
GM-CSF Protects Macrophages from DNA Damage by Inducing Differentiation

 ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Mice
2.3. Cell Culture
2.4. RNA Extraction and Real-Time RT-PCR
2.5. Proliferation Assay
2.6. Cytometry
2.7. Apoptosis
2.8. Cell Cycle Analysis
2.9. Western Blot Protein Analysis
2.10. DNA Damage Susceptibility Assay
2.11. Test Fraction of Activity Released (FAR) Assay
2.12. Senescence-Associated β-Galactosidase Staining
2.13. Quantification and Statistical Analysis
3. Results
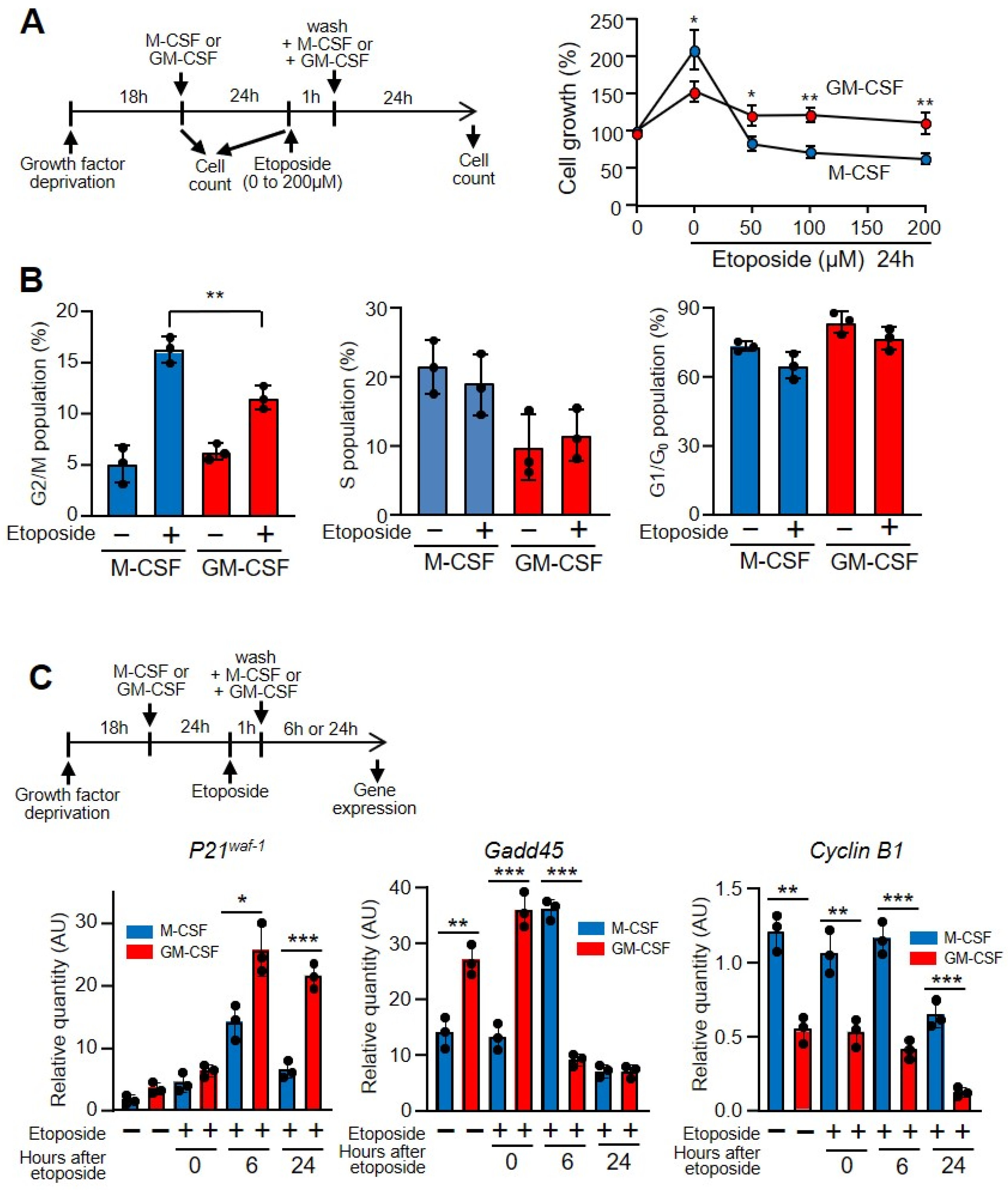
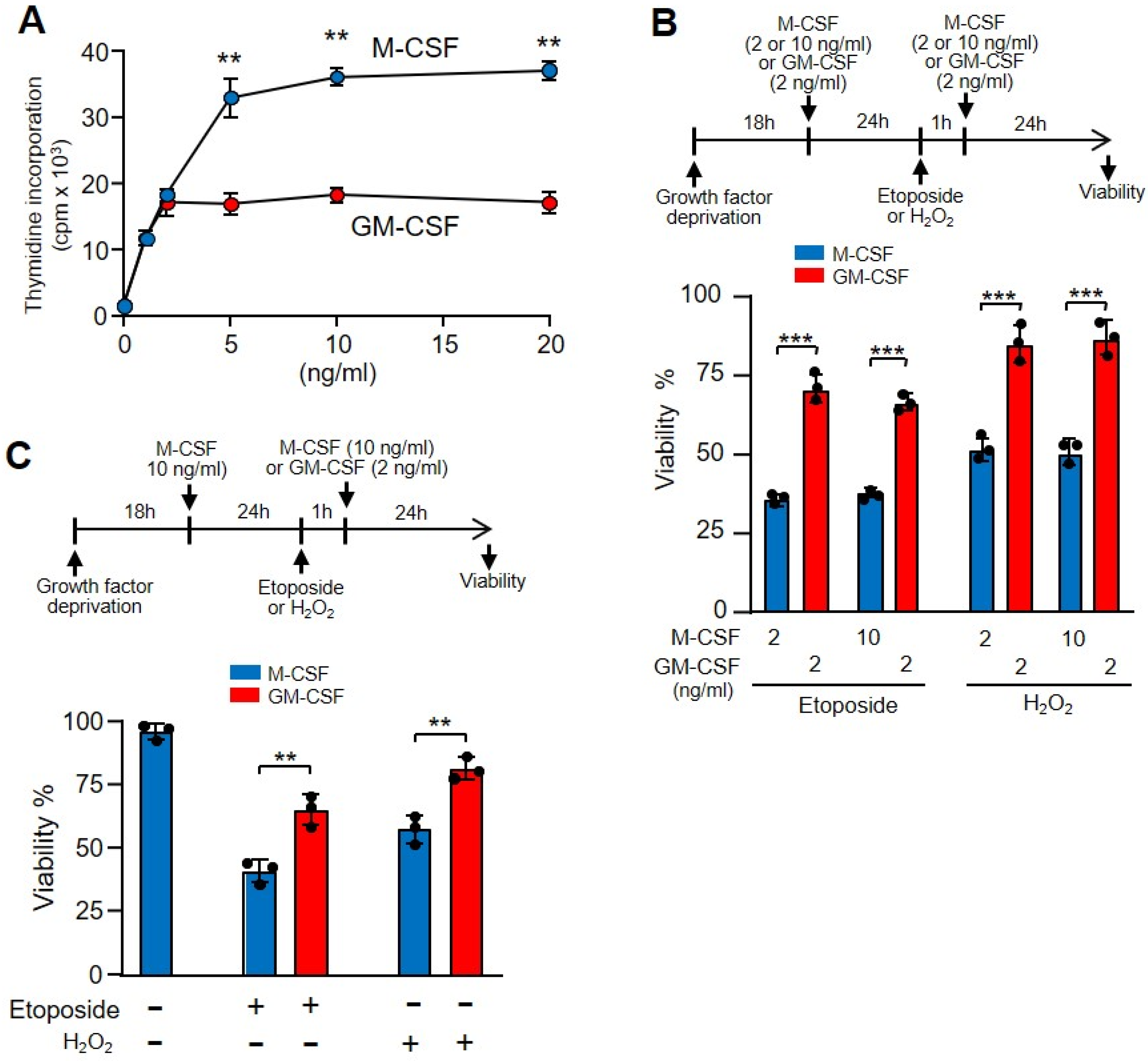
3.1. GM-CSF Induced Increased Protection against DNA Damage in Relation to M-CSF
3.2. GM-CSF Induced More Rapid Recovery from DNA Damage Than M-CSF
3.3. DNA Damage Induced Cell Cycle Impairment
3.4. GM-CSF Induced the Expression of Anti-Apoptotic B-Cell Lymphoma 2 (Bcl-2) A1
3.5. DNA Damage Was Independent of Proliferation
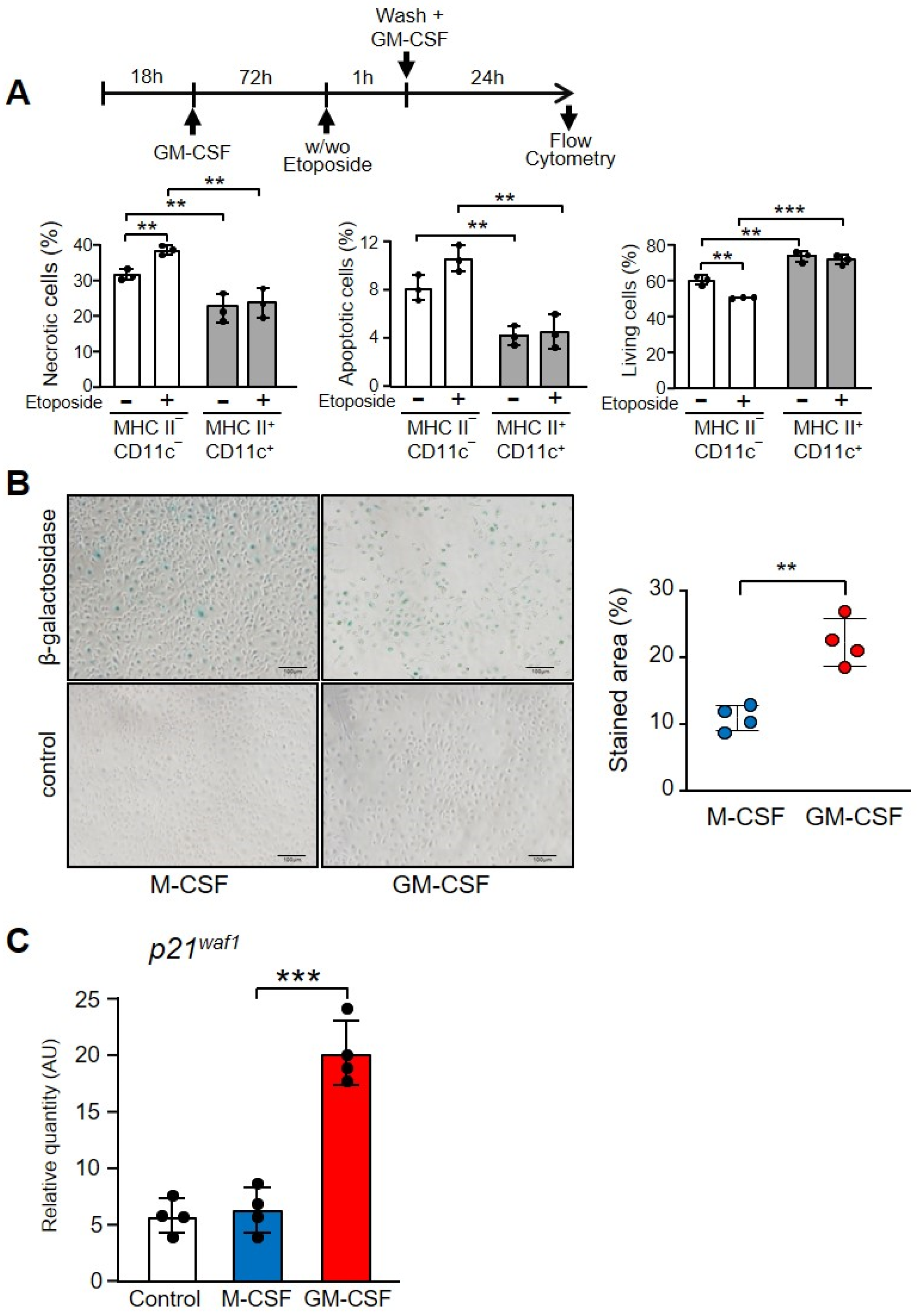
3.6. GM-CSF Induced CD11c+ MHC II+ Cells That Were Resistant to the Effect of Etoposide
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lawrence, T.; Willoughby, D.A.; Gilroy, D.W. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat. Rev. Immunol. 2002, 2, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Tur, J.; Pereira-Lopes, S.; Vico, T.; Marin, E.A.; Munoz, J.P.; Hernandez-Alvarez, M.; Cardona, P.J.; Zorzano, A.; Lloberas, J.; Celada, A. Mitofusin 2 in Macrophages Links Mitochondrial ROS Production, Cytokine Release, Phagocytosis, Autophagy, and Bactericidal Activity. Cell Rep. 2020, 32, 108079. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, M.; Goldstein, M.; Christmann, M.; Becker, H.; Heylmann, D.; Kaina, B. Human monocytes are severely impaired in base and DNA double-strand break repair that renders them vulnerable to oxidative stress. Proc. Natl. Acad. Sci. USA 2011, 108, 21105–21110. [Google Scholar] [CrossRef] [Green Version]
- Pereira-Lopes, S.; Tur, J.; Calatayud-Subias, J.A.; Lloberas, J.; Stracker, T.H.; Celada, A. NBS1 is required for macrophage homeostasis and functional activity in mice. Blood 2015, 126, 2502–2510. [Google Scholar] [CrossRef] [Green Version]
