
Role of the Circadian Clock “Death-Loop” in the DNA Damage Response Underpinning Cancer Treatment Resistance

 , ,
, ,  and
and 
Abstract
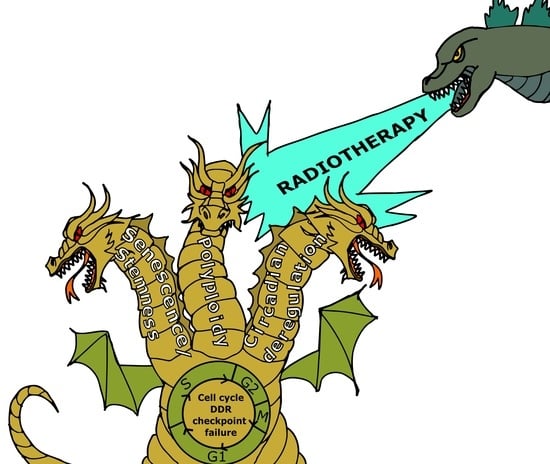
1. Introduction

{kind=link}
{kind=link}
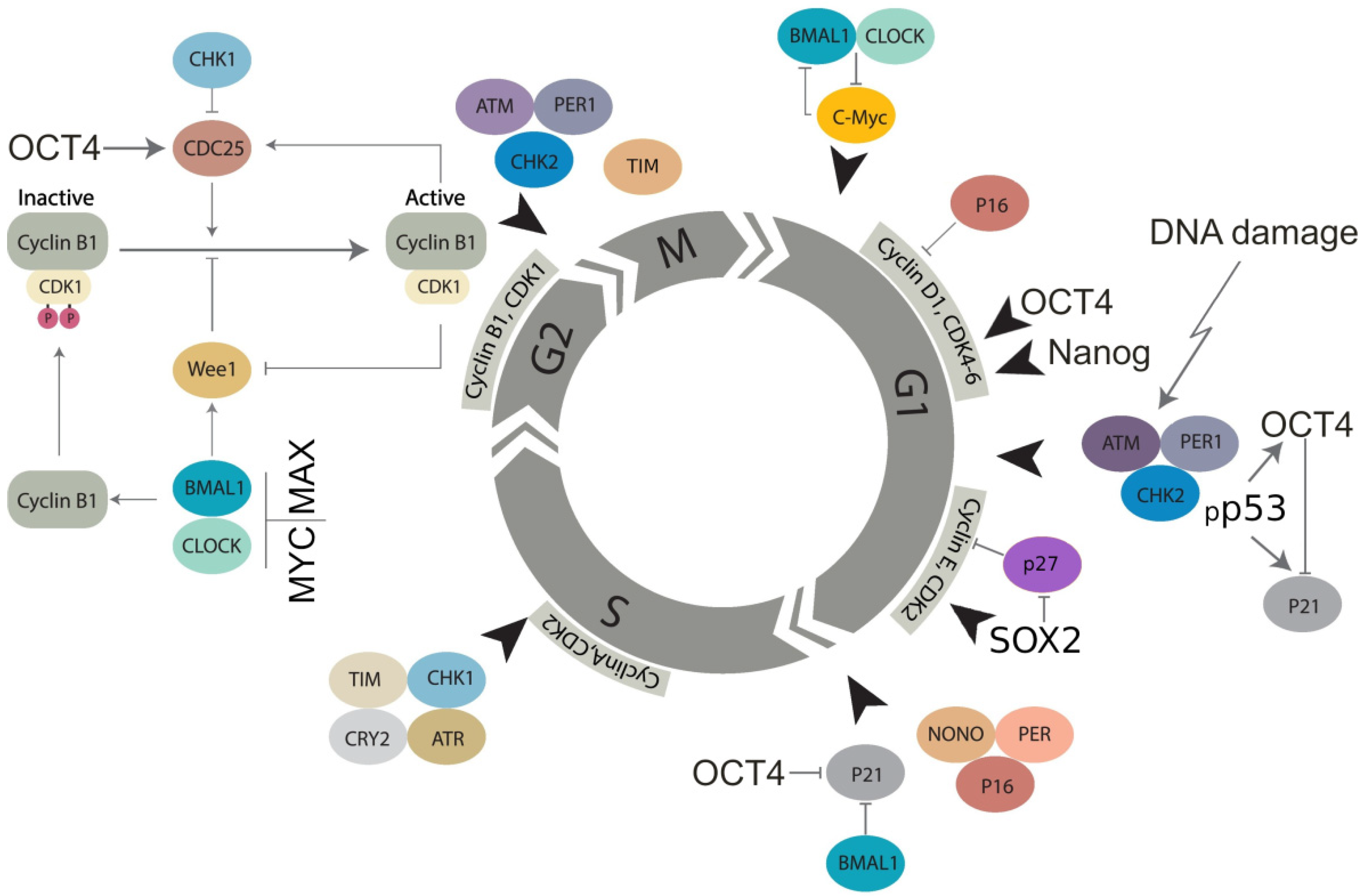{kind=link}
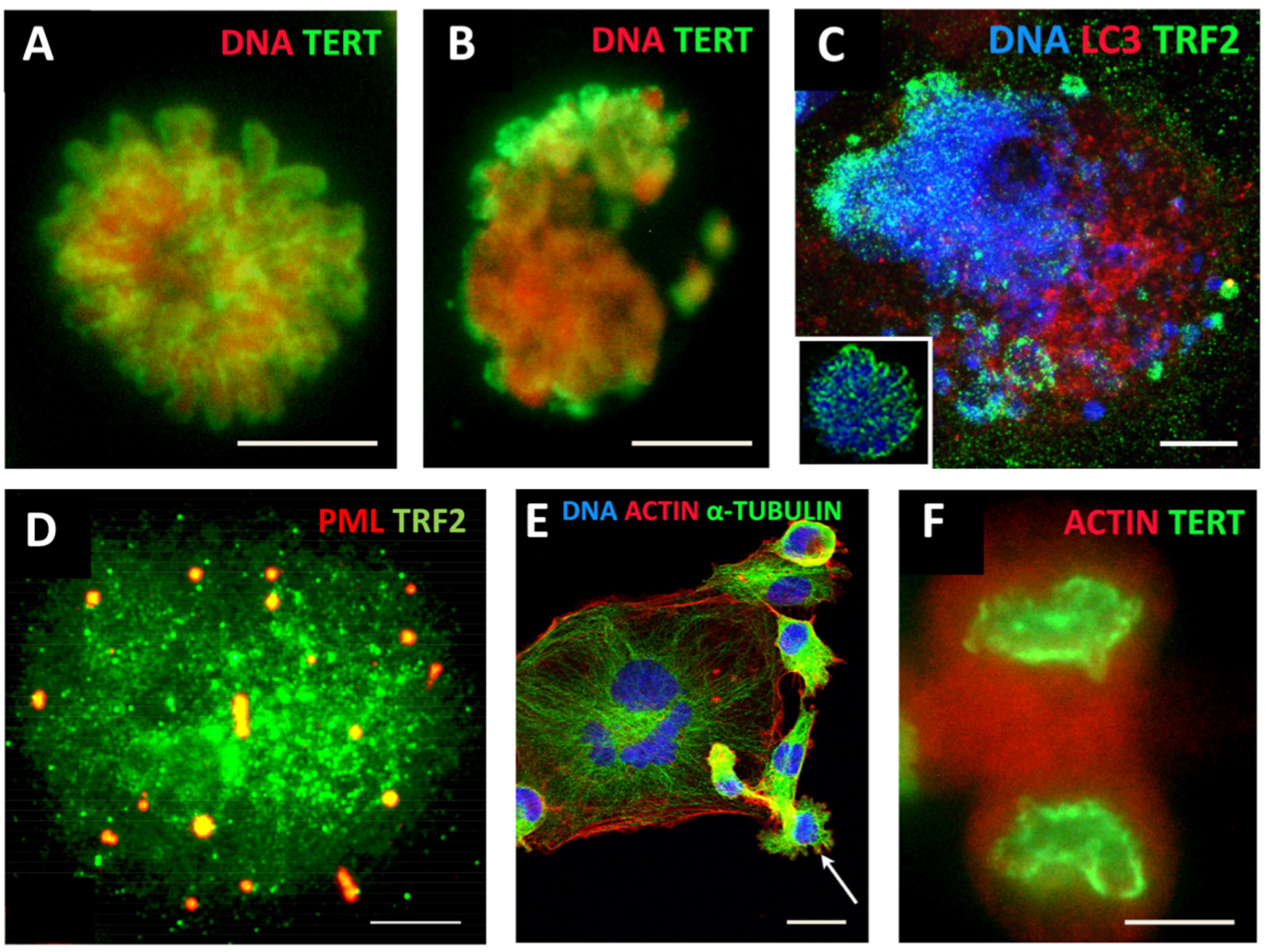{kind=link}
{kind=link}
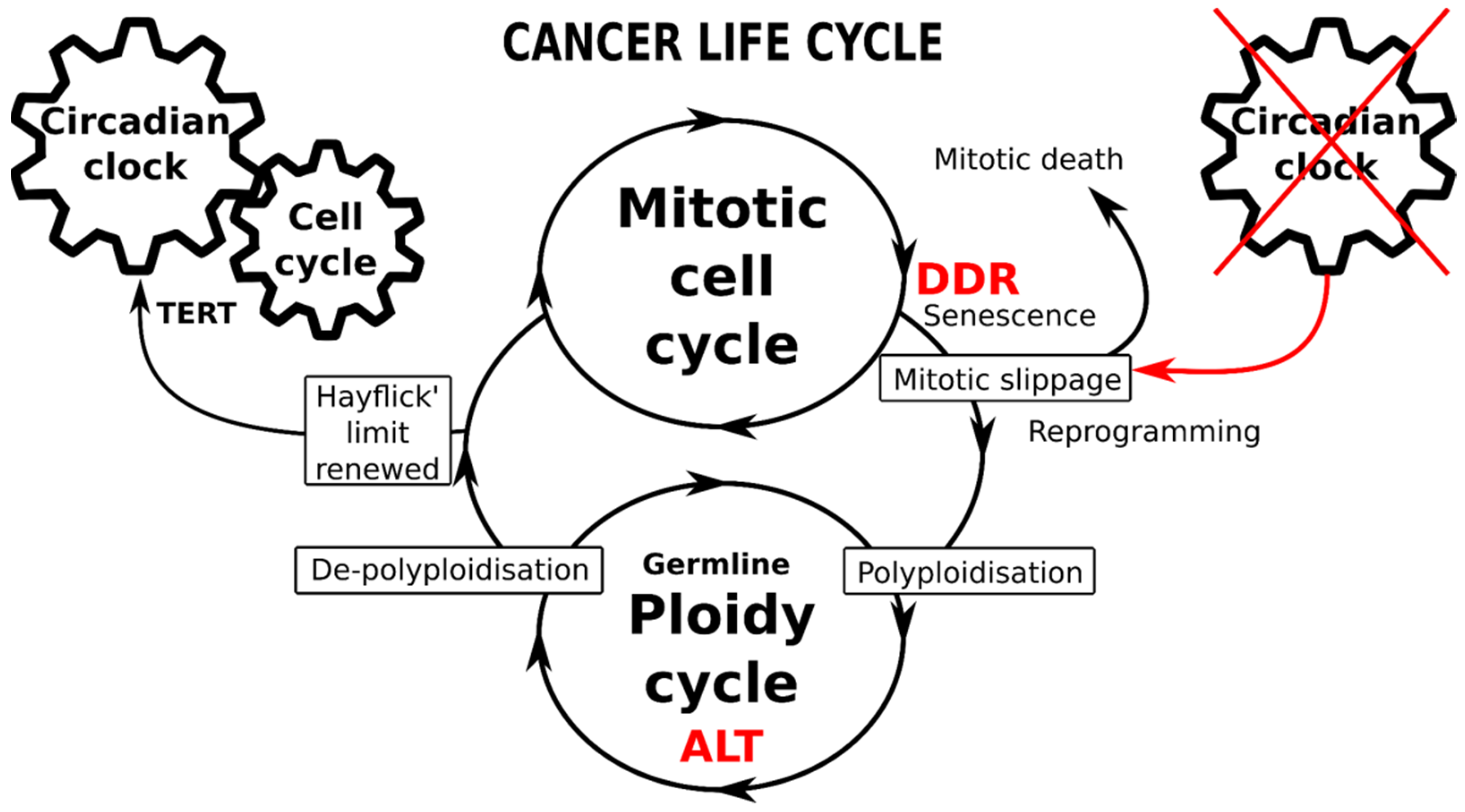{kind=link}
| Cancer Type | Anticancer Treatments | Experiment Type and The Results | Source |
|---|---|---|---|
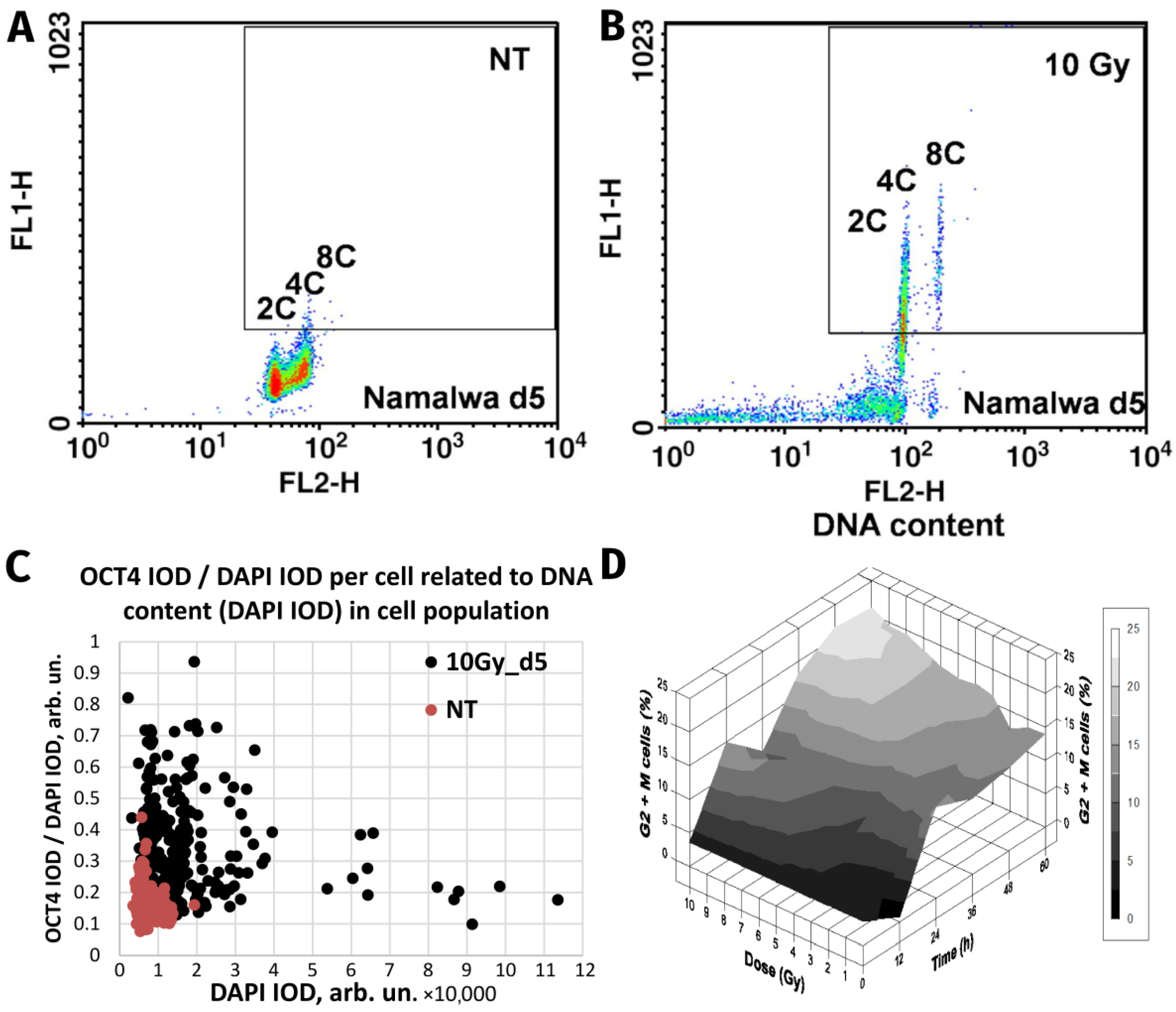
| Burkitt’s lymphoma Namalwa and Ramos | Ionising radiation (single dose of 10 Gy) | In vitro. DNA flow cytometry of induced reversible polyploidy; separation of >4C DNA by FACS, clonogenicity of the labelled polyploid fraction; detailed microscopy. | [9,59] |
| Transformed cell lines, cervical carcinoma, renal adenocarcinoma, neuro-blastoma | Ionising radiation, etoposide | In vitro. Computerised video-time-lapse microscopy recording of polyploidisation followed by bursting or budding of small cells restarting mitosis | [10] |
| Colon carcinoma DHD-K12-TRb (PROb) (rat) | Cisplatin | In vitro. Prolonged observation revealed delayed emergence of a limited number of extensive colonies which originate from polyploid cells, as demonstrated by cell sorting analysis. These colonies are made of small diploid cells which differ from parental cells by increased resistance to cytotoxic drugs. | [55] |
| Colorectal carcinoma HT 116 | Nocodazole | In vitro. Fluorescence-activated cell FACS-purified cells with an 8n DNA content formed colonies that gave rise to a ~2n generation, which was followed by video-microscopy; the plating efficiency was higher for the TP53−/− subline. | [60] |
| Lymphoblastoma (WI-L2-NS, TK6), Burkitt’s lymphoma (Namalwa) | Ionising radiation (single dose of 10 Gy) | In vitro. Induction of reversible polyploidy upregulates OCT4, NANOG, and SOX2), which facilitate survival suppressed by retinoic acid. Dependence on mutant TP53 status. | [34] |
| Fibrosarcoma (mouse) | Doxorubicin | In vitro. Induced and isolated single giant cell allografts cause metastatic cancer. | [21] |
| NK/Ly lymphoma mouse | Vinblastine | In vivo. An increased number of giant cells were induced by vinblastine treatment and observed microscopically in tumour-bearing mice. | [61] |
| Colorectal carcinoma HCT116 modified lines | H2O2 | Tetraploid cell line established from parental diploid HCT116 via cell fusion revealed the superiority of tetraploidy over p53 for cell survival when compared by cell viability, cell cycle, and apoptotic response to H2O2 with parental HCT116 and p53- inactivated sublines. | [62] |
| Breast carcinoma | Ionising radiation (single dose of 4 and 8 Gy) | Ex vivo. patient samples, ionising radiation reprogrammed differentiated breast cancer cells into induced stem cells. They showed increased mammosphere formation and increased tumorigenicity in xenografts. Reprogramming occurred in a polyploid subpopulation of cells, coinciding with re-expression of the transcription factors Oct4, SOX2, Nanog, and Klf4, and could be partially prevented by Notch inhibition. | [13] |
| Non-small cell lung cancer in patients, NCI-H1299 cell line | Camptothecin, doxorubicin, cisplatin | Ex vivo: Clinicopathological study in patients with locally advanced non-small-cell lung cancer demonstrate that therapy-induced senescent cells following neoadjuvant therapy are prognostic of an adverse clinical outcome. In vitro: polyploid senescent cells represent transition states through which escape preferentially occurs. | [63] |
| Breast carcinoma T-47D and ZR-75-1 | Genotoxic drugs and mTOR inhibitors | In vitro. Inhibition of mTOR signalling prevents the polyploidy/senescence induced by genotoxic drugs and increases cell chemosensitivity. | [64] |
| Colorectal carcinoma HCT-116 and Caco-2 cell lines | 5-fluorouracil and oxaliplatin | In vitro. CoCl2 induction of hypoxia in colon cancer cells causes the formation of PGCCs, the expansion of a cell subpopulation with CSC characteristics and chemoresistance. | [65] |
