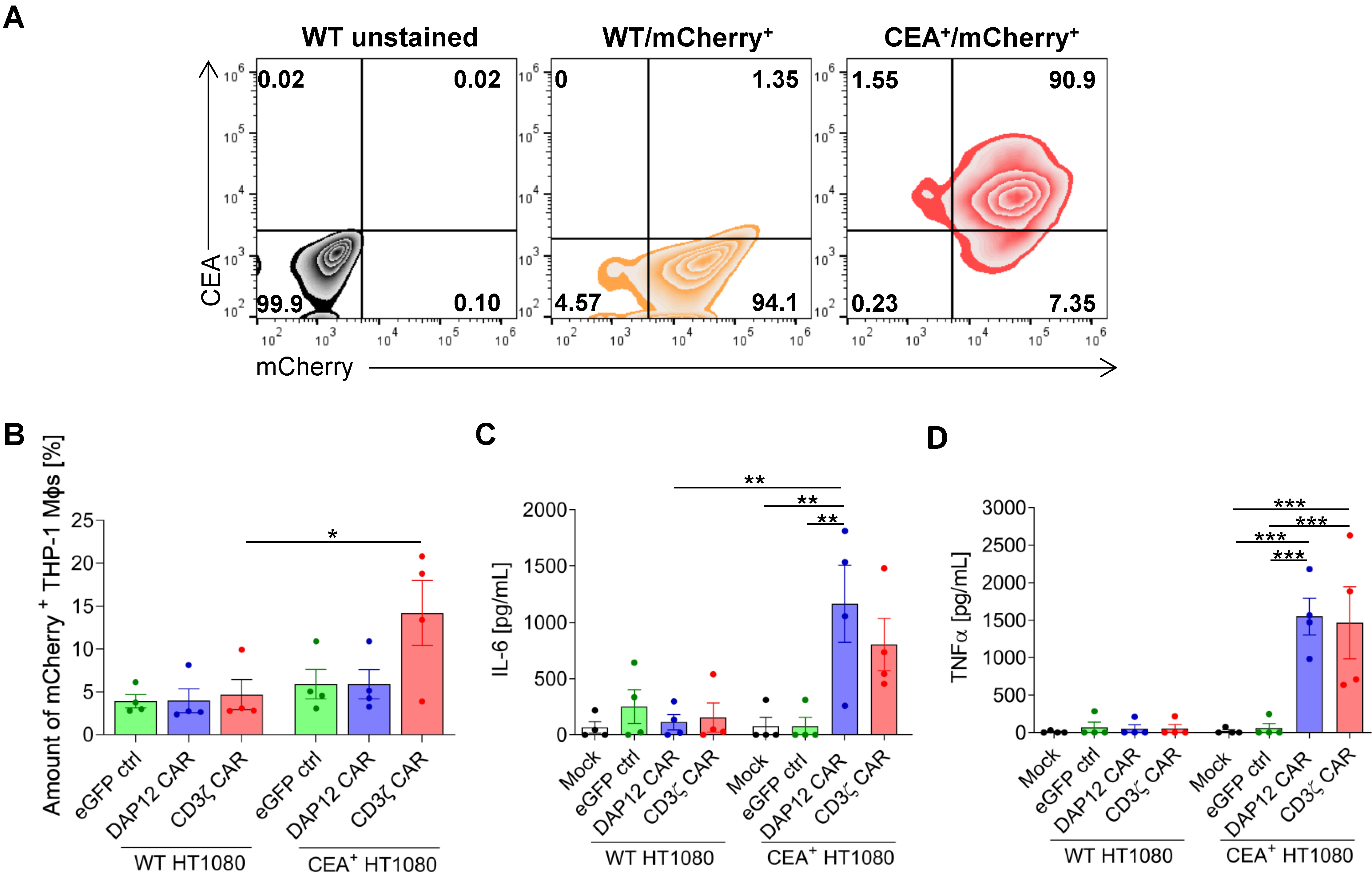{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Line Culture Conditions
2.2. Differentiation of THP-1 Cells to MΦs
2.3. Generation of Lentiviral CAR Constructs and Viral Vector Production
2.4. Transduction of Cell Lines with Lentiviral Constructs
2.5. Isolation and Cultivation of Human CD34+ Cells from Umbilical Cord Blood
2.6. Transduction of CD34+ Cells and Differentiation into MΦs
2.7. Cytospins
2.8. Phagocytosis Assay (pHrodo)
2.9. Flow Cytometry
2.10. Quantification of Vector Copy Number
2.11. CAR MΦ Stimulation with the Anti-Idiotypic Anti-BW2064 Antibody
2.12. Quantification of MΦ Phagocytic Activity against HT1080 Target Cells by Flow Cytometry
2.13. Confocal Microscopy and Quantitative Analysis of mCherry Expression
2.14. Cytokine Secretion
2.15. Viability and Apoptosis Assay
2.16. Statistical Analysis
3. Results
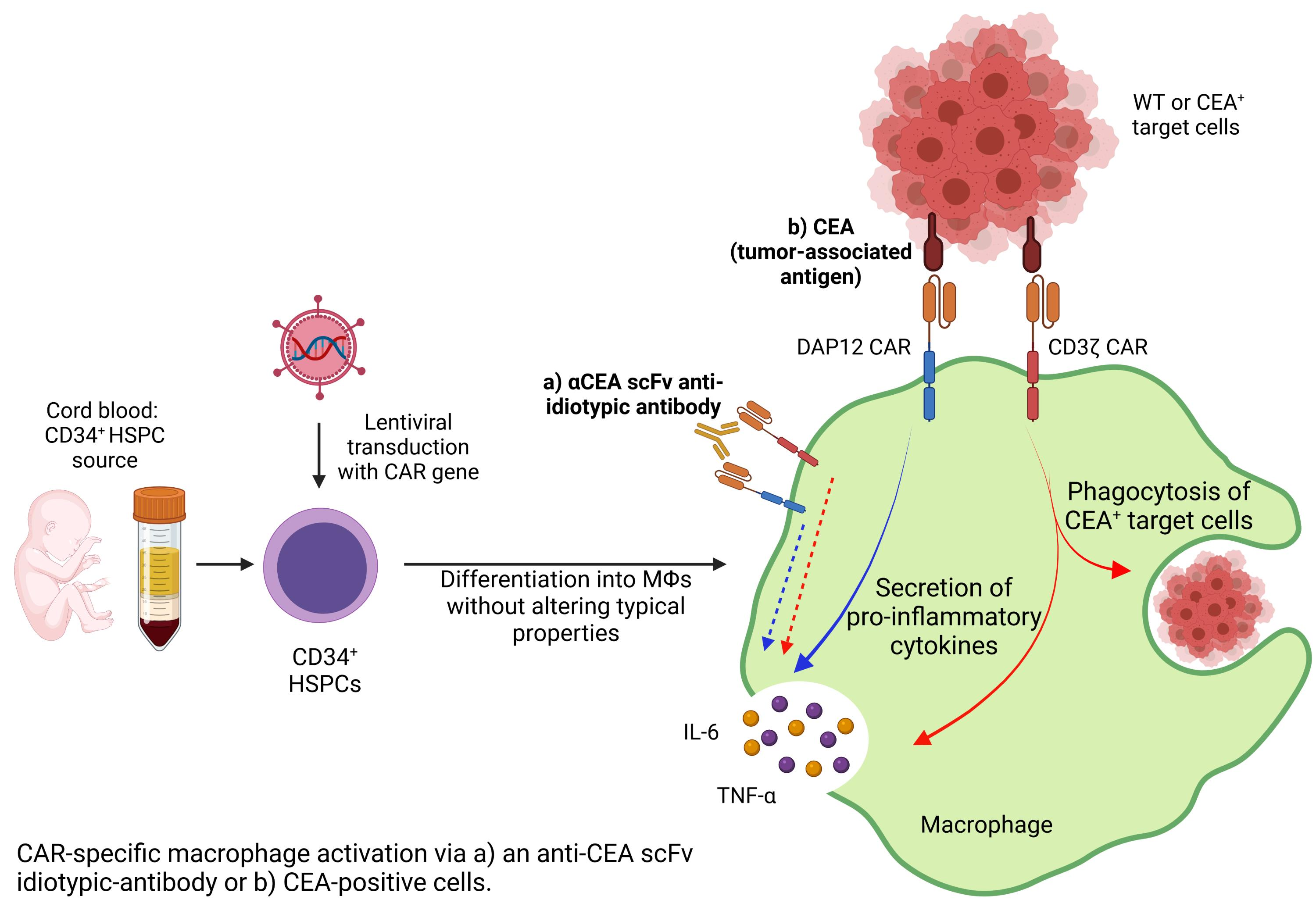
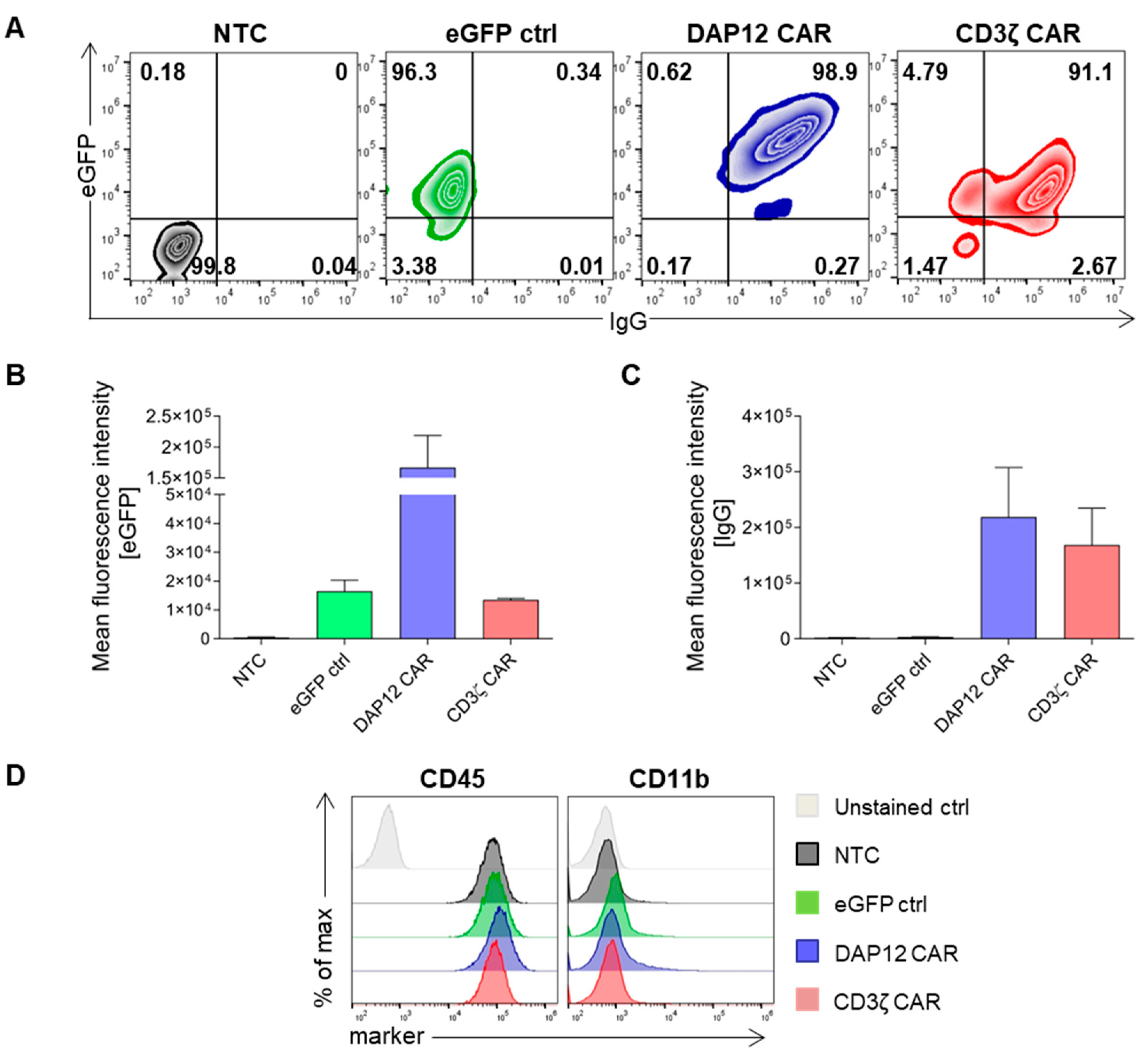
3.1. Validation of Anti-CEA CAR Vectors in Myeloid Cell Lines
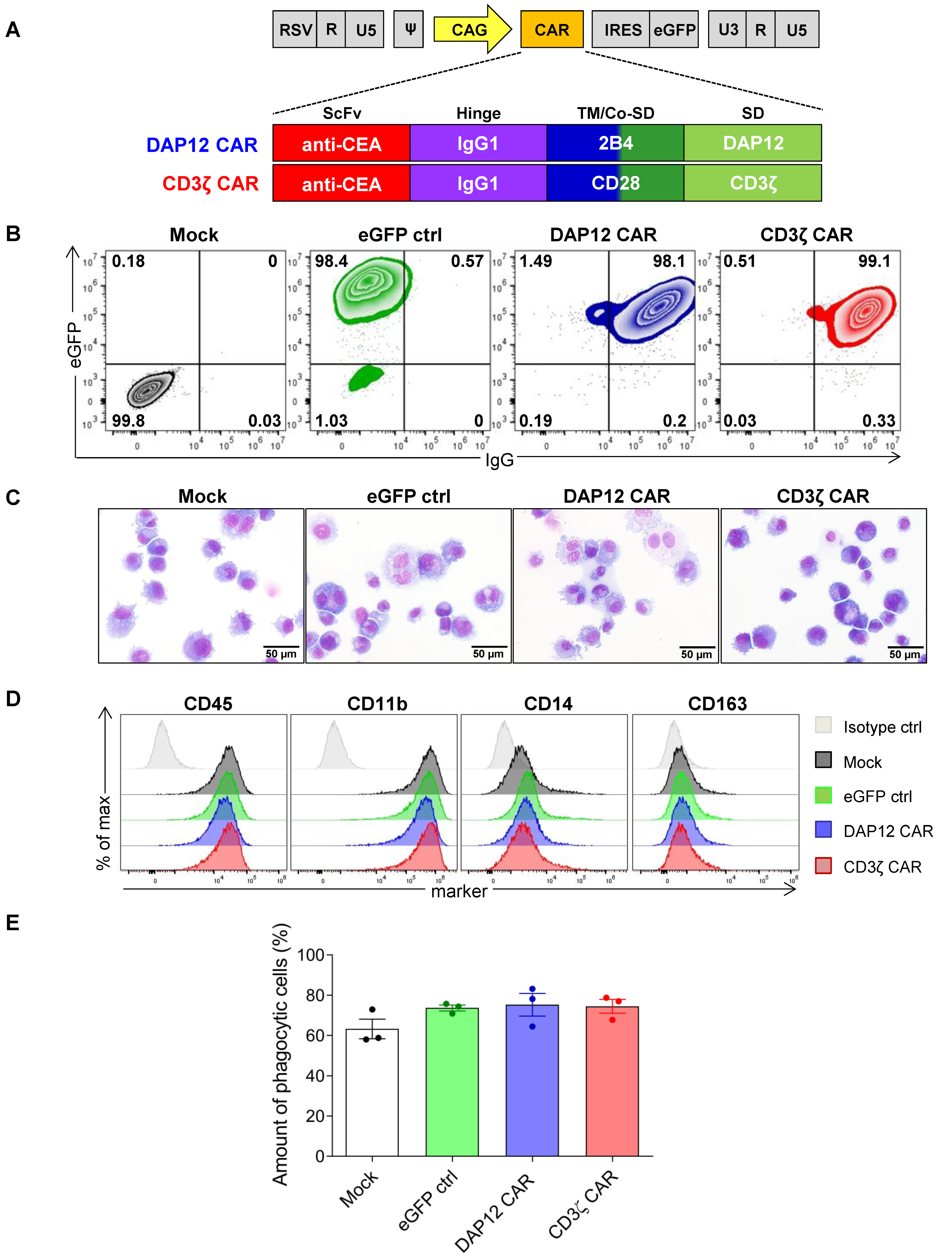
3.2. Differentiated CAR THP-1 Cells Showed Enhanced Cytokine Secretion upon Co-Culture with HT1080 Cells Engineered to Express CEA
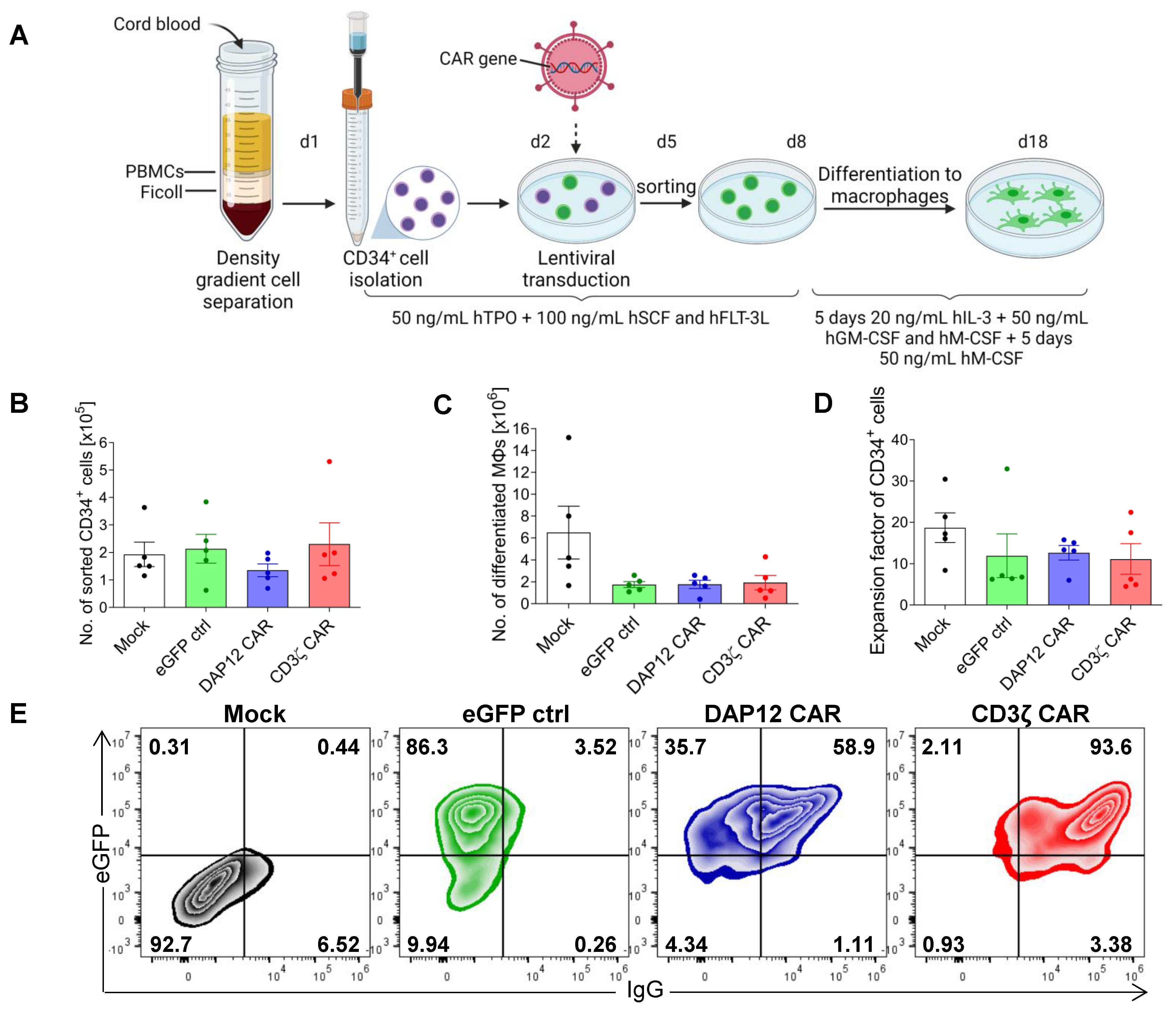
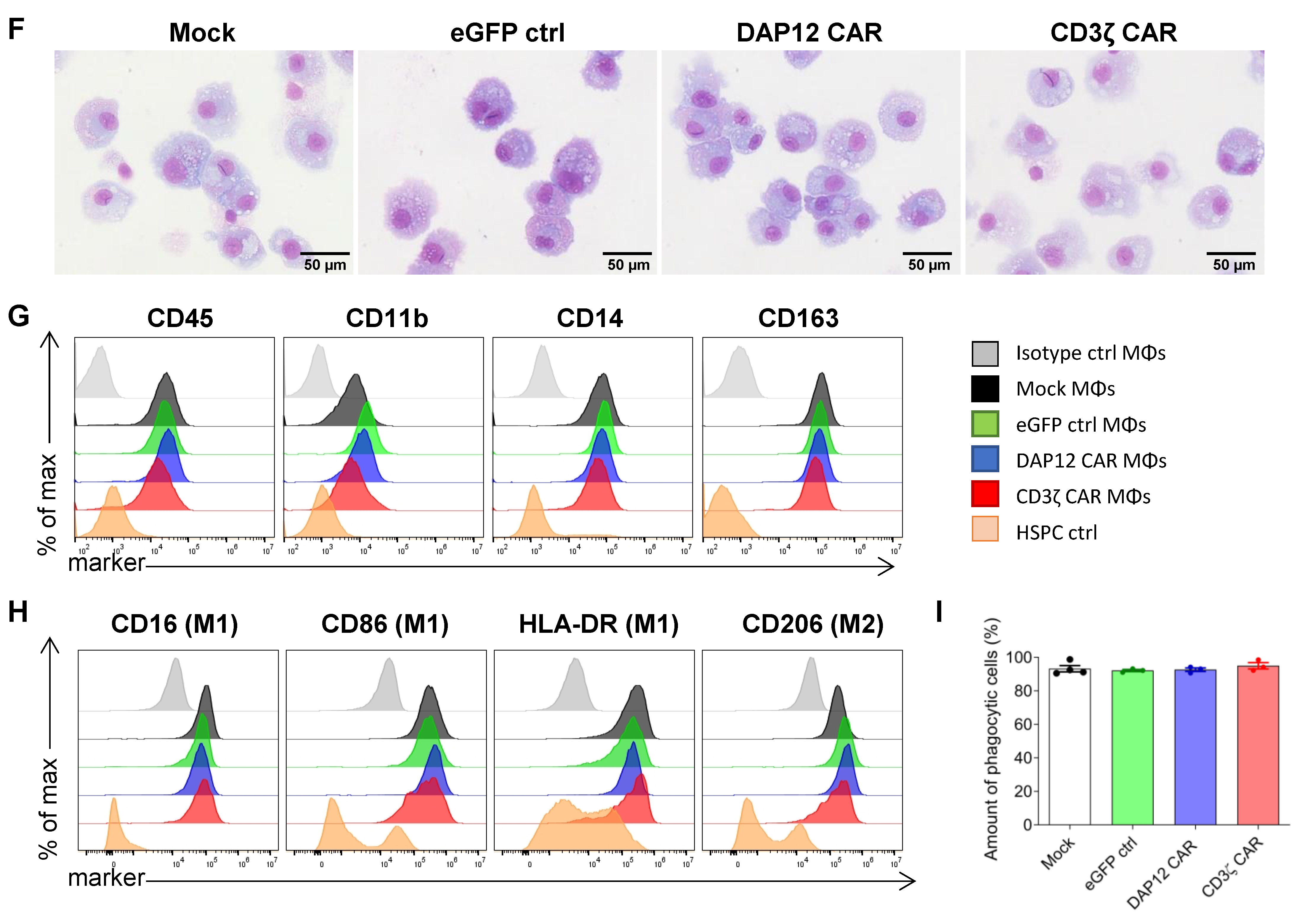
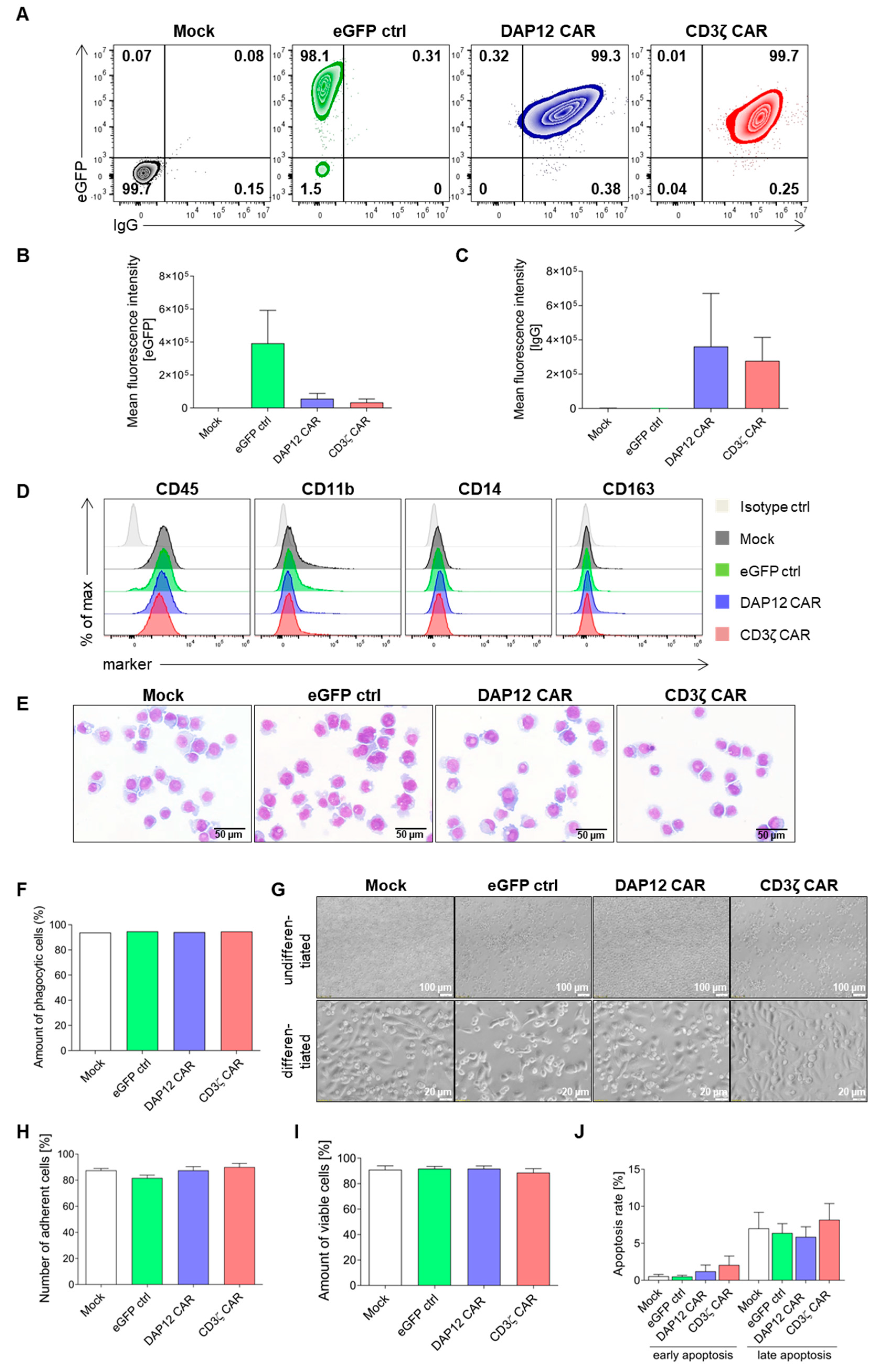
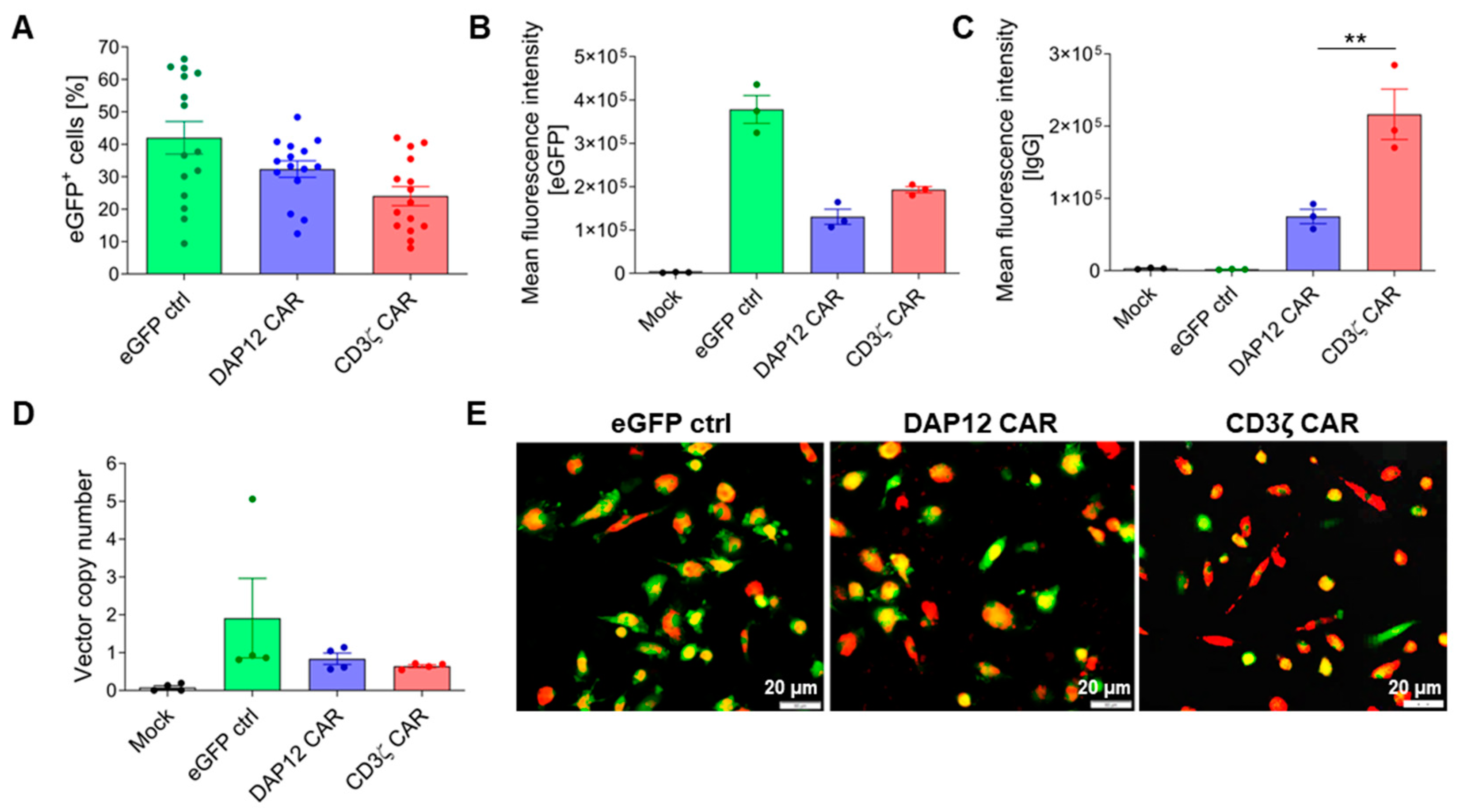
3.3. CAR Expression Did Not Interfere with the Induced Differentiation of Cord Blood-Derived CD34+ Cells into Functional Macrophages
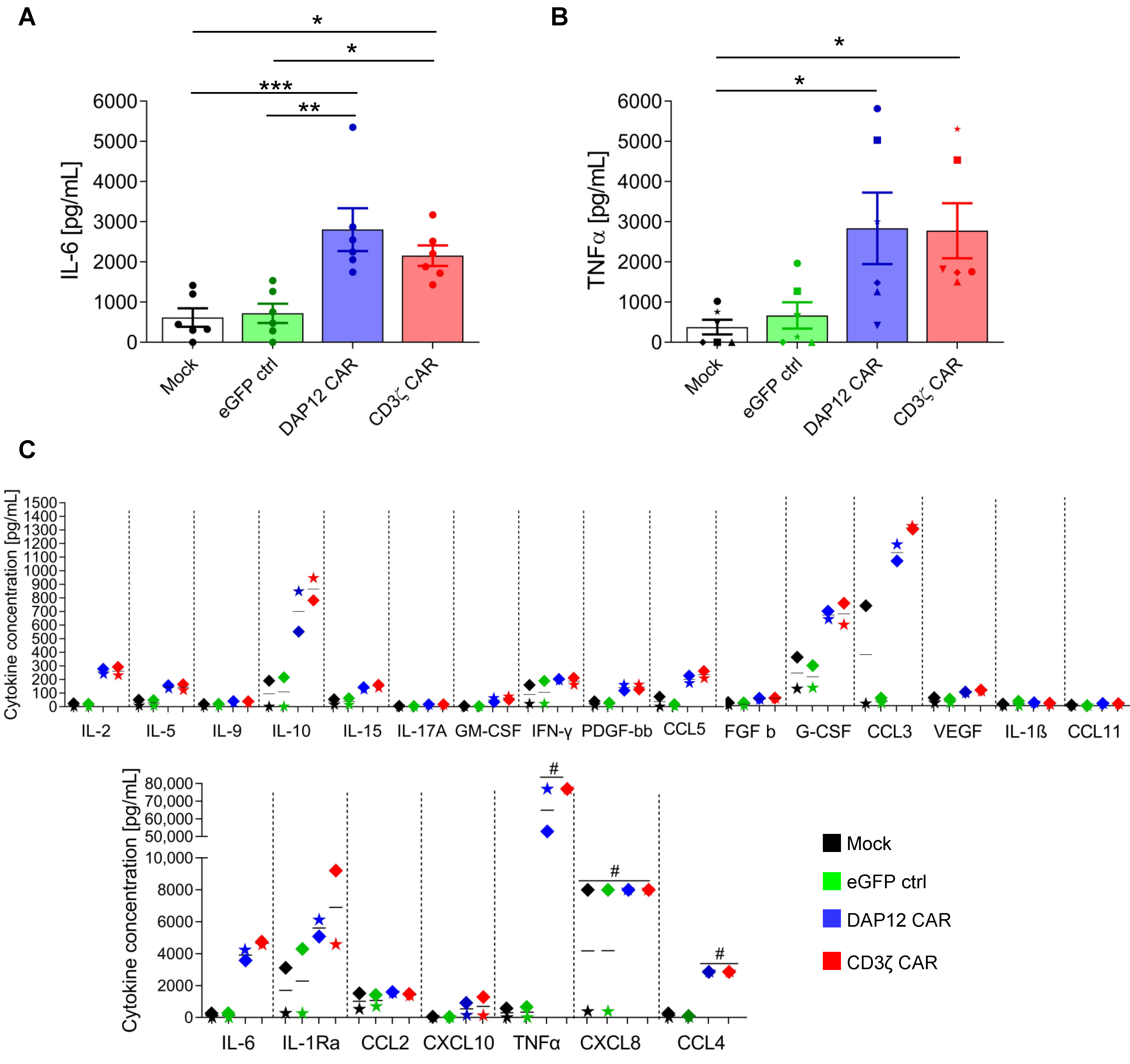
3.4. CD34+ Cell-Derived CAR MΦs Showed Cytokine Secretion upon CAR Stimulation
3.5. CD34+ Cell-Derived CAR MΦs Showed Enhanced Phagocytosis and Cytokine Secretion upon Contact with CEA+ HT1080 Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