- Serra, M.; Forcales, S.V.; Pereira-Lopes, S.; Lloberas, J.; Celada, A. Characterization of Trex1 induction by IFN-gamma in murine macrophages. J. Immunol. 2011, 186, 2299–2308. [Google Scholar] [CrossRef] [Green Version]
- Valverde-Estrella, L.; Lopez-Serrat, M.; Sanchez-Sanchez, G.; Vico, T.; Lloberas, J.; Celada, A. Induction of Samhd1 by interferon gamma and lipopolysaccharide in murine macrophages requires IRF1. Eur. J. Immunol. 2020, 50, 1321–1334. [Google Scholar] [CrossRef]
- Crow, Y.J.; Stetson, D.B. The type I interferonopathies: 10 years on. Nat. Rev. Immunol. 2021, 20, 131. [Google Scholar] [CrossRef]
- Lasry, A.; Ben-Neriah, Y. Senescence-associated inflammatory responses: Aging and cancer perspectives. Trends Immunol. 2015, 36, 217–228. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Nyberg, K.A.; Michelson, R.J.; Putnam, C.W.; Weinert, T.A. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 2002, 36, 617–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darzynkiewicz, Z.; Zhao, H.; Halicka, H.D.; Rybak, P.; Dobrucki, J.; Wlodkowic, D. DNA damage signaling assessed in individual cells in relation to the cell cycle phase and induction of apoptosis. Crit. Rev. Clin. Lab. Sci. 2012, 49, 199–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozzi, P.; Mongelli, N.; Suarato, A. Recent anticancer cytotoxic agents. Curr. Med. Chem. Anti-Cancer Agents 2004, 4, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef]
- Barreda, D.R.; Hanington, P.C.; Belosevic, M. Regulation of myeloid development and function by colony stimulating factors. Dev. Comp. Immunol. 2004, 28, 509–554. [Google Scholar] [CrossRef]
- Stanley, E.R.; Berg, K.L.; Einstein, D.B.; Lee, P.S.; Pixley, F.J.; Wang, Y.; Yeung, Y.G. Biology and action of colony-stimulating factor-1. Mol. Reprod. Dev. 1997, 46, 4–10. [Google Scholar] [CrossRef]
- Comalada, M.; Xaus, J.; Sanchez, E.; Valledor, A.F.; Celada, A. Macrophage colony-stimulating factor-, granulocyte-macrophage colony-stimulating factor-, or IL-3-dependent survival of macrophages, but not proliferation, requires the expression of p21(Waf1) through the phosphatidylinositol 3-kinase/Akt pathway. Eur. J. Immunol. 2004, 34, 2257–2267. [Google Scholar] [CrossRef]
