
Computational Workflow to Study the Diversity of Secondary Metabolites in Fourteen Different Isatis Species

Abstract
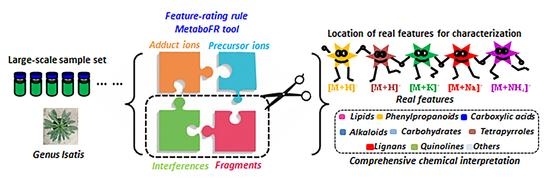
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Standard Solution Preparation
2.3. Sample Preparation
2.4. LC-MS Analysis
2.5. Data Processing
2.6. Statistical Analysis
2.7. Metabolite Identification
3. Results and Discussion
3.1. The Workflow of “Feature-Rating” Rule and MetaboFR
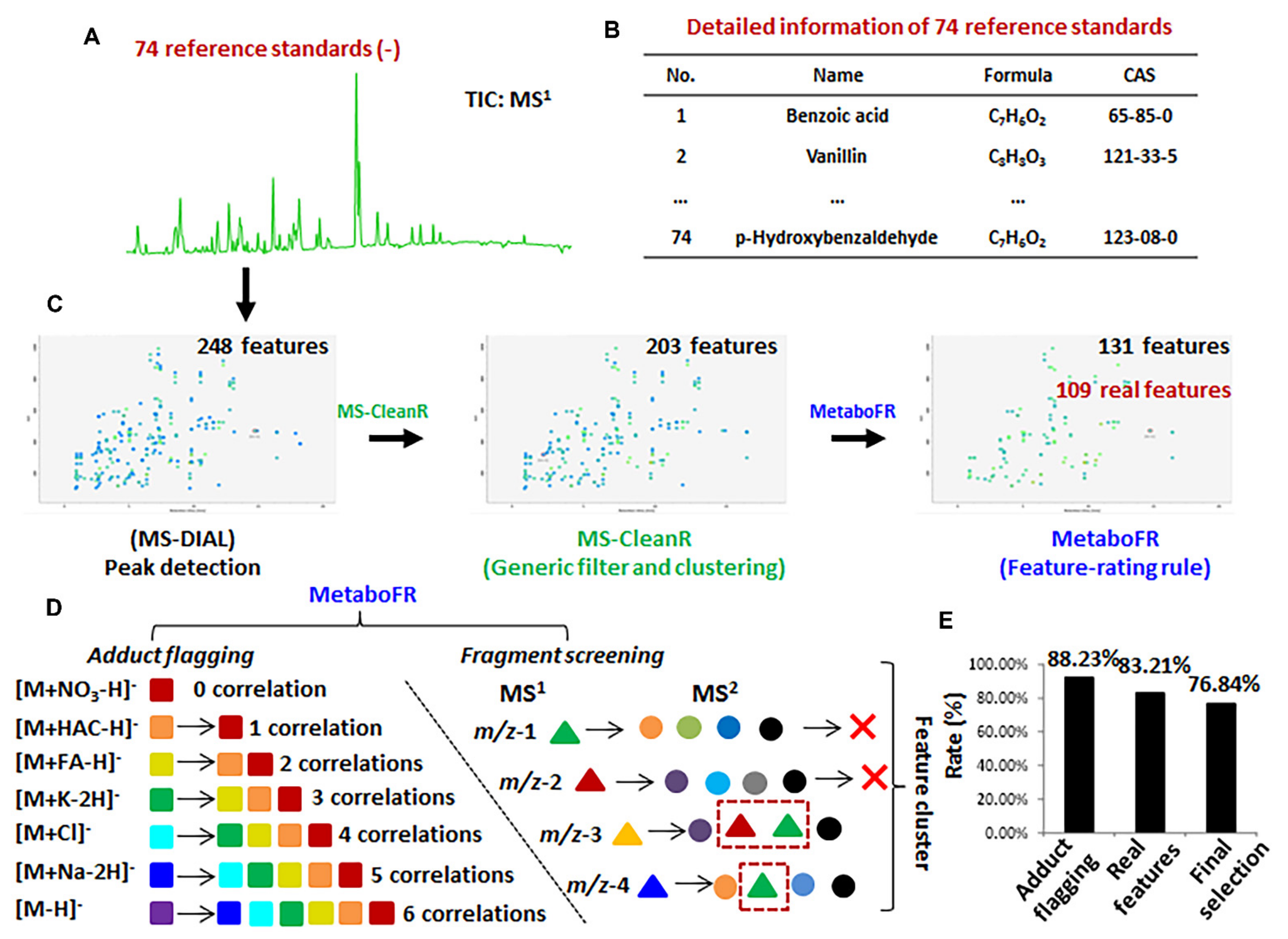
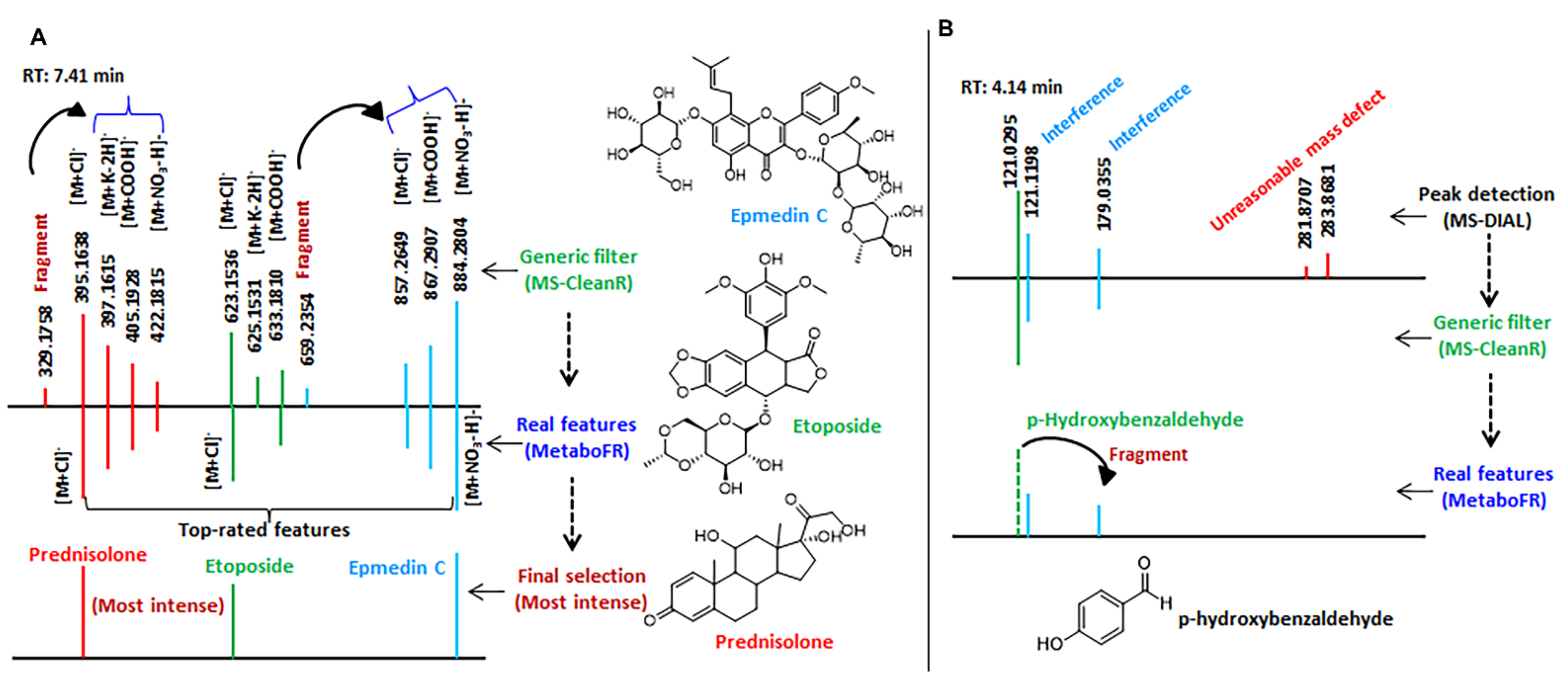
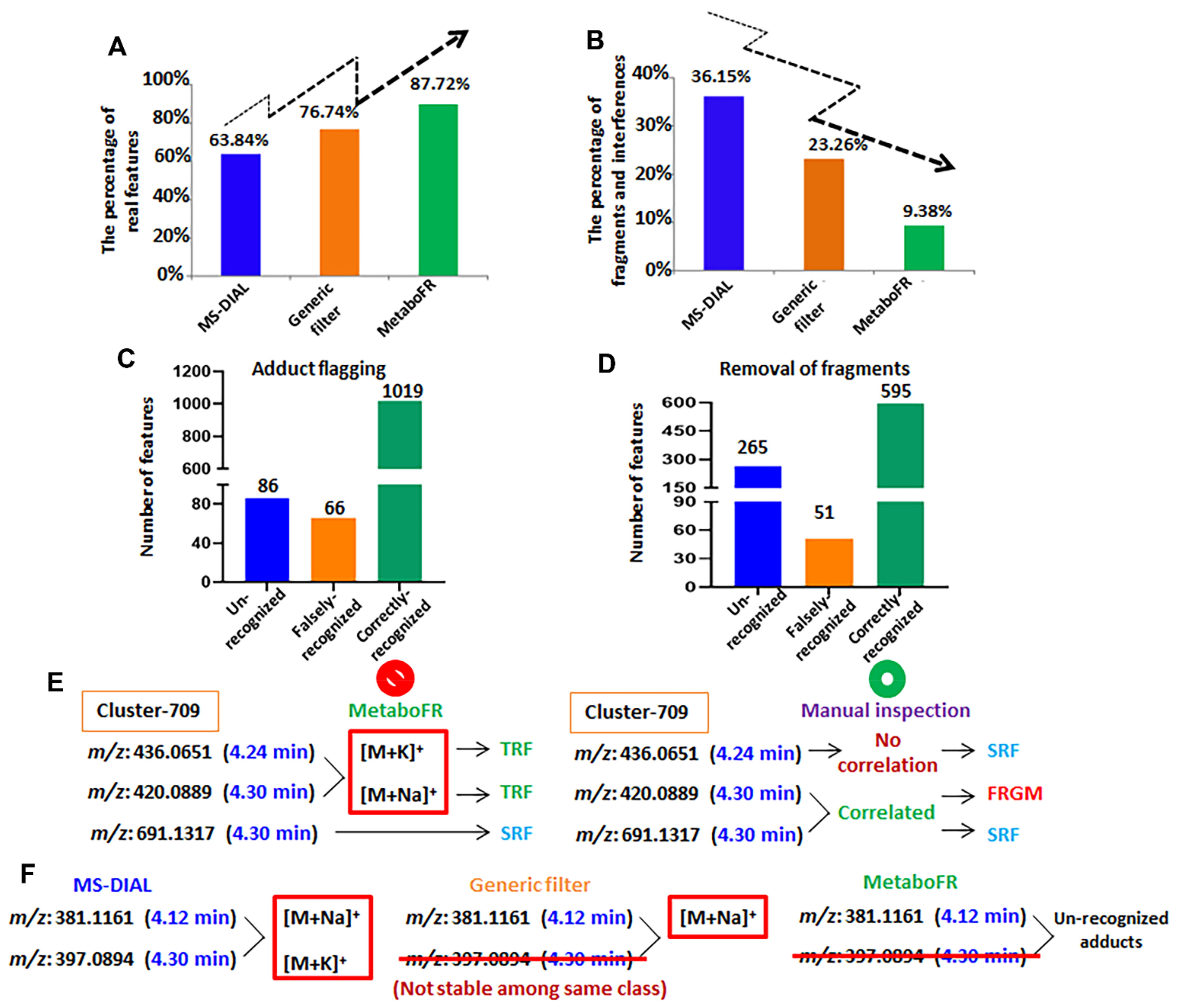
3.2. Validation of Workflow by Applying Reference Standards
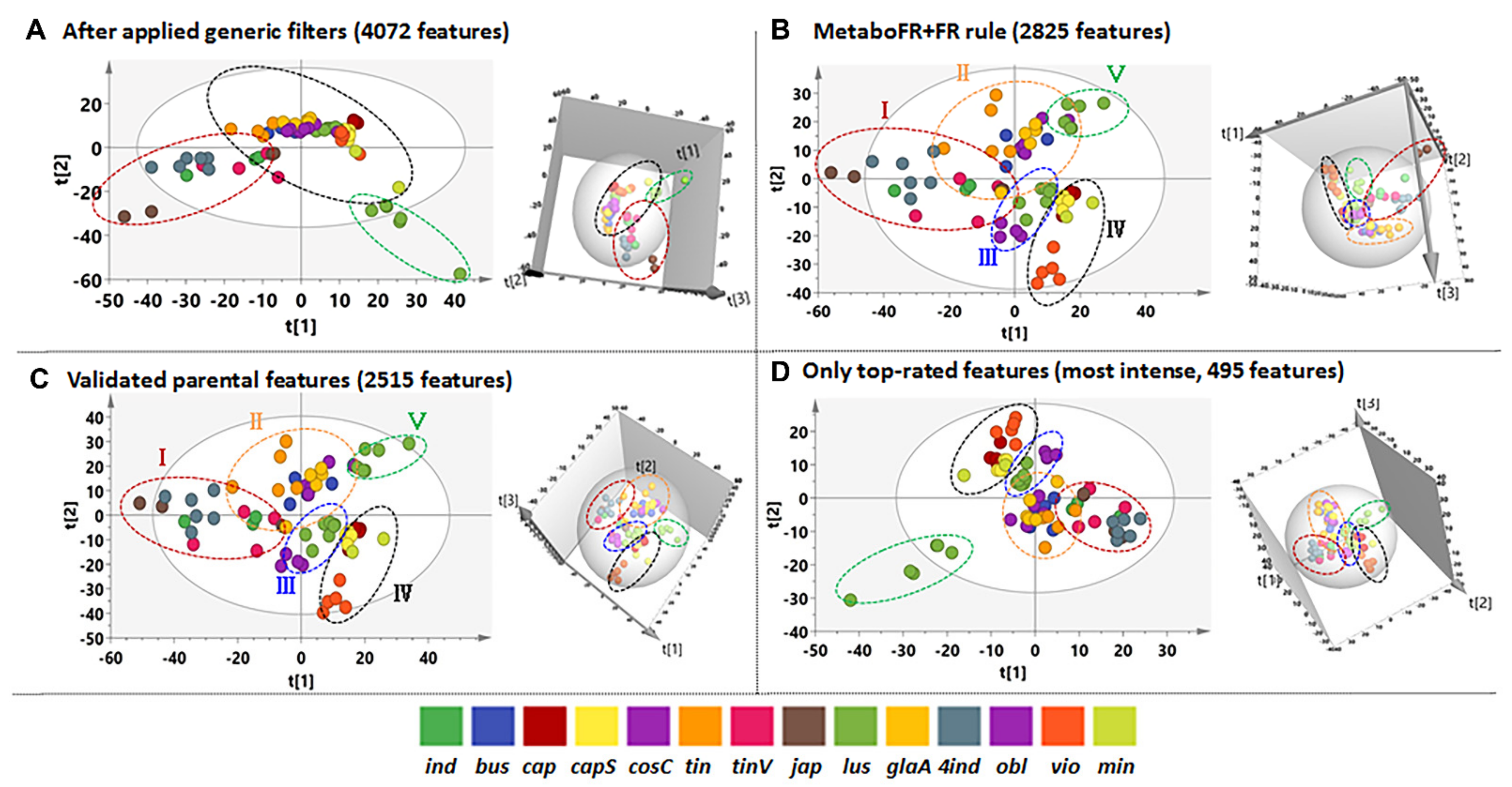
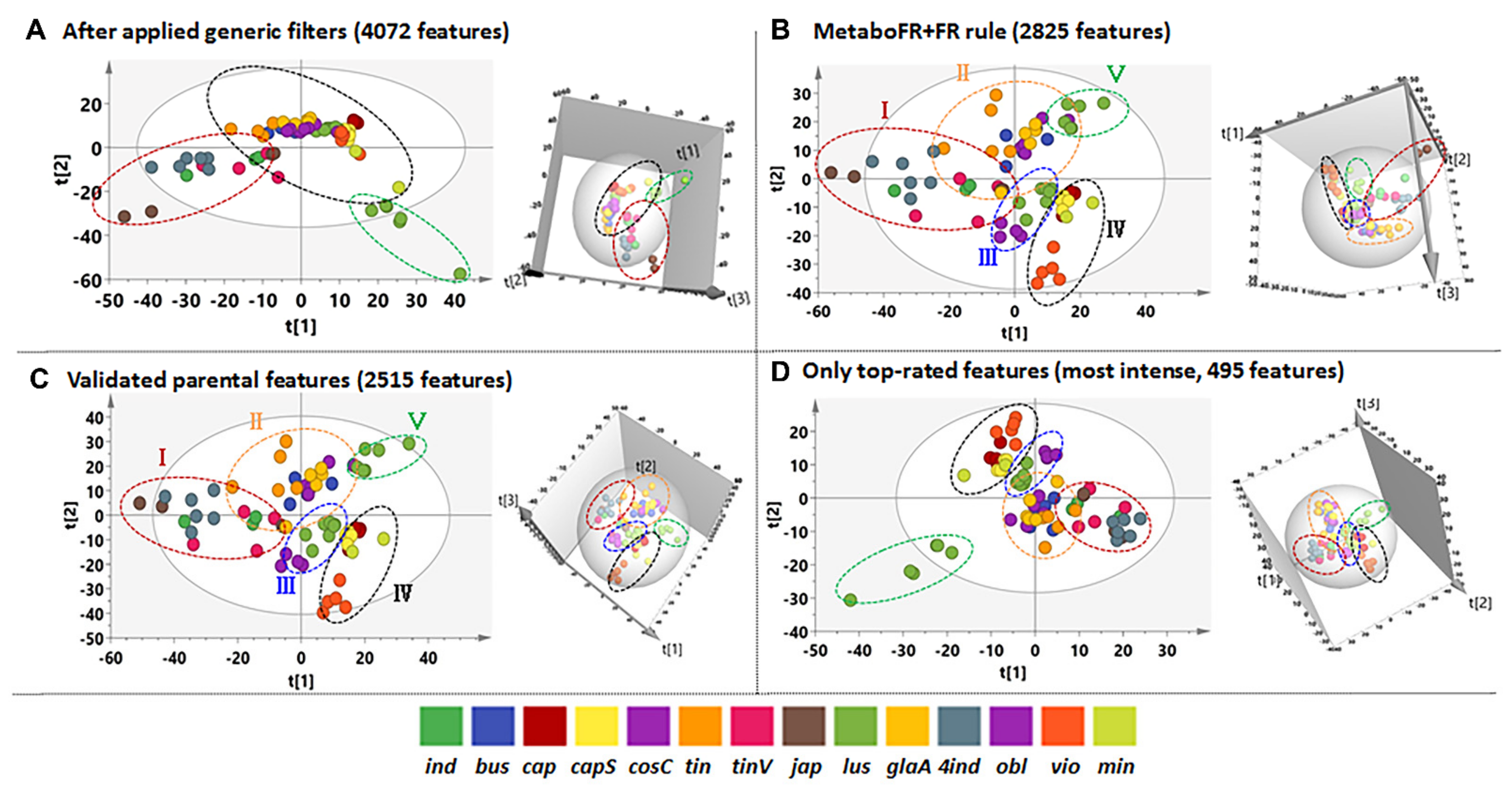
3.3. Application of Workflow on Multicomplex Samples
3.4. Detailed Annotation of Peak Tables after MetaboFR and FR Analysis of Genus Isatis
3.5. Chemical Analysis of Genus Isatis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, C.; Zhang, J.M.; Wu, R.J.; Liu, Y.; Hu, X.; Yan, Y.Q.; Ling, X.M. A novel strategy for rapidly and accurately screening biomarkers based on ultraperformance liquid chromatography-mass spectrometry metabolomics data. Anal. Chim. Acta 2019, 31, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Treutler, H.; Tsugawa, H.; Porzel, A.; Gorzolka, K.; Tissier, A.; Neumann, S.; Balcke, G.U. Discovering regulated metabolite families in untargeted metabolomics studies. Anal. Chem. 2016, 88, 8082–8090. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef]