| Virally transformed rat fibroblasts with suppressed apoptosis in E1A + E1B cell lines | Ionising radiation | In vitro. Permanent activation of DDR signalling due to impaired DNA repair results in the induction of cellular senescence in E1A + E1B cells. However, irradiated cells bypass senescence and restore the population by dividing cells, which have a near-normal size and ploidy and do not express senescence markers. | [66] |
| Ovarian adenocarcinoma, breast carcinoma (HEY, SKOv3, and MDA-MB-231) | Cisplatin | In vitro and in vivo. Separation of induced PGCCs by CoCl2; characterisation of stemness, observation of budding offspring, A single PGCC formed cancer spheroids in vitro and generated tumorigenic xenografts. | [11] |
| Multiple human tumour types | Etoposide, doxorubicin, ionising radiation | In vitro and in vivo. Cell lines, time-lapse video microscopy observing budding of survivors from giant tumour cells; tumour xenografts. | [22,38] |
| Ovarian carcinoma (SKVO3, IGROV-1 cell lines) | carboplatin | In vitro. Generation and depolyploidisation of PGCCs by multipolar divisions and budding (time-lapse life cell imaging). Induction of EMT and senescence markers. | [67] |
| N-RA(61K)-mutant pigment cell culture cell | Doxycycline-inducible activation of oncogenic N-RAS | In vitro. Multinuclear senescent cells are induced, giving rise to mononuclear tumour progeny observed by time-lapse microscopy. The progeny is tumorigenic in xenografts. | [68] |
| Colorectal carcinoma (HC116) | Doxorubicin | In vitro. The cells which, along with therapy-induced senescence, undergo polyploidisation are prone to regaining the ability to proliferate. | [53] |
| Ovarian carcinoma (Hey, SKOV3, OVCAR433) | Paclitaxel | In vitro. Generation of genomically altered tumour-initiating cells through a giant cell cycle that contributes to tumour relapse was observed using live-cell fluorescence time-lapse microscopy. PGCCs were shown to self-renew via endoreplication and divide by nuclear budding or fragmentation. | [69] |
| Breast carcinoma | Doxorubicin + paclitaxel | Ex vivo. Sampling before and after neoadjuvant therapy. Induction of depolyploidising PGCCs positive for OCT4, SOX2, NANOG, and CD44 was mainly observed in near-triploid resistant cases. | [70] |
| Ovarian carcinoma (Hey, SKOV3, and MDA-HGSC-1 cell lines) | Paclitaxel | In vitro and in vivo. The obtained single PGCCs formed spheroids with the properties of blastomeres, including differentiation into three germ layers and formation of carcinoma, germ cell tumours, as well as benign tissue, in xenografts. | [37] |
| Prostate carcinoma PC3 line | Docetaxel | In vitro. A micro-fabricated “evolution accelerator” environment for controllable in vitro with a spatially varying drug concentration. The authors observed the rapid emergence of a large number of PGCCs with EMT marks at a very high drug concentration. | [15] |
| Glioblastoma T98G, A172, R2, T1 cell lines | Ionising radiation; Fotemustine | In vitro. The resistant cell lines displayed the PGCCs and high activity of tumour and microenvironment promoting genes. | [71] |
| Breast carcinoma and mouse melanoma | 5-fluorouracil | In vitro and in vivo. The authors found IL 33 to be a key driver of cancer resistance through polyploidy. | [72] |
| Breast carcinoma (MDA MB 231 cell line) | Doxorubicin | In vitro. Resistant reversible polyploidisation registered by DNA cytometry; 7-week follow-up; IF, microscopy. Transient ALT in mitotic slippage; Budding of mitotic progeny from PGCCs. | [73] |
| Ovarian carcinoma (SCOV-3 and A2780 cell lines) | Cisplatin | In vitro. Bioinformatic analysis of induced PGCCs—upregulation of genes mainly related to gene regulatory mechanisms and nuclear processes, including negative chromatid segregation, microtubule polymerization and membrane budding. | [74] |
2. Transcriptome Analysis of Polyploidy versus Diploidy in Normal Mammalian Tissues Reveals a c-Myc-Targeted Shift to Stemness and Other Known Mechanisms of Cancer Origin and Resistance
3. Resistance to Ionising Irradiation in Malignant Tumours and Tissue Stem Cells Is Associated with Induced ESC Stemness Concurrent with Senescence, Weak DNA Damage Checkpoints, and Polyploidy