References
- Arruebo, M.; Vilaboa, N.; Sáez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; González-Fernández, A. Assessment of the evolution of cancer treatment therapies. Cancers 2011, 3, 3279–3330. [Google Scholar] [CrossRef] [PubMed]
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med. J. 2015, 6, e0004. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Tahmasebi, S.; Elahi, R.; Esmaeilzadeh, A. Solid Tumors Challenges and New Insights of CAR T Cell Engineering. Stem Cell Rev. Rep. 2019, 15, 619–636. [Google Scholar] [CrossRef]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef]
- Randolph, G.J.; Jakubzick, C.; Qu, C. Antigen presentation by monocytes and monocyte-derived cells. Curr. Opin. Immunol. 2008, 20, 52–60. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, M.; Tang, W.; Wen, R.; Zhou, S.; Lee, C.; Wang, H.; Jiang, W.; Delahunty, I.M.; Zhen, Z.; et al. Nanoparticle-Laden Macrophages for Tumor-Tropic Drug Delivery. Adv. Mater. 2018, 30, e1805557. [Google Scholar] [CrossRef] [PubMed]
- Faradji, A.; Bohbot, A.; Schmitt-Goguel, M.; Roeslin, N.; Dumont, S.; Wiesel, M.L.; Lallot, C.; Eber, M.; Bartholeyns, J.; Poindron, P. Phase I trial of intravenous infusion of ex-vivo-activated autologous blood-derived macrophages in patients with non-small-cell lung cancer: Toxicity and immunomodulatory effects. Cancer Immunol. Immunother. 1991, 33, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Andreesen, R.; Scheibenbogen, C.; Brugger, W.; Krause, S.; Meerpohl, H.G.; Leser, H.G.; Engler, H.; Löhr, G.W. Adoptive transfer of tumor cytotoxic macrophages generated in vitro from circulating blood monocytes: A new approach to cancer immunotherapy. Cancer Res. 1990, 50, 7450–7456. [Google Scholar] [PubMed]
