
T Cells, Interleukin-2 and Systemic Lupus Erythematosus—From Pathophysiology to Therapy


{kind=link}
Abstract
:1. Introduction
2. Abnormalities of Lupus T Cells
3. Physiology and Action of IL-2
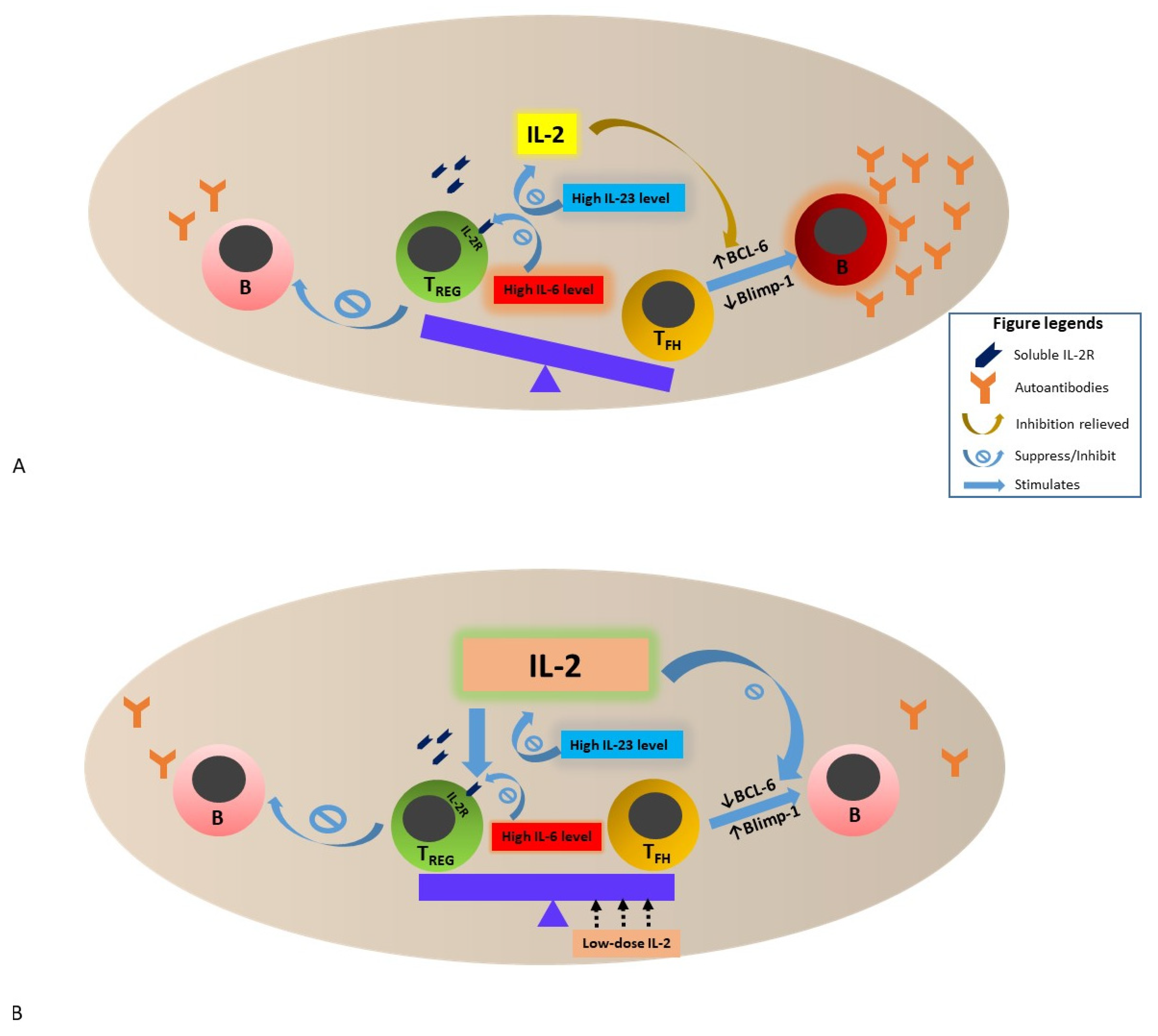
4. IL-2 Deficiency and Hypo-Responsiveness in SLE—Potential Mechanisms and Impact
5. Restoration of IL-2 in Murine Lupus Model
6. Clinical Observation and Therapeutic Trials of Low-Dose IL-2 Therapy in Patients with SLE
7. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, A.; Kow, N.Y. The pathology of T cells in systemic lupus erythematosus. J. Immunol. Res. 2014, 2014, 419029. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.E.; Clark, M.R. Compartments and Connections within the Germinal Center. Front. Immunol. 2021, 12, 659151. [Google Scholar] [CrossRef] [PubMed]
- Reijm, S.; Kissel, T.; Toes, R.E.M. Checkpoints controlling the induction of B cell mediated autoimmunity in human autoimmune diseases. Eur. J. Immunol. 2020, 50, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Kanta, H.; Mohan, C. Three checkpoints in lupus development: Central tolerance in adaptive immunity, peripheral amplification by innate immunity and end-organ inflammation. Genes Immun. 2009, 10, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Ma, Y.; Grant, C.R.; Samyn, M.; Gordon, P.; Mieli-Vergani, G.; Vergani, D. T-regs in autoimmune hepatitis-systemic lupus erythematosus/mixed connective tissue disease overlap syndrome are functionally defective and display a Th1 cytokine profile. J. Autoimmun. 2013, 41, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.G.; Jones, R.B.; Burns, S.M.; Jayne, D.R. Long-term comparison of rituximab treatment for refractory systemic lupus erythematosus and vasculitis: Remission, relapse, and re-treatment. Arthritis Rheum. 2006, 54, 2970–2982. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012, 64, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010, 62, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.C.; Ying, K.Y.; Yim, C.W.; Siu, Y.P.; Tong, K.H.; To, C.H.; Ng, W.L. Tacrolimus versus mycophenolate mofetil for induction therapy of lupus nephritis: A randomised controlled trial and long-term follow-up. Ann. Rheum. Dis. 2016, 75, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.C.; Ho, L.Y.; Ying, S.K.Y.; Leung, M.C.; To, C.H.; Ng, W.L. Long-term outcome of a randomised controlled trial comparing tacrolimus with mycophenolate mofetil as induction therapy for active lupus nephritis. Ann. Rheum. Dis. 2020, 79, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Austin, H.A., III; Illei, G.G.; Braun, M.J.; Balow, J.E. Randomized, controlled trial of prednisone, cyclophosphamide, and cyclosporine in lupus membranous nephropathy. J. Am. Soc. Nephrol. 2009, 20, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahvildari, M.; Dana, R. Low-Dose IL-2 Therapy in Transplantation, Autoimmunity, and Inflammatory Diseases. J. Immunol. 2019, 203, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- Diaz-de-Durana, Y.; Lau, J.; Knee, D.; Filippi, C.; Londei, M.; McNamara, P.; Nasoff, M.; DiDonato, M.; Glynne, R.; Herman, A.E. IL-2 immunotherapy reveals potential for innate beta cell regeneration in the non-obese diabetic mouse model of autoimmune diabetes. PLoS ONE 2013, 8, e78483. [Google Scholar] [CrossRef] [PubMed]