4. Embryonic Stem Cells (ESCs) Have Defective Cell Cycle Checkpoints That Favour DNA Damage Tolerance and a Shift to Polyploidy
5. The Hyperactivated Hippo-YAP Pathway Relieves Control of Karyo-Cytokinesis, Reciprocally Favours MS, ACS, cGAS-STING Signalling and Polyploidy, and Enables Cell Fate Change
6. Under-Replication, Erosion, and Recovery of ACS-Compromised Telomeres in Mitotic Slippage and Transient Polyploidy through Transient Alternative Telomere Lengthening
7. The Circadian Clock (CC) Paces the Mitotic Cell Cycle, DDR Checkpoints, and Reciprocally, the TERT-Dependent Hayflick Limit Count. It Is Absent in ESC, Early Embryo, and Germ Cells and Likely Becomes Dis-Engaged and Then Restored (By Reversible Polyploidy) in Cancer Cells
8. The Circadian Clock Is Deregulated in Mammalian Polyploidy and Cancer
8.1. The Reciprocal Regulation of Polyploidy and CC Activity in Non-Malignant Tissues
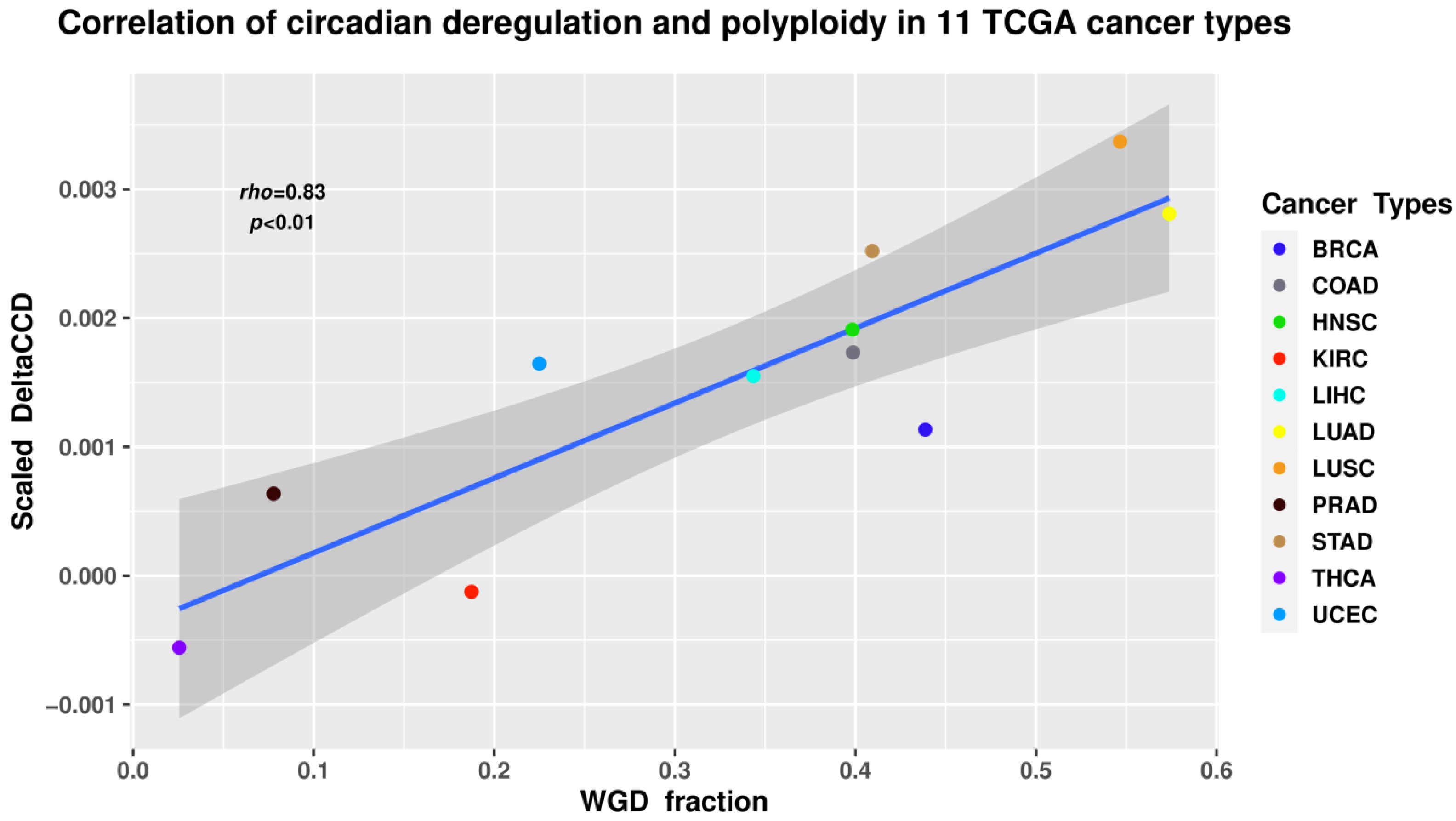
8.2. Circadian Deregulation Correlates with Polyploidisation (Whole-Genome Doubling) in Malignant Tumour Patient Samples
9. Conclusions, Hypothesis, Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bauke, J.; Schöffling, K. Polyploidy in Human Malignancy. Hypopentaploid Chromosome Pattern in Malignant Reticulosis with Secondary Sideroachrestic Anemia. Cancer 1968, 22, 686–694. [Google Scholar] [CrossRef]
- Moein, S.; Adibi, R.; da Silva Meirelles, L.; Nardi, N.B.; Gheisari, Y. Cancer Regeneration: Polyploid Cells Are the Key Drivers of Tumor Progression. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188408. [Google Scholar] [CrossRef] [PubMed]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome Doubling Shapes the Evolution and Prognosis of Advanced Cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.O.; Davidson, J.M.; Duronio, R.J. Endoreplication: Polyploidy with Purpose. Genes Dev. 2009, 23, 2461–2477. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The Evolutionary Significance of Polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- Pienta, K.J.; Hammarlund, E.U.; Axelrod, R.; Brown, J.S.; Amend, S.R. Poly-Aneuploid Cancer Cells Promote Evolvability, Generating Lethal Cancer. Evol. Appl. 2020, 13, 1626–1634. [Google Scholar] [CrossRef]
- Christodoulidou, A.; Raftopoulou, C.; Chiourea, M.; Papaioannou, G.K.; Hoshiyama, H.; Wright, W.E.; Shay, J.W.; Gagos, S. The Roles of Telomerase in the Generation of Polyploidy during Neoplastic Cell Growth. Neoplasia 2013, 15, 156–168. [Google Scholar] [CrossRef]
- Coward, J.; Harding, A. Size Does Matter: Why Polyploid Tumor Cells Are Critical Drug Targets in the War on Cancer. Front. Oncol. 2014, 4, 123. [Google Scholar] [CrossRef]
- Illidge, T.M.; Cragg, M.S.; Fringes, B.; Olive, P.; Erenpreisa, J.A. Polyploid Giant Cells Provide a Survival Mechanism for p53 Mutant Cells after DNA Damage. Cell Biol. Int. 2000, 24, 621–633. [Google Scholar] [CrossRef]
- Sundaram, M.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, R. Neosis: A Novel Type of Cell Division in Cancer. Cancer Biol. Ther. 2004, 3, 207–218. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of Cancer Stem-like Cells through the Formation of Polyploid Giant Cancer Cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-Induced Reprogramming of Breast Cancer Cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Cragg, M.S.; Erenpreisa, J.; Emzinsh, D.; Lukman, H.; Illidge, T.M. Endopolyploid Cells Produced after Severe Genotoxic Damage Have the Potential to Repair DNA Double Strand Breaks. J. Cell Sci. 2003, 116, 4095–4106. [Google Scholar] [CrossRef]
- Lin, K.-C.; Torga, G.; Sun, Y.; Axelrod, R.; Pienta, K.J.; Sturm, J.C.; Austin, R.H. The Role of Heterogeneous Environment and Docetaxel Gradient in the Emergence of Polyploid, Mesenchymal and Resistant Prostate Cancer Cells. Clin. Exp. Metastasis 2019, 36, 97–108. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmina, K.; Anatskaya, O.; Cragg, M.S. Paradoxes of Cancer: Survival at the Brink. Semin. Cancer Biol. 2020, S1044. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Cancer: A Matter of Life Cycle? Cell Biol. Int. 2007, 31, 1507–1510. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. MOS, Aneuploidy and the Ploidy Cycle of Cancer Cells. Oncogene 2010, 29, 5447–5451. [Google Scholar] [CrossRef]