- Catarinella, M.; Monestiroli, A.; Escobar, G.; Fiocchi, A.; Tran, N.L.; Aiolfi, R.; Marra, P.; Esposito, A.; Cipriani, F.; Aldrighetti, L.; et al. IFNα gene/cell therapy curbs colorectal cancer colonization of the liver by acting on the hepatic microenvironment. EMBO Mol. Med. 2016, 8, 155–170. [Google Scholar] [CrossRef]
- Escobar, G.; Gentner, B.; Naldini, L.; Mazzieri, R. Engineered tumor-infiltrating macrophages as gene delivery vehicles for interferon-α activates immunity and inhibits breast cancer progression. OncoImmunology 2014, 3, e28696. [Google Scholar] [CrossRef]
- Moyes, K.W.; Lieberman, N.A.P.; Kreuser, S.A.; Chinn, H.; Winter, C.; Deutsch, G.; Hoglund, V.; Watson, R.; Crane, C.A. Genetically Engineered Macrophages: A Potential Platform for Cancer Immunotherapy. Hum. Gene Ther. 2017, 28, 200–215. [Google Scholar] [CrossRef]
- Kaczanowska, S.; Beury, D.W.; Gopalan, V.; Tycko, A.K.; Qin, H.; Clements, M.E.; Drake, J.; Nwanze, C.; Murgai, M.; Rae, Z.; et al. Genetically engineered myeloid cells rebalance the core immune suppression program in metastasis. Cell 2021, 184, 2033–2052.e21. [Google Scholar] [CrossRef]
- Escobar, G.; Moi, D.; Ranghetti, A.; Ozkal-Baydin, P.; Squadrito, M.L.; Kajaste-Rudnitski, A.; Bondanza, A.; Gentner, B.; de Palma, M.; Mazzieri, R.; et al. Genetic engineering of hematopoiesis for targeted IFN-α delivery inhibits breast cancer progression. Sci. Transl. Med. 2014, 6, 217ra3. [Google Scholar] [CrossRef]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7, e36688. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Kiss, M.; Caro, A.A.; Raes, G.; Laoui, D. Systemic Reprogramming of Monocytes in Cancer. Front. Oncol. 2020, 10, 1399. [Google Scholar] [CrossRef]