- Ushach, I.; Zlotnik, A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J. Leukoc. Biol. 2016, 100, 481–489. [Google Scholar] [CrossRef]
- Fleetwood, A.J.; Cook, A.D.; Hamilton, J.A. Functions of granulocyte-macrophage colony-stimulating factor. Crit. Rev. Immunol. 2005, 25, 405–428. [Google Scholar] [CrossRef]
- Lacey, D.C.; Achuthan, A.; Fleetwood, A.J.; Dinh, H.; Roiniotis, J.; Scholz, G.M.; Chang, M.W.; Beckman, S.K.; Cook, A.D.; Hamilton, J.A. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J. Immunol. 2012, 188, 5752–5765. [Google Scholar] [CrossRef]
- Celada, A.; Gray, P.W.; Rinderknecht, E.; Schreiber, R.D. Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J. Exp. Med. 1984, 160, 55–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celada, A.; Borras, F.E.; Soler, C.; Lloberas, J.; Klemsz, M.; van Beveren, C.; McKercher, S.; Maki, R.A. The transcription factor PU.1 is involved in macrophage proliferation. J. Exp. Med. 1996, 184, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, pdb.prot087379. [Google Scholar] [CrossRef]
- Xaus, J.; Cardo, M.; Valledor, A.F.; Soler, C.; Lloberas, J.; Celada, A. Interferon gamma induces the expression of p21waf-1 and arrests macrophage cell cycle, preventing induction of apoptosis. Immunity 1999, 11, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Pereira-Lopes, S.; Celhar, T.; Sans-Fons, G.; Serra, M.; Fairhurst, A.M.; Lloberas, J.; Celada, A. The exonuclease Trex1 restrains macrophage proinflammatory activation. J. Immunol. 2013, 191, 6128–6135. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Ismail, I.H.; Nystrom, S.; Nygren, J.; Hammarsten, O. Activation of ataxia telangiectasia mutated by DNA strand break-inducing agents correlates closely with the number of DNA double strand breaks. J. Biol. Chem. 2005, 280, 4649–4655. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.; et al. Aging of mice is associated with p16(Ink4a)- and beta-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 2016, 8, 1294–1315. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anti-Cancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, C.; Herrero, C.; Serra, M.; Lloberas, J.; Blasco, M.A.; Celada, A. Telomere shortening and oxidative stress in aged macrophages results in impaired STAT5a phosphorylation. J. Immunol. 