- Liu, J. The Dualistic Origin of Human Tumors. Semin. Cancer Biol. 2018, 53, 1–16. [Google Scholar] [CrossRef]
- Rajaraman, R.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, S.R. Stem Cells, Senescence, Neosis and Self-Renewal in Cancer. Cancer Cell Int. 2006, 6, 25. [Google Scholar] [CrossRef]
- Weihua, Z.; Lin, Q.; Ramoth, A.J.; Fan, D.; Fidler, I.J. Formation of Solid Tumors by a Single Multinucleated Cancer Cell. Cancer 2011, 117, 4092–4099. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Carballo, D.; Saka, S.; Klein, J.; Rennkamp, T.; Acikelli, A.H.; Malak, S.; Jastrow, H.; Wennemuth, G.; Tempfer, C.; Schmitz, I.; et al. A Distinct Oncogenerative Multinucleated Cancer Cell Serves as a Source of Stemness and Tumor Heterogeneity. Cancer Res. 2018, 78, 2318–2331. [Google Scholar] [CrossRef]
- Chen, J.; Niu, N.; Zhang, J.; Qi, L.; Shen, W.; Donkena, K.V.; Feng, Z.; Liu, J. Polyploid Giant Cancer Cells (PGCCs): The Evil Roots of Cancer. Curr. Cancer Drug Targets 2019, 19, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Amend, S.R.; Torga, G.; Lin, K.-C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid Giant Cancer Cells: Unrecognized Actuators of Tumorigenesis, Metastasis, and Resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef]
- Liu, J.; Erenpreisa, J.; Sikora, E. Polyploid Giant Cancer Cells: An Emerging New Field of Cancer Biology. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Comai, L. The Advantages and Disadvantages of Being Polyploid. Nat. Rev. Genet. 2005, 6, 836–846. [Google Scholar] [CrossRef]
- Otto, S.P. The Evolutionary Consequences of Polyploidy. Cell 2007, 131, 452–462. [Google Scholar] [CrossRef]
- Edgar, B.A.; Orr-Weaver, T.L. Endoreplication Cell Cycles. Cell 2001, 105, 297–306. [Google Scholar] [CrossRef]
- Gerashchenko, B.I.; Salmina, K.; Krigerts, J.; Erenpreisa, J.; Babsky, A.M. Induced Polyploidy and Sorting of Damaged Dna by Micronucleation in Radioresistant Rat Liver Epithelial Stem-Like Cells Exposed to X-rays. Probl. Radiat. Med. Radiobiol. 2019, 24, 220–234. [Google Scholar] [CrossRef]
- Holland, A.J.; Cleveland, D.W. Boveri Revisited: Chromosomal Instability, Aneuploidy and Tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef]
- Ben-David, U.; Amon, A. Context Is Everything: Aneuploidy in Cancer. Nat. Rev. Genet. 2020, 21, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Vainshelbaum, N.M.; Zayakin, P.; Kleina, R.; Giuliani, A.; Erenpreisa, J. Meta-Analysis of Cancer Triploidy: Rearrangements of Genome Complements in Male Human Tumors Are Characterized by XXY Karyotypes. Genes 2019, 10, 613. [Google Scholar] [CrossRef] [PubMed]
- Salmina, K.; Gerashchenko, B.I.; Hausmann, M.; Vainshelbaum, N.M.; Zayakin, P.; Erenpreiss, J.; Freivalds, T.; Cragg, M.S.; Erenpreisa, J. When Three Isn’t a Crowd: A Digyny Concept for Treatment-Resistant, Near-Triploid Human Cancers. Genes 2019, 10, 551. [Google Scholar] [CrossRef] [PubMed]
- Salmina, K.; Jankevics, E.; Huna, A.; Perminov, D.; Radovica, I.; Klymenko, T.; Ivanov, A.; Jascenko, E.; Scherthan, H.; Cragg, M.; et al. Up-Regulation of the Embryonic Self-Renewal Network through Reversible Polyploidy in Irradiated p53-Mutant Tumour Cells. Exp. Cell Res. 2010, 316, 2099–2112. [Google Scholar] [CrossRef]
- Liu, J. The “life Code”: A Theory That Unifies the Human Life Cycle and the Origin of Human Tumors. Semin. Cancer Biol. 2020, 60, 380–397. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The “Virgin Birth”, Polyploidy, and the Origin of Cancer. Oncoscience 2015, 2, 3–14. [Google Scholar] [CrossRef]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into Blastomere-like Cancer Stem Cells via Formation of Polyploid Giant Cancer Cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef]
- Díaz-Carballo, D.; Gustmann, S.; Jastrow, H.; Acikelli, A.H.; Dammann, P.; Klein, J.; Dembinski, U.; Bardenheuer, W.; Malak, S.; Araúzo-Bravo, M.J.; et al. Atypical Cell Populations Associated with Acquired Resistance to Cytostatics and Cancer Stem Cell Features: The Role of Mitochondria in Nuclear Encapsulation. DNA Cell Biol. 2014, 33, 749–774. [Google Scholar] [CrossRef]