- Lower, E.E.; Baughman, R.P. The effect of cancer and chemotherapy on monocyte function. J. Clin. Lab. Immunol. 1990, 31, 121–125. [Google Scholar]
- Ouyang, W.; Liu, Y.; Deng, D.; Zhou, F.; Xie, C. The change in peripheral blood monocyte count: A predictor to make the management of chemotherapy-induced neutropenia. J. Can. Res. Ther. 2018, 14, S565–S570. [Google Scholar] [CrossRef]
- Tiwari, A.; Moeneclaey, G.; Jenkin, G.; Kirkland, M.A. Exploring Life Saving Potential of Umbilical Cord Blood Derived Hematopoietic Stem Cells. Insights Stem Cells 2016, 2, 1–4. [Google Scholar]
- Harris, D.T. Stem Cell Banking for Regenerative and Personalized Medicine. Biomedicines 2014, 2, 50–79. [Google Scholar] [CrossRef]
- Dolscheid-Pommerich, R.C.; Manekeller, S.; Walgenbach-Brünagel, G.; Kalff, J.C.; Hartmann, G.; Wagner, B.S.; Holdenrieder, S. Clinical Performance of CEA, CA19-9, CA15-3, CA125 and AFP in Gastrointestinal Cancer Using LOCI™-based Assays. Anticancer Res. 2017, 37, 353–359. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bethigh FoxO1low Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef]
- Hetzel, M.; Suzuki, T.; Hashtchin, A.R.; Arumugam, P.; Carey, B.; Schwabbauer, M.; Kuhn, A.; Meyer, J.; Schambach, A.; van der Loo, J.; et al. Function and Safety of Lentivirus-Mediated Gene Transfer for CSF2RA-Deficiency. Hum. Gene Ther. Methods 2017, 28, 318–329. [Google Scholar] [CrossRef]
- Hombach, A.; Sent, D.; Schneider, C.; Heuser, C.; Koch, D.; Pohl, C.; Seliger, B.; Abken, H. T-cell activation by recombinant receptors: CD28 costimulation is required for interleukin 2 secretion and receptor-mediated T-cell proliferation but does not affect receptor-mediated target cell lysis. Cancer Res. 2001, 61, 1976–1982. [Google Scholar]