- Gorrochategui, E.; Jaumot, J.; Lacorte, S.; Tauler, R. Data analysis strategies for targeted and untargeted LC-MS metabolomics studies: Overview and workflow. Trends Anal. Chem. 2016, 82, 425–442. [Google Scholar] [CrossRef]
- Wolfender, J.J.; Nuzillard, J.M.; Van der Hooft, J.J.J.; Renault, J.H.; Bertrand, S. Accelerating metabolite identification in natural product research: Toward an ideal combination of liquid chromatography-high-resolution tandem mass spectrometry and NMR profiling, in silico databases, and chemometrics. Anal. Chem. 2019, 91, 704–742. [Google Scholar] [CrossRef]
- Lai, Z.J.; Tsugawa, H.; Wohlgemuth, G.; Mehta, S.; Mueller, M.; Zheng, Y.X.; Ogiwara, A.; Meissen, J.; Showalter, M.; Takeuchi, K.; et al. Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nat. Methods 2018, 15, 53–56. [Google Scholar] [CrossRef]
- Kind, T.; Tsugawa, H.; Cajka, T.; Ma, Y.; Lai, Z.J.; Mehta, S.S.; Wohlgemuth, G.; Barupal, D.K.; Showalter, M.R.; Arita, M.; et al. Identification of small molecules using accurate mass MS/MS search. Mass Spec. Rev. 2017, 37, 513–532. [Google Scholar] [CrossRef]