- Pjanova, D.; Vainshelbaum, N.M.; Salmina, K.; Erenpreisa, J. The Role of the Meiotic Component in Reproduction of B-RAF-Mutated Melanoma: A Review and “Brainstorming” Session. In Melanoma; Lasfar, A., Cohen-Solal, K., Eds.; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- McFarlane, R.J.; Wakeman, J.A. Meiosis-like Functions in Oncogenesis: A New View of Cancer. Cancer Res. 2017, 77, 5712–5716. [Google Scholar] [CrossRef]
- Ianzini, F.; Kosmacek, E.A.; Nelson, E.S.; Napoli, E.; Erenpreisa, J.; Kalejs, M.; Mackey, M.A. Activation of Meiosis-Specific Genes Is Associated with Depolyploidisation of Human Tumor Cells Following Radiation-Induced Mitotic Catastrophe. Cancer Res. 2009, 69, 2296–2304. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S.; Salmina, K.; Hausmann, M.; Scherthan, H. The Role of Meiotic Cohesin REC8 in Chromosome Segregation in Gamma Irradiation-Induced Endopolyploid Tumour Cells. Exp. Cell Res. 2009, 315, 2593–2603. [Google Scholar] [CrossRef] [PubMed]
- Bomken, S.; Fiser, K.; Heidenreich, O.; Vormoor, J. Understanding the Cancer Stem Cell. Br. J. Cancer 2010, 103, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer Stem Cells as Key Drivers of Tumour Progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Anatskaya, O.V.; Giuliani, A.; Erenpreisa, J.; Huang, S.; Salmina, K.; Inashkina, I.; Huna, A.; Nikolsky, N.N.; Vinogradov, A.E. Somatic Polyploidy Is Associated with the Upregulation of c-MYC Interacting Genes and EMT-like Signature. Oncotarget 2016, 7, 75235–75260. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E.; Vainshelbaum, N.M.; Giuliani, A.; Erenpreisa, J. Phylostratic Shift of Whole-Genome Duplications in Normal Mammalian Tissues towards Unicellularity Is Driven by Developmental Bivalent Genes and Reveals a Link to Cancer. Int. J. Mol. Sci. 2020, 21, 8759. [Google Scholar] [CrossRef]
- Roninson, I.B.; Broude, E.V.; Chang, B.-D. If Not Apoptosis, Then What? Treatment-Induced Senescence and Mitotic Catastrophe in Tumor Cells. Drug Resist. Updates 2001, 4, 303–313. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; de Martino, A.; Serrano, M. Senescence Promotes in vivo Reprogramming through p16 and IL-6. Aging Cell 2018, 17, e12711. [Google Scholar] [CrossRef]
- Lee, S.; Schmitt, C.A. The Dynamic Nature of Senescence in Cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef]
- Davoli, T.; de Lange, T. Telomere-Driven Tetraploidization Occurs in Human Cells Undergoing Crisis and Promotes Transformation of Mouse Cells. Cancer Cell 2012, 21, 765–776. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Three Steps to the Immortality of Cancer Cells: Senescence, Polyploidy and Self-Renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Mosieniak, G.; Sikora, E. Polyploidy: The Link between Senescence and Cancer. Curr. Pharm. Des. 2010, 16, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Puig, P.-E.; Guilly, M.-N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor Cells Can Escape DNA-Damaging Cisplatin through DNA Endoreduplication and Reversible Polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- Jackson, T.R.; Salmina, K.; Huna, A.; Inashkina, I.; Jankevics, E.; Riekstina, U.; Kalnina, Z.; Ivanov, A.; Townsend, P.A.; Cragg, M.S.; et al. DNA Damage Causes TP53-Dependent Coupling of Self-Renewal and Senescence Pathways in Embryonal Carcinoma Cells. Cell Cycle 2013, 12, 430–441. [Google Scholar] [CrossRef]
- Huna, A.; Salmina, K.; Erenpreisa, J.; Vazquez-Martin, A.; Krigerts, J.; Inashkina, I.; Gerashchenko, B.I.; Townsend, P.A.; Cragg, M.S.; Jackson, T.R. Role of Stress-Activated OCT4A in the Cell Fate Decisions of Embryonal Carcinoma Cells Treated with Etoposide. Cell Cycle 2015, 14, 2969–2984. [Google Scholar] [CrossRef]
- Suzuki, M.; Boothman, D.A. Stress-Induced Premature Senescence (SIPS)-Influence of SIPS on Radiotherapy. J. Radiat. Res. 2008, 49, 105–112. [Google Scholar] [CrossRef]
- Erenpreisa, J.A.; Cragg, M.S.; Fringes, B.; Sharakhov, I.; Illidge, T.M. Release of Mitotic Descendants by Giant Cells from Irradiated Burkitt’s Lymphoma Cell Line. Cell Biol. Int. 2000, 24, 635–648. [Google Scholar] [CrossRef]