- Wang, E.; Wang, L.-C.; Tsai, C.-Y.; Bhoj, V.; Gershenson, Z.; Moon, E.; Newick, K.; Sun, J.; Lo, A.; Baradet, T.; et al. Generation of Potent T-cell Immunotherapy for Cancer Using DAP12-Based, Multichain, Chimeric Immunoreceptors. Cancer Immunol. Res. 2015, 3, 815–826. [Google Scholar] [CrossRef]
- Töpfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Müller, N.; Lindemann, D.; Bachmann, M.; Füssel, M.; Schackert, G.; Temme, A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef]
- Starr, T.; Bauler, T.J.; Malik-Kale, P.; Steele-Mortimer, O. The phorbol 12-myristate-13-acetate differentiation protocol is critical to the interaction of THP-1 macrophages with Salmonella Typhimurium. PLoS ONE 2018, 13, e0193601. [Google Scholar] [CrossRef]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef]
- Brown, C.E.; Adusumilli, P.S. Next frontiers in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16028. [Google Scholar] [CrossRef][Green Version]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef]
- Suh, H.C.; Pohl, K.; Javier, A.P.L.; Slamon, D.J.; Chute, J.P. Effect of dendritic cells (DC) transduced with chimeric antigen receptor (CAR) on CAR T-cell cytotoxicity. JCO 2017, 35, 144. [Google Scholar] [CrossRef]
- Andreesen, R.; Hennemann, B.; Krause, S.W. Adoptive immunotherapy of cancer using monocyte-derived macrophages: Rationale, current status, and perspectives. J. Leukoc. Biol. 1998, 64, 419–426. [Google Scholar] [CrossRef]
- Lacerna, L.V.; Stevenson, G.W.; Stevenson, H.C. Adoptive cancer immunotherapy utilizing lymphokine activated killer cells and gamma interferon activated killer monocytes. Pharmacol. Ther. 1988, 38, 453–465. [Google Scholar] [CrossRef]