- Gowda, H.; Ivanisevic, J.; Johnson, C.H.; Kurczy, M.E.; Benton, H.P.; Rinehart, D.; Nguyen, T.; Ray, J.; Kuehl, J.; Arevalo, B.; et al. Interactive XCMS Online: Simplifying Advanced Metabolomic Data Processing and Subsequent Statistical Analyses. Anal. Chem. 2014, 86, 6931–6939. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Plyushchenko, I.; Shakhmatov, D.; Bolotnik, T.; Baygildiev, T.; Nesterenko, P.N.; Rodin, I. An approach for feature selection with data modelling in LC-MS metabolomics. Anal. Methods 2020, 12, 3582–3591. [Google Scholar] [CrossRef] [PubMed]
- Ran, J.; Liu, X.Y.; Zheng, F.J.; Zhao, X.J.; Lu, X.; Zeng, Z.D.; Lin, X.H.; Xu, G.W. Removal of false positive features to generate authentic peak table for high-resolution mass spectrometry-based metabolomics study. Anal. Chim. Acta 2019, 1067, 79–87. [Google Scholar]
- Tian, L.; Lin, L.; Bayen, S. Optimization of the post-acquisition data processing for the non-targeted screening of trace leachable residues from reusable plastic bottles by high performance liquid chromatography coupled to hybrid quadrupole time of flight mass spectrometry. Talanta 2019, 193, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Marney, L.C.; Siegler, W.C.; Parsons, B.A.; Hoggard, J.C.; Wright, B.W.; Synovec, R.E. Tile-based Fisher-ratio software for improved feature selection analysis of comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry data. Talanta 2013, 115, 887–895. [Google