- Kitas, G.D.; Salmon, M.; Farr, M.; Gaston, J.S.; Bacon, P.A. Deficient interleukin 2 production in rheumatoid arthritis: Association with active disease and systemic complications. Clin. Exp. Immunol. 1988, 73, 242–249. [Google Scholar] [PubMed]
- Hulme, M.A.; Wasserfall, C.H.; Atkinson, M.A.; Brusko, T.M. Central role for interleukin-2 in type 1 diabetes. Diabetes 2012, 61, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linker-Israeli, M.; Bakke, A.C.; Kitridou, R.C.; Gendler, S.; Gillis, S.; Horwitz, D.A. Defective production of interleukin 1 and interleukin 2 in patients with systemic lupus erythematosus (SLE). J. Immunol. 1983, 130, 2651–2655. [Google Scholar] [PubMed]
- Nagy, N.; Kaber, G.; Kratochvil, M.J.; Kuipers, H.F.; Ruppert, S.M.; Yadava, K.; Yang, J.; Heilshorn, S.C.; Long, S.A.; Pugliese, A.; et al. Weekly injection of IL-2 using an injectable hydrogel reduces autoimmune diabetes incidence in NOD mice. Diabetologia 2021, 64, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; He, J.; Wei, Y.; Zeng, Q.; Gong, D.; Qin, J.; Ding, H.; Chen, Z.; Zhou, P.; Niu, P.; et al. Sustained low-dose interleukin-2 therapy alleviates pathogenic humoral immunity via elevating the Tfr/Tfh ratio in lupus. Clin. Transl. Immunol. 2021, 10, e1293. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, D.; Rosenzwajg, M.; Joly, F.; Six, A.; Carrat, F.; Thibault, V.; Sene, D.; Cacoub, P.; Klatzmann, D. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N. Engl. J. Med. 2011, 365, 2067–2077. [Google Scholar] [CrossRef]
- Koreth, J.; Matsuoka, K.; Kim, H.T.; McDonough, S.M.; Bindra, B.; Alyea, E.P., III; Armand, P.; Cutler, C.; Ho, V.T.; Treister, N.S.; et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N. Engl. J. Med. 2011, 365, 2055–2066. [Google Scholar] [CrossRef] [Green Version]
- Humrich, J.Y.; Riemekasten, G. Low-dose interleukin-2 therapy for the treatment of systemic lupus erythematosus. Curr. Opin. Rheumatol. 2019, 31, 208–212. [Google Scholar] [CrossRef]
- Kow, N.Y.; Mak, A. Costimulatory pathways: Physiology and potential therapeutic manipulation in systemic lupus erythematosus. Clin. Dev. Immunol. 2013, 2013, 245928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, D.B. T Cell Receptors and MHC Molecules. In Immunology; Male, D., Brostoff, J., Roth, D.B., Roitt, I.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 89–106. [Google Scholar]
- Krishnan, S.; Warke, V.G.; Nambiar, M.P.; Tsokos, G.C.; Farber, D.L. The FcR gamma subunit and Syk kinase replace the CD3 zeta-chain and ZAP-70 kinase in the TCR signaling complex of human effector CD4 T cells. J. Immunol. 2003, 170, 4189–4195. [Google Scholar] [CrossRef] [Green Version]
- Liossis, S.N.; Ding, X.Z.; Dennis, G.J.; Tsokos, G.C. Altered pattern of TCR/CD3-mediated protein-tyrosyl phosphorylation in T cells from patients with systemic lupus erythematosus. Deficient expression of the T cell receptor zeta chain. J. Clin. Investig. 1998, 101, 1448–1457. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, S.; Farber, D.L.; Tsokos, G.C. T cell rewiring in differentiation and disease. J. Immunol. 2003, 171, 3325–3331. [Google Scholar] [CrossRef] [Green Version]