- Niu, Z.; Chen, G.; Chang, W.; Sun, P.; Luo, Z.; Zhang, H.; Zhi, L.; Guo, C.; Chen, H.; Yin, M.; et al. Chimeric antigen receptor-modified macrophages trigger systemic anti-tumour immunity. J. Pathol. 2021, 253, 247–257. [Google Scholar] [CrossRef]
- Hordyjewska, A.; Popiołek, Ł.; Horecka, A. Characteristics of hematopoietic stem cells of umbilical cord blood. Cytotechnology 2015, 67, 387–396. [Google Scholar] [CrossRef]
- Hölig, K.; Kramer, M.; Kroschinsky, F.; Bornhäuser, M.; Mengling, T.; Schmidt, A.H.; Rutt, C.; Ehninger, G. Safety and efficacy of hematopoietic stem cell collection from mobilized peripheral blood in unrelated volunteers: 12 years of single-center experience in 3928 donors. Blood 2009, 114, 3757–3763. [Google Scholar] [CrossRef]
- Shiozawa, M.; Chang, C.-H.; Huang, Y.-C.; Chen, Y.-C.; Chi, M.-S.; Hao, H.-C.; Chang, Y.-C.; Takeda, S.; Chi, K.-H.; Wang, Y.-S. Pharmacologically upregulated carcinoembryonic antigen-expression enhances the cytolytic activity of genetically-modified chimeric antigen receptor NK-92MI against colorectal cancer cells. BMC Immunol. 2018, 19, 27. [Google Scholar] [CrossRef]
- Barckhausen, C.; Rice, B.; Baila, S.; Sensebé, L.; Schrezenmeier, H.; Nold, P.; Hackstein, H.; Rojewski, M.T. GMP-Compliant Expansion of Clinical-Grade Human Mesenchymal Stromal/Stem Cells Using a Closed Hollow Fiber Bioreactor. Methods Mol. Biol. 2016, 1416, 389–412. [Google Scholar] [CrossRef]