Scholar] [CrossRef]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Broeckling, C.D.; Afsar, F.A.; Neumann, S.; Ben-Hur, A.; Prenni, J.E. RAMClust: A Novel Feature Clustering Method Enables Spectral-Matching-Based Annotation for Metabolomics Data. Anal. Chem. 2014, 86, 6812–6817. [Google Scholar] [CrossRef]
- Fraisier-Vannier, O.; Chervin, J.; Cabanac, G.; Puech, V.; Fournier, S.; Durand, V.; Amiel, A.; André, O.; Benamar, O.A.; Dumas, B.; et al. MS-CleanR: A feature-filtering workflow for untargeted LC-MS based metabolomics. Anal. Chem. 2020, 92, 9971–9981. [Google Scholar] [CrossRef]
- Kang, K.B.; Ernst, M.; van der Hooft, J.J.; da Silva, R.R.; Park, J.; Medema, M.H.; Sung, S.H.; Dorrestein, P.C. Comprehensive mass spectrometry-guided phenotyping of plant specialized metabolites reveals metabolic diversity in the cosmopolitan plant family Rhamnaceae. Plant J. 2019, 98, 1134–1144. [Google Scholar] [CrossRef] [Green Version]
- Speranza, J.; Miceli, N.; Taviano, M.F.; Ragusa, S.; Kwiecien, I.; Szopa, A.; Ekiert, H. Isatis tinctoria L. (Woad): A review of its botany, ethnobotanical uses, phytochemistry, biological activities, and biotechnological studies. Plants 2020, 9, 298–337. [Google Scholar]