- Vitale, I.; Senovilla, L.; Jemaà, M.; Michaud, M.; Galluzzi, L.; Kepp, O.; Nanty, L.; Criollo, A.; Rello-Varona, S.; Manic, G.; et al. Multipolar Mitosis of Tetraploid Cells: Inhibition by p53 and Dependency on Mos. EMBO J. 2010, 29, 1272–1284. [Google Scholar] [CrossRef]
- Horbay, R.; Stoika, R. Giant Cell Formation: The Way to Cell Death or Cell Survival? Open Life Sci. 2011, 6, 675–684. [Google Scholar] [CrossRef]
- Park, S.U.; Choi, E.S.; Jang, Y.S.; Hong, S.-H.; Kim, I.-H.; Chang, D.K. Effects of chromosomal polyploidy on survival of colon cancer cells. Korean J. Gastroenterol. 2011, 57, 150. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy Road to Therapy-Induced Cellular Senescence and Escape. Int. J. Cancer 2013, 132, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Yao, H.-P.; Zhou, Y.-Q.; Zhou, J.; Zhang, R.; Wang, M.-H. Prevention of BMS-777607-Induced Polyploidy/senescence by mTOR Inhibitor AZD8055 Sensitizes Breast Cancer Cells to Cytotoxic Chemotherapeutics. Mol. Oncol. 2014, 8, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sánchez, L.M.; Jimenez, C.; Valverde, A.; Hernandez, V.; Peñarando, J.; Martinez, A.; Lopez-Pedrera, C.; Muñoz-Castañeda, J.R.; De la Haba-Rodríguez, J.R.; Aranda, E.; et al. CoCl2, a Mimic of Hypoxia, Induces Formation of Polyploid Giant Cells with Stem Characteristics in Colon Cancer. PLoS ONE 2014, 9, e99143. [Google Scholar] [CrossRef] [PubMed]
- Chitikova, Z.V.; Gordeev, S.A.; Bykova, T.V.; Zubova, S.G.; Pospelov, V.A.; Pospelova, T.V. Sustained Activation of DNA Damage Response in Irradiated Apoptosis-Resistant Cells Induces Reversible Senescence Associated with mTOR Downregulation and Expression of Stem Cell Markers. Cell Cycle 2014, 13, 1424–1439. [Google Scholar] [CrossRef]
- Rohnalter, V.; Roth, K.; Finkernagel, F.; Adhikary, T.; Obert, J.; Dorzweiler, K.; Bensberg, M.; Müller-Brüsselbach, S.; Müller, R. A Multi-Stage Process Including Transient Polyploidisation and EMT Precedes the Emergence of Chemoresistent Ovarian Carcinoma Cells with a Dedifferentiated and pro-Inflammatory Secretory Phenotype. Oncotarget 2015, 6, 40005–40025. [Google Scholar] [CrossRef]
- Leikam, C.; Hufnagel, A.L.; Otto, C.; Murphy, D.J.; Mühling, B.; Kneitz, S.; Nanda, I.; Schmid, M.; Wagner, T.U.; Haferkamp, S.; et al. In Vitro Evidence for Senescent Multinucleated Melanocytes as a Source for Tumor-Initiating Cells. Cell Death Dis. 2015, 6, e1711. [Google Scholar] [CrossRef]
- Niu, N.; Zhang, J.; Zhang, N.; Mercado-Uribe, I.; Tao, F.; Han, Z.; Pathak, S.; Multani, A.S.; Kuang, J.; Yao, J.; et al. Linking Genomic Reorganization to Tumor Initiation via the Giant Cell Cycle. Oncogenesis 2016, 5, e281. [Google Scholar] [CrossRef]
- Gerashchenko, B.I.; Salmina, K.; Eglitis, J.; Huna, A.; Grjunberga, V.; Erenpreisa, J. Disentangling the Aneuploidy and Senescence Paradoxes: A Study of Triploid Breast Cancers Non-Responsive to Neoadjuvant Therapy. Histochem. Cell Biol. 2016, 145, 497–508. [Google Scholar] [CrossRef]
- Kiseleva, L.N.; Kartashev, A.V.; Vartanyan, N.L.; Pinevich, A.A.; Samoilovich, M.P. Multinucleated Cells Resistant to Genotoxic Factors within Human Glioblastoma Cell Lines. Cell Tissue Biol. 2019, 13, 1–7. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Miyamoto, T.; Imazeki, H.; Shoji, H.; Aoki, K.; Boku, N. IL33 Is a Key Driver of Treatment Resistance of Cancer. Cancer Res. 2020, 80, 1981–1990. [Google Scholar] [CrossRef]
- Salmina, K.; Bojko, A.; Inashkina, I.; Staniak, K.; Dudkowska, M.; Podlesniy, P.; Rumnieks, F.; Vainshelbaum, N.M.; Pjanova, D.; Sikora, E.; et al. “Mitotic Slippage” and Extranuclear DNA in Cancer Chemoresistance: A Focus on Telomeres. Int. J. Mol. Sci. 2020, 21, 2779. [Google Scholar] [CrossRef] [PubMed]
- Adibi, R.; Moein, S.; Gheisari, Y. Cisplatin Resistant Ovarian Cancer Cells Reveal a Polyploid Phenotype with Remarkable Activation of Nuclear Processes. Res. Square 2021. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wood, P.A.; Hrushesky, W.J.M. Mammalian TIMELESS Is Required for ATM-Dependent CHK2 Activation and G2/M Checkpoint Control. J. Biol. Chem. 2010, 285, 3030–3034. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Pajonk, F. Cancer Stem Cells, Cancer Cell Plasticity and Radiation Therapy. Semin. Cancer Biol. 2015, 31, 28–35. [Google Scholar] [CrossRef]