- Fekete, N.; Rojewski, M.T.; Fürst, D.; Kreja, L.; Ignatius, A.; Dausend, J.; Schrezenmeier, H. GMP-compliant isolation and large-scale expansion of bone marrow-derived MSC. PLoS ONE 2012, 7, e43255. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J.; et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA+ Metastatic Colorectal Cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hahn, O.; Rappl, G.; Nowak, M.; Schmidt-Wolf, I.H.; Hombach, A.A.; Abken, H. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology 2012, 143, 1095–1107.e2. [Google Scholar] [CrossRef]
- Tessarz, A.S.; Cerwenka, A. The TREM-1/DAP12 pathway. Immunol. Lett. 2008, 116, 111–116. [Google Scholar] [CrossRef]
- Koth, L.L.; Cambier, C.J.; Ellwanger, A.; Solon, M.; Hou, L.; Lanier, L.L.; Abram, C.L.; Hamerman, J.A.; Woodruff, P.G. DAP12 is required for macrophage recruitment to the lung in response to cigarette smoke and chemotaxis toward CCL2. J. Immunol. 2010, 184, 6522–6528. [Google Scholar] [CrossRef]
- Dalton, R.; Calescibetta, A.; Zhou, J.M.; Maurin, M.; Ward, G.; Le Trinh, T.; Tu, N.; Gilvary, D.; Chen, X.; Cheng, P.; et al. Constitutively Activated DAP12 Induces Functional Anti-Tumor Activation and Maturation of Human Monocyte-Derived DC. Int. J. Mol. Sci. 2021, 22, 1241. [Google Scholar] [CrossRef]
- Underhill, D.M.; Goodridge, H.S. Information processing during phagocytosis. Nat. Rev. Immunol. 2012, 12, 492–502. [Google Scholar] [CrossRef]
- James, J.R. Tuning ITAM multiplicity on T cell receptors can control potency and selectivity to ligand density. Sci. Signal. 2018, 11, eaan1088. [Google Scholar] [CrossRef]
- Johnson, S.A.; Pleiman, C.M.; Pao, L.; Schneringer, J.; Hippen, K.; Cambier, J.C. Phosphorylated immunoreceptor signaling motifs (ITAMs) exhibit unique abilities to bind and activate Lyn and Syk tyrosine kinases. J. Immunol. 1995, 155, 4596–4603. [Google Scholar]