2009, 183, 2356–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanning, E.; Klimovich, V.; Nager, A.R. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006, 34, 4126–4137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleaver, J.E.; Feeney, L.; Revet, I. Phosphorylated H2Ax is not an unambiguous marker for DNA double-strand breaks. Cell Cycle 2011, 10, 3223–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkevich, A.N.; Martin, O.A.; Smith, A.J.; Redon, C.E.; Bonner, W.M.; Martin, R.F.; Lobachevsky, P.N. gammaH2AX foci as a measure of DNA damage: A computational approach to automatic analysis. Mutat. Res. 2011, 711, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Riches, L.C.; Lynch, A.M.; Gooderham, N.J. Early events in the mammalian response to DNA double-strand breaks. Mutagenesis 2008, 23, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Stracker, T.H.; Roig, I.; Knobel, P.A.; Marjanovic, M. The ATM signaling network in development and disease. Front. Genet. 2013, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Stracker, T.H.; Petrini, J.H. Working together and apart: The twisted relationship of the Mre11 complex and Chk2 in apoptosis and tumor suppression. Cell Cycle 2008, 7, 3618–3621. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, X.; Teng, L.; Legerski, R.J. DNA damage checkpoint recovery and cancer development. Exp. Cell Res. 2015, 334, 350–358. [Google Scholar] [CrossRef]
- Lopez-Contreras, A.J.; Fernandez-Capetillo, O. The ATR barrier to replication-born DNA damage. DNA Repair 2010, 9, 1249–1255. [Google Scholar] [CrossRef] [Green Version]
- Salvador, J.M.; Brown-Clay, J.D.; Fornace, A.J., Jr. Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv. Exp. Med. Biol. 2013, 793, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Yamaguchi, N. Role of cyclin B1 levels in DNA damage and DNA damage-induced senescence. Int. Rev. Cell Mol. Biol. 2013, 305, 303–337. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M. BCL2A1: The underdog in the BCL2 family. Cell Death Differ. 