- Gerashchenko, B.I.; Azzam, E.I.; Howell, R.W. Characterization of Cell-Cycle Progression and Growth of WB-F344 Normal Rat Liver Epithelial Cells Following Gamma-Ray Exposure. Cytometry A 2004, 61, 134–141. [Google Scholar] [CrossRef]
- Neganova, I.; Lako, M. G1 to S Phase Cell Cycle Transition in Somatic and Embryonic Stem Cells. J. Anat. 2008, 213, 30–44. [Google Scholar] [CrossRef]
- Suvorova, I.I.; Grigorash, B.B.; Chuykin, I.A.; Pospelova, T.V.; Pospelov, V.A. G1 Checkpoint Is Compromised in Mouse ESCs due to Functional Uncoupling of p53-p21Waf1 Signaling. Cell Cycle 2016, 15, 52–63. [Google Scholar] [CrossRef]
- Liu, L.; Michowski, W.; Kolodziejczyk, A.; Sicinski, P. The Cell Cycle in Stem Cell Proliferation, Pluripotency and Differentiation. Nat. Cell Biol. 2019, 21, 1060–1067. [Google Scholar] [CrossRef]
- Mantel, C.; Guo, Y.; Lee, M.R.; Han, M.K.; Rhorabough, S.; Kim, K.S.; Broxmeyer, H.E. Cells Enter a Unique Intermediate 4N Stage, Not 4N-G1, after Aborted Mitosis. Cell Cycle 2008, 7, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Kalejs, M.; Cragg, M.S. Mitotic Catastrophe and Endomitosis in Tumour Cells: An Evolutionary Key to a Molecular Solution. Cell Biol. Int. 2005, 29, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B. Wild-Type p53: Tumors Can’t Stand It. Cell 2007, 128, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. Reprogramming Cell Fates: Reconciling Rarity with Robustness. Bioessays 2009, 31, 546–560. [Google Scholar] [CrossRef]
- Zhang, X.; Neganova, I.; Przyborski, S.; Yang, C.; Cooke, M.; Atkinson, S.P.; Anyfantis, G.; Fenyk, S.; Keith, W.N.; Hoare, S.F.; et al. A Role for NANOG in G1 to S Transition in Human Embryonic Stem Cells through Direct Binding of CDK6 and CDC25A. J. Cell Biol. 2009, 184, 67–82. [Google Scholar] [CrossRef]
- Neganova, I.; Vilella, F.; Atkinson, S.P.; Lloret, M.; Passos, J.F.; von Zglinicki, T.; O’Connor, J.-E.; Burks, D.; Jones, R.; Armstrong, L.; et al. An Important Role for CDK2 in G1 to S Checkpoint Activation and DNA Damage Response in Human Embryonic Stem Cells. Stem Cells 2011, 29, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.J.; Liu, K.; Rameshwar, P. Functional Similarities among Genes Regulated by OCT4 in Human Mesenchymal and Embryonic Stem Cells. Stem Cells 2007, 25, 3143–3154. [Google Scholar] [CrossRef] [PubMed]
- Keyes, W.M.; Wu, Y.; Vogel, H.; Guo, X.; Lowe, S.W.; Mills, A.A. p63 Deficiency Activates a Program of Cellular Senescence and Leads to Accelerated Aging. Genes Dev. 2005, 19, 1986–1999. [Google Scholar] [CrossRef] [PubMed]
- Baryshev, M.; Inashkina, I.; Salmina, K.; Huna, A.; Jackson, T.R.; Erenpreisa, J. DNA Methylation of the Oct4A Enhancers in Embryonal Carcinoma Cells after Etoposide Treatment Is Associated with Alternative Splicing and Altered Pluripotency in Reversibly Senescent Cells. Cell Cycle 2018, 17, 362–366. [Google Scholar] [CrossRef]
- Kulaberoglu, Y.; Gundogdu, R.; Hergovich, A. The Role of p53/p21/p16 in DNA-Damage Signaling and DNA Repair. In Genome Stability; Elsevier: Amsterdam, The Netherlands, 2016; pp. 243–256. ISBN 9780128033098. [Google Scholar]
- Li, H.; Collado, M.; Villasante, A.; Matheu, A.; Lynch, C.J.; Cañamero, M.; Rizzoti, K.; Carneiro, C.; Martínez, G.; Vidal, A.; et al. p27(Kip1) Directly Represses Sox2 during Embryonic Stem Cell Differentiation. Cell Stem Cell 2012, 11, 845–852. [Google Scholar] [CrossRef]
- She, S.; Wei, Q.; Kang, B.; Wang, Y.-J. Cell Cycle and Pluripotency: Convergence on Octamer-binding Transcription Factor 4 (Review). Mol. Med. Rep. 2017, 16, 6459–6466. [Google Scholar] [CrossRef] [PubMed]
- Farshadi, E.; van der Horst, G.T.J.; Chaves, I. Molecular Links between the Circadian Clock and the Cell Cycle. J. Mol. Biol. 2020, 432, 3515–3524. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Guan, K.-L. The Hippo Pathway: Regulators and Regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Steigemann, P.; Wurzenberger, C.; Schmitz, M.H.A.; Held, M.; Guizetti, J.; Maar, S.; Gerlich, D.W. Aurora B-Mediated Abscission Checkpoint Protects against Tetraploidization. Cell 2009, 136, 473–484. [Google Scholar] [CrossRef]