- Juang, Y.T.; Wang, Y.; Jiang, G.; Peng, H.B.; Ergin, S.; Finnell, M.; Magilavy, A.; Kyttaris, V.C.; Tsokos, G.C. PP2A dephosphorylates Elf-1 and determines the expression of CD3zeta and FcRgamma in human systemic lupus erythematosus T cells. J. Immunol. 2008, 181, 3658–3664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, C.; Brink, R. The unique biology of germinal center B cells. Immunity 2021, 54, 1652–1664. [Google Scholar] [CrossRef]
- Qiu, H.; Wu, H.; Chan, V.; Lau, C.S.; Lu, Q. Transcriptional and epigenetic regulation of follicular T-helper cells and their role in autoimmunity. Autoimmunity 2017, 50, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Tsai, H.F.; Tzeng, H.T.; Liao, H.J.; Hsu, P.N. Lipid raft assembly and Lck recruitment in TRAIL costimulation mediates NF-κB activation and T cell proliferation. J. Immunol. 2011, 186, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Jury, E.C.; Kabouridis, P.S.; Flores-Borja, F.; Mageed, R.A.; Isenberg, D.A. Altered lipid raft-associated signaling and ganglioside expression in T lymphocytes from patients with systemic lupus erythematosus. J. Clin. Investig. 2004, 113, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Nambiar, M.P.; Warke, V.G.; Fisher, C.U.; Mitchell, J.; Delaney, N.; Tsokos, G.C. Alterations in lipid raft composition and dynamics contribute to abnormal T cell responses in systemic lupus erythematosus. J. Immunol. 2004, 172, 7821–7831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Fu, J.; Zhou, Y. Metabolism Controls the Balance of Th17/T-Regulatory Cells. Front. Immunol. 2017, 8, 1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolios, A.G.A.; Tsokos, G.C.; Klatzmann, D. Interleukin-2 and regulatory T cells in rheumatic diseases. Nat. Rev. Rheumatol. 2021, 17, 749–766. [Google Scholar] [CrossRef] [PubMed]
- Votavova, P.; Tomala, J.; Kovar, M. Increasing the biological activity of IL-2 and IL-15 through complexing with anti-IL-2 mAbs and IL-15Rα-Fc chimera. Immunol. Lett. 2014, 159, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rickert, M.; Garcia, K.C. Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gammac receptors. Science 2005, 310, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.H.; Cantrell, D.A. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu. Rev. Immunol. 2018, 36, 411–433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Miao, J.; Zhu, P. Regulatory T cell heterogeneity and therapy in autoimmune diseases. Autoimmun. Rev. 2021, 20, 102715. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef] [PubMed]
- Zeiser, R.; Negrin, R.S. Interleukin-2 receptor downstream events in regulatory T cells: Implications for the choice of immunosuppressive drug therapy. Cell Cycle 2008, 7, 458–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, T.R.; Yu, A.; Vincek, V.; Scibelli, P.; Kong, L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity 2002, 17, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Sadlack, B.; Merz, H.; Schorle, H.; Schimpl, A.; Feller, A.C.; Horak, I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 1993, 75, 253–261. [Google Scholar] [CrossRef]