2012, 19, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Trapnell, B.C.; Carey, B.C.; Uchida, K.; Suzuki, T. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr. Opin. Immunol. 2009, 21, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Helft, J.; Bottcher, J.; Chakravarty, P.; Zelenay, S.; Huotari, J.; Schraml, B.U.; Goubau, D.; Reis e Sousa, C. GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c(+)MHCII(+) Macrophages and Dendritic Cells. Immunity 2015, 42, 1197–1211. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- Kaul, S.C.; Hasan, K.; Wadhwa, R. CARF regulates p19ARF-p53-p21WAF1 senescence pathway by multiple checkpoints. Ann. N. Y. Acad. Sci. 2006, 1067, 217–219. [Google Scholar] [CrossRef]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakubzick, C.V.; Randolph, G.J.; Henson, P.M. Monocyte differentiation and antigen-presenting functions. Nat. Rev. Immunol. 2017, 17, 349–362. [Google Scholar] [CrossRef]
- So, E.Y.; Ouchi, T. Translational initiation regulated by ATM in dendritic cells development. Cell Death Dis. 2014, 5, e1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar] [CrossRef]
- Bauer, M.; Goldstein, M.; Heylmann, D.; Kaina, B. Human monocytes undergo excessive apoptosis following temozolomide activating the ATM/ATR pathway while dendritic cells and macrophages are resistant. PLoS ONE 2012, 7, e39956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinam, C.; Poueymirou, W.T.; Rojas, J.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Rongvaux, A.; Eynon, E.E.; Manz, M.G.; Flavell, R.A. Efficient differentiation and function of human macrophages in humanized CSF-1 mice. Blood 2011, 118, 3119–3128. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.A.; Kingston, D.; Boddupalli, S.; Manz, M.G. Instructive cytokine signals in dendritic cell lineage commitment. Immunol. Rev. 2010, 234, 32–44. [Google Scholar] [CrossRef]
- Yeramian, A.; Martin, L.; Serrat, N.; Arpa, L.; Soler, C.; Bertran, J.; McLeod, C.; Palacin, M.; Modolell, M.; Lloberas, J.; et al. Arginine transport via cationic amino acid transporter 2 plays a critical regulatory role in classical or alternative activation of macrophages. J. Immunol. 2006, 176, 5918–5924. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, A.; Garcia-Muse, T. Causes of genome instability. Annu. Rev. Genet. 2013, 47, 1–32. [Google Scholar] [CrossRef]