- Lanier, L.L. DAP10- and DAP12-associated receptors in innate immunity. Immunol. Rev. 2009, 227, 150–160. [Google Scholar] [CrossRef]
- Hombach, A.; Hombach, A.A.; Abken, H. Adoptive immunotherapy with genetically engineered T cells: Modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010, 17, 1206–1213. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paasch, D.; Meyer, J.; Stamopoulou, A.; Lenz, D.; Kuehle, J.; Kloos, D.; Buchegger, T.; Holzinger, A.; Falk, C.S.; Kloth, C.; et al. Ex Vivo Generation of CAR Macrophages from Hematopoietic Stem and Progenitor Cells for Use in Cancer Therapy. Cells 2022, 11, 994. https://doi.org/10.3390/cells11060994
Paasch D, Meyer J, Stamopoulou A, Lenz D, Kuehle J, Kloos D, Buchegger T, Holzinger A, Falk CS, Kloth C, et al. Ex Vivo Generation of CAR Macrophages from Hematopoietic Stem and Progenitor Cells for Use in Cancer Therapy. Cells. 2022; 11(6):994. https://doi.org/10.3390/cells11060994
Chicago/Turabian StylePaasch, Daniela, Johann Meyer, Andriana Stamopoulou, Daniela Lenz, Johannes Kuehle, Doreen Kloos, Theresa Buchegger, Astrid Holzinger, Christine S. Falk, Christina Kloth, and et al. 2022. "Ex Vivo Generation of CAR Macrophages from Hematopoietic Stem and Progenitor Cells for Use in Cancer Therapy" Cells 11, no. 6: 994. https://doi.org/10.3390/cells11060994
APA StylePaasch, D., Meyer, J., Stamopoulou, A., Lenz, D., Kuehle, J., Kloos, D., Buchegger, T., Holzinger, A., Falk, C. S., Kloth, C., von Kaisenberg, C. S., Abken, H., Schambach, A., Lachmann, N., Morgan, M., & Moritz, T. (2022). Ex Vivo Generation of CAR Macrophages from Hematopoietic Stem and Progenitor Cells for Use in Cancer Therapy. Cells, 11(6), 994. https://doi.org/10.3390/cells11060994