- Alcocer-Varela, J.; Alarcón-Segovia, D. Decreased production of and response to interleukin-2 by cultured lymphocytes from patients with systemic lupus erythematosus. J. Clin. Investig. 1982, 69, 1388–1392. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, E.; Yan, M.; Watanabe, K.S.; Narazaki, M.; Matsushima, S.; Yamamura, Y.; Hiramatsu, S.; Ohashi, K.; Watanabe, H.; Katsuyama, T.; et al. Downregulation of miR-200a-3p, Targeting CtBP2 Complex, Is Involved in the Hypoproduction of IL-2 in Systemic Lupus Erythematosus-Derived T Cells. J. Immunol. 2017, 198, 4268–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharabi, A.; Kasper, I.R.; Tsokos, G.C. The serine/threonine protein phosphatase 2A controls autoimmunity. Clin. Immunol. 2018, 186, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Solomou, E.E.; Juang, Y.T.; Gourley, M.F.; Kammer, G.M.; Tsokos, G.C. Molecular basis of deficient IL-2 production in T cells from patients with systemic lupus erythematosus. J. Immunol. 2001, 166, 4216–4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudensky, A.Y.; Gavin, M.; Zheng, Y. FOXP3 and NFAT: Partners in tolerance. Cell 2006, 126, 253–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, H.; He, F.; Tsokos, G.C.; Kyttaris, V.C. IL-23 Limits the Production of IL-2 and Promotes Autoimmunity in Lupus. J. Immunol. 2017, 199, 903–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyttaris, V.C.; Zhang, Z.; Kuchroo, V.K.; Oukka, M.; Tsokos, G.C. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J. Immunol. 2010, 184, 4605–4609. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros-Tato, A.; Papillion, A. Mechanisms of action of low-dose IL-2 restoration therapies in SLE. Curr. Opin. Immunol. 2019, 61, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Papillion, A.; Powell, M.D.; Chisolm, D.A.; Bachus, H.; Fuller, M.J.; Weinmann, A.S.; Villarino, A.; O’Shea, J.J.; León, B.; Oestreich, K.J.; et al. Inhibition of IL-2 responsiveness by IL-6 is required for the generation of GC-TFH cells. Sci. Immunol. 2019, 4, eaaw7636. [Google Scholar] [CrossRef] [PubMed]
- Tokano, Y.; Murashima, A.; Takasaki, Y.; Hashimoto, H.; Okumura, K.; Hirose, S. Relation between soluble interleukin 2 receptor and clinical findings in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 1989, 48, 803–809. [Google Scholar] [CrossRef]
- Nurieva, R.I.; Chung, Y.; Martinez, G.J.; Yang, X.O.; Tanaka, S.; Matskevitch, T.D.; Wang, Y.H.; Dong, C. Bcl6 mediates the development of T follicular helper cells. Science 2009, 325, 1001–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mountz, J.D.; Hsu, H.C.; Ballesteros-Tato, A. Dysregulation of T Follicular Helper Cells in Lupus. J. Immunol. 2019, 202, 1649–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humrich, J.Y.; Morbach, H.; Undeutsch, R.; Enghard, P.; Rosenberger, S.; Weigert, O.; Kloke, L.; Heimann, J.; Gaber, T.; Brandenburg, S.; et al. Homeostatic imbalance of regulatory and effector T cells due to IL-2 deprivation amplifies murine lupus. Proc. Natl. Acad. Sci. USA 2010, 107, 204–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, A.; von Spee-Mayer, C.; Kloke, L.; Wu, K.; Kühl, A.; Enghard, P.; Burmester, G.R.; Riemekasten, G.; Humrich, J.Y. IL-2 Therapy Diminishes Renal Inflammation and the Activity of Kidney-Infiltrating CD4+ T Cells in Murine Lupus Nephritis. Cells 2019, 8, 1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.J.; Lee, J.G.; Jang, J.Y.; Koo, T.Y.; Ahn, C.; Yang, J. IL-2/anti-IL-2 complexes ameliorate lupus nephritis by expansion of CD4+ CD25+ Foxp3+ regulatory T cells. Kidney Int. 2017, 91, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Mizui, M.; Koga, T.; Lieberman, L.A.; Beltran, J.; Yoshida, N.; Johnson, M.C.; Tisch, R.; Tsokos, G.C. IL-2 protects lupus-prone mice from multiple end-organ damage by limiting CD4-CD8- IL-17-producing T cells. J. Immunol. 