- Qin, G.W.; Xu, R.S. Recent advances on bioactive natural products from Chinese medicinal plants. Med. Res. Rev. 1998, 6, 375–382. [Google Scholar] [CrossRef]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Depicting the inhibitory potential of polyphenols from Isatis indigotica root against the main protease of SARS CoV-2 using computational approaches. J. Biomol. Struct. Dyn. 2020, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Feng, J.X.; Li, Q.; Zhou, Y.Y.; Bu, Q.T.; Zhou, J.H.; Tan, H.X.; Yang, Y.B.; Zhang, L.; Chen, W.S. IiWRKY34 positively regulates yield, lignan biosynthesis and stress tolerance in Isatis indigotica. Acta Pharm. Sin. B 2020, 10, 2417–2432. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.L.; Ma, Y.; He, Y.Q.; Yang, H.; Chen, Y.H.; Wang, L.; Huang, D.D.; Qiu, S.; Tao, X.; Chen, W.S. A Network Pharmacology-Based Investigation to the Pharmacodynamic Material Basis and Mechanisms of the Anti-Inflammatory and Anti-Viral Effect of Isatis indigotica. Drug. Des. Dev. Ther. 2021, 15, 3193–3206. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Zhang, C.; Tu, Y.; Chen, J.F.; Li, Q.; Zeng, Z.; Wang, F.Y.; Sun, L.N.; Huang, D.D.; Li, M.M.; et al. Biosynthesis-based spatial metabolome of Salvia miltiorrhiza Bunge by combining metabolomics approaches with mass spectrometry-imaging. Talanta 2022, 238, 123045. [Google Scholar] [CrossRef]
- Tsugawa, H.; Kind, T.; Nakabayashi, R.; Yukihira, D.; Tanaka, W.; Cajka, T.; Saito, K.; Fiehn, O.; Arita, M. Hydrogen rearrangement rules: Computational MS/MS fragmentation and structure elucidation using MS-FINDER software. Anal. Chem. 2016, 88, 7946–7958. [Google Scholar] [CrossRef] [PubMed]