- Sebastian, C.; Serra, M.; Yeramian, A.; Serrat, N.; Lloberas, J.; Celada, A. Deacetylase activity is required for STAT5-dependent GM-CSF functional activity in macrophages and differentiation to dendritic cells. J. Immunol. 2008, 180, 5898–5906. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Orlofsky, A.; Berger, M.S.; Prystowsky, M.B. Characterization of A1, a novel hemopoietic-specific early-response gene with sequence similarity to bcl-2. J. Immunol. 1993, 151, 1979–1988. [Google Scholar] [PubMed]
- Wang, C.Y.; Guttridge, D.C.; Mayo, M.W.; Baldwin, A.S., Jr. NF-kappaB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol. Cell. Biol. 1999, 19, 5923–5929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karrich, J.J.; Balzarolo, M.; Schmidlin, H.; Libouban, M.; Nagasawa, M.; Gentek, R.; Kamihira, S.; Maeda, T.; Amsen, D.; Wolkers, M.C.; et al. The transcription factor Spi-B regulates human plasmacytoid dendritic cell survival through direct induction of the antiapoptotic gene BCL2-A1. Blood 2012, 119, 5191–5200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, M.J.; Fan, G.; Zong, W.X.; Degenhardt, K.; White, E.; Gelinas, C. Bfl-1/A1 functions, similar to Mcl-1, as a selective tBid and Bak antagonist. Oncogene 2008, 27, 1421–1428. [Google Scholar] [CrossRef] [Green Version]
- Lukens, J.R.; Barr, M.J.; Chaplin, D.D.; Chi, H.; Kanneganti, T.D. Inflammasome-derived IL-1beta regulates the production of GM-CSF by CD4(+) T cells and gammadelta T cells. J. Immunol. 2012, 188, 3107–3115. [Google Scholar] [CrossRef] [Green Version]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 2011, 12, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Kaur, K.; Bachus, H.; Lewis, C.; Papillion, A.M.; Rosenberg, A.F.; Ballesteros-Tato, A.; Leon, B. GM-CSF production by non-classical monocytes controls antagonistic LPS-driven functions in allergic inflammation. Cell Rep. 2021, 37, 110178. [Google Scholar] [CrossRef]
- Hespel, C.; Moser, M. Role of inflammatory dendritic cells in innate and adaptive immunity. Eur. J. Immunol. 2012, 42, 2535–2543. [Google Scholar] [CrossRef]