2014, 193, 2168–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humrich, J.Y.; von Spee-Mayer, C.; Siegert, E.; Alexander, T.; Hiepe, F.; Radbruch, A.; Burmester, G.R.; Riemekasten, G. Rapid induction of clinical remission by low-dose interleukin-2 in a patient with refractory SLE. Ann. Rheum. Dis. 2015, 74, 791–792. [Google Scholar] [CrossRef]
- von Spee-Mayer, C.; Siegert, E.; Abdirama, D.; Rose, A.; Klaus, A.; Alexander, T.; Enghard, P.; Sawitzki, B.; Hiepe, F.; Radbruch, A.; et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2016, 75, 1407–1415. [Google Scholar] [CrossRef]
- Zhao, C.; Chu, Y.; Liang, Z.; Zhang, B.; Wang, X.; Jing, X.; Hao, M.; Wang, Y.; An, J.; Zhang, X.; et al. Low dose of IL-2 combined with rapamycin restores and maintains the long-term balance of Th17/Treg cells in refractory SLE patients. BMC Immunol. 2019, 20, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Zhang, X.; Wei, Y.; Sun, X.; Chen, Y.; Deng, J.; Jin, Y.; Gan, Y.; Hu, X.; Jia, R.; et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat. Med. 2016, 22, 991–993. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhang, R.; Shao, M.; Zhao, X.; Miao, M.; Chen, J.; Liu, J.; Zhang, X.; Zhang, X.; Jin, Y.; et al. Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: A randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2020, 79, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Odler, B.; Windpessl, M.; Krall, M.; Steiner, M.; Riedl, R.; Hebesberger, C.; Ursli, M.; Zitt, E.; Lhotta, K.; Antlanger, M.; et al. The Risk of Severe Infections Following Rituximab Administration in Patients with Autoimmune Kidney Diseases: Austrian ABCDE Registry Analysis. Front. Immunol. 2021, 12, 760708. [Google Scholar] [CrossRef] [PubMed]
- Tummala, R.; Abreu, G.; Pineda, L.; Michaels, M.A.; Kalyani, R.N.; Furie, R.A.; Morand, E.F. Safety profile of anifrolumab in patients with active SLE: An integrated analysis of phase II and III trials. Lupus Sci. Med. 2021, 8, e000464. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Gordon, C.; Crow, M.K.; Touma, Z.; Urowitz, M.B.; van Vollenhoven, R.; Ruiz-Irastorza, G.; Hughes, G. Systemic lupus erythematosus. Nat. Rev. Dis. Primers 2016, 2, 16039. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mak, A. T Cells, Interleukin-2 and Systemic Lupus Erythematosus—From Pathophysiology to Therapy. Cells 2022, 11, 980. https://doi.org/10.3390/cells11060980
Mak A. T Cells, Interleukin-2 and Systemic Lupus Erythematosus—From Pathophysiology to Therapy. Cells. 2022; 11(6):980. https://doi.org/10.3390/cells11060980
Chicago/Turabian StyleMak, Anselm. 2022. "T Cells, Interleukin-2 and Systemic Lupus Erythematosus—From Pathophysiology to Therapy" Cells 11, no. 6: 980. https://doi.org/10.3390/cells11060980
APA StyleMak, A. (2022). T Cells, Interleukin-2 and Systemic Lupus Erythematosus—From Pathophysiology to Therapy. Cells, 11(6), 980. https://doi.org/10.3390/cells11060980