- Feunang, Y.D.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 61–80. [Google Scholar] [CrossRef] [Green Version]
- Broadhurst, D.; Kell, B. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Nakabayashi, R.; Mori, T.; Yamada, Y.; Takahashi, M.; Rai, A.; Sugiyama, R.; Yamamoto, H.; Nakaya, T.; Yamazaki, M.; et al. A cheminformatics approach to characterize metabolomes in stable-isotope-labeled organisms. Nat. Methods 2019, 16, 295–298. [Google Scholar] [CrossRef]
- Nguyen, T.; Marcelo, P.; Gontier, E.; Dauwe, R. Metabolic markers for the yield of lipophilic indole alkaloids in dried woad leaves (Isatis tinctoria L.). Phytochemistry 2019, 163, 89–98. [Google Scholar] [CrossRef]
- Nguyen, T.K.O.; Jamali, A.; Grand, E.; Morreel, K.; Marcelo, P.; Gontier, E.; Dauwe, R. Phenylpropanoid profiling reveals a class of hydroxycinnamoyl glucaric acid conjugates in Isatis tinctoria leaves. Phytochemistry 2017, 144, 127–140. [Google Scholar] [CrossRef]
- Mohn, T.; Plitzko, I.; Hamburger, M. A comprehensive metabolite profiling of Isatis tinctoria leaf extracts. Phytochemistry 2009, 70, 924–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, E.; Nisani, S.; Chamovitz, D.A. Indole-3-carbinol: A plant hormone combatting cancer. F1000Research 