- Bui, D.A.; Lee, W.; White, A.E.; Harper, J.W.; Schackmann, R.C.J.; Overholtzer, M.; Selfors, L.M.; Brugge, J.S. Cytokinesis Involves a Nontranscriptional Function of the Hippo Pathway Effector YAP. Sci. Signal. 2016, 9, ra23. [Google Scholar] [CrossRef]
- Yabuta, N.; Mukai, S.; Okada, N.; Aylon, Y.; Nojima, H. The Tumor Suppressor Lats2 Is Pivotal in Aurora A and Aurora B Signaling during Mitosis. Cell Cycle 2011, 10, 2724–2736. [Google Scholar] [CrossRef]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic Catastrophe: A Mechanism for Avoiding Genomic Instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef]
- Ganem, N.J.; Cornils, H.; Chiu, S.-Y.; O’Rourke, K.P.; Arnaud, J.; Yimlamai, D.; Théry, M.; Camargo, F.D.; Pellman, D. Cytokinesis Failure Triggers Hippo Tumor Suppressor Pathway Activation. Cell 2014, 158, 833–848. [Google Scholar] [CrossRef]
- Lavado, A.; Park, J.Y.; Paré, J.; Finkelstein, D.; Pan, H.; Xu, B.; Fan, Y.; Kumar, R.P.; Neale, G.; Kwak, Y.D.; et al. The Hippo Pathway Prevents YAP/TAZ-Driven Hypertranscription and Controls Neural Progenitor Number. Dev. Cell 2018, 47, 576–591.e8. [Google Scholar] [CrossRef]
- Kim, T.; Yang, S.-J.; Hwang, D.; Song, J.; Kim, M.; Kyum Kim, S.; Kang, K.; Ahn, J.; Lee, D.; Kim, M.-Y.; et al. A Basal-like Breast Cancer-Specific Role for SRF-IL6 in YAP-Induced Cancer Stemness. Nat. Commun. 2015, 6, 10186. [Google Scholar] [CrossRef]
- He, C.; Lv, X.; Huang, C.; Hua, G.; Ma, B.; Chen, X.; Angeletti, P.C.; Dong, J.; Zhou, J.; Wang, Z.; et al. YAP1-LATS2 Feedback Loop Dictates Senescent or Malignant Cell Fate to Maintain Tissue Homeostasis. EMBO Rep. 2019, 20, e44948. [Google Scholar] [CrossRef]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-Mediated Processing of Chromatin in Senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Lezaja, A.; Altmeyer, M. Dealing with DNA Lesions: When One Cell Cycle Is Not Enough. Curr. Opin. Cell Biol. 2021, 70, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhou, L.; Ling, L.; Meng, X.; Chu, F.; Zhang, S.; Zhou, F. The Crosstalk Between Hippo-YAP Pathway and Innate Immunity. Front. Immunol. 2020, 11, 323. [Google Scholar] [CrossRef]
- Furth, N.; Bossel Ben-Moshe, N.; Pozniak, Y.; Porat, Z.; Geiger, T.; Domany, E.; Aylon, Y.; Oren, M. Down-Regulation of LATS Kinases Alters p53 to Promote Cell Migration. Genes Dev. 2015, 29, 2325–2330. [Google Scholar] [CrossRef]
- Raj, N.; Bam, R. Reciprocal Crosstalk Between YAP1/Hippo Pathway and the p53 Family Proteins: Mechanisms and Outcomes in Cancer. Front. Cell Dev. Biol. 2019, 7, 159. [Google Scholar] [CrossRef]
- Franklin, J.M.; Ghosh, R.P.; Shi, Q.; Reddick, M.P.; Liphardt, J.T. Concerted Localization-Resets Precede YAP-Dependent Transcription. Nat. Commun. 2020, 11, 4581. [Google Scholar] [CrossRef]
- Lahav, G. Oscillations by the p53-Mdm2 Feedback Loop. Adv. Exp. Med. Biol. 2008, 641, 28–38. [Google Scholar]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. p53 Dynamics Control Cell Fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Giuliani, A. Resolution of Complex Issues in Genome Regulation and Cancer Requires Non-Linear and Network-Based Thermodynamics. Int. J. Mol. Sci. 2019, 21, 240. [Google Scholar] [CrossRef]
- Nagl, W. Endopolyploidy and Polyteny in Differentiation and Evolution: Towards an Understanding of Quantitative and Qualitative Variation of Nuclear DNA in Ontogeny and Phylogeny; North-Holland Publish: Amsterdam, The Netherlands; New York, NY, USA; Oxford, UK, 1978; 283p. [Google Scholar]
- Munden, A.; Rong, Z.; Sun, A.; Gangula, R.; Mallal, S.; Nordman, J.T. Rif1 Inhibits Replication Fork Progression and Controls DNA Copy Number in Drosophila. Elife 2018, 7, e39140. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Caballero, M.; Kolesnikova, T.; Zhimulev, I.; Koren, A.; Nordman, J. Replication Timing Analysis in Polyploid Cells Reveals Rif1 Uses Multiple Mechanisms to Promote Underreplication in Drosophila. Genetics 2021, 219, iyab147. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Mistrik, M.; Bartkova, J. Thresholds of Replication