- Peschke, K.; Achleitner, M.; Frenzel, K.; Gerbaulet, A.; Ada, S.R.; Zeller, N.; Lienenklaus, S.; Lesche, M.; Poulet, C.; Naumann, R.; et al. Loss of Trex1 in Dendritic Cells Is Sufficient To Trigger Systemic Autoimmunity. J. Immunol. 2016, 197, 2157–2166. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Tak, P.P. The dynamics of macrophage lineage populations in inflammatory and autoimmune diseases. Arthritis Rheum. 2009, 60, 1210–1221. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.N.; Chen, M.; Jongstra-Bilen, J.; Cybulsky, M.I. GM-CSF regulates intimal cell proliferation in nascent atherosclerotic lesions. J. Exp. Med. 2009, 206, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.D.; Turner, A.L.; Braine, E.L.; Pobjoy, J.; Lenzo, J.C.; Hamilton, J.A. Regulation of systemic and local myeloid cell subpopulations by bone marrow cell-derived granulocyte-macrophage colony-stimulating factor in experimental inflammatory arthritis. Arthritis Rheum. 2011, 63, 2340–2351. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, C.; Espia, M.; Serra, M.; Celada, A.; Lloberas, J. MacrophAging: A cellular and molecular review. Immunobiology 2005, 210, 121–126. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vico, T.; Youssif, C.; Zare, F.; Comalada, M.; Sebastian, C.; Lloberas, J.; Celada, A. GM-CSF Protects Macrophages from DNA Damage by Inducing Differentiation. Cells 2022, 11, 935. https://doi.org/10.3390/cells11060935
Vico T, Youssif C, Zare F, Comalada M, Sebastian C, Lloberas J, Celada A. GM-CSF Protects Macrophages from DNA Damage by Inducing Differentiation. Cells. 2022; 11(6):935. https://doi.org/10.3390/cells11060935
Chicago/Turabian StyleVico, Tania, Catrin Youssif, Fathema Zare, Mònica Comalada, Carlos Sebastian, Jorge Lloberas, and Antonio Celada. 2022. "GM-CSF Protects Macrophages from DNA Damage by Inducing Differentiation" Cells 11, no. 6: 935. https://doi.org/10.3390/cells11060935
APA StyleVico, T., Youssif, C., Zare, F., Comalada, M., Sebastian, C., Lloberas, J., & Celada, A. (2022). GM-CSF Protects Macrophages from DNA Damage by Inducing Differentiation. Cells, 11(6), 935. https://doi.org/10.3390/cells11060935