2018, 7, 689–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Lan, H.Y.; Peng, W.; Rahman, K.; Liu, Q.C.; Luan, X.; Zhang, H. Isatis indigotica: A review of phytochemistry, pharmacological activities and clinical applications. J. Pharm. Pharmacol. 2021, 73, 1137–1150. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, D.; Zhang, C.; Chen, J.; Xiao, Y.; Li, M.; Sun, L.; Qiu, S.; Chen, W. Computational Workflow to Study the Diversity of Secondary Metabolites in Fourteen Different Isatis Species. Cells 2022, 11, 907. https://doi.org/10.3390/cells11050907
Huang D, Zhang C, Chen J, Xiao Y, Li M, Sun L, Qiu S, Chen W. Computational Workflow to Study the Diversity of Secondary Metabolites in Fourteen Different Isatis Species. Cells. 2022; 11(5):907. https://doi.org/10.3390/cells11050907
Chicago/Turabian StyleHuang, Doudou, Chen Zhang, Junfeng Chen, Ying Xiao, Mingming Li, Lianna Sun, Shi Qiu, and Wansheng Chen. 2022. "Computational Workflow to Study the Diversity of Secondary Metabolites in Fourteen Different Isatis Species" Cells 11, no. 5: 907. https://doi.org/10.3390/cells11050907
APA StyleHuang, D., Zhang, C., Chen, J., Xiao, Y., Li, M., Sun, L., Qiu, S., & Chen, W. (2022). Computational Workflow to Study the Diversity of Secondary Metabolites in Fourteen Different Isatis Species. Cells, 11(5), 907. https://doi.org/10.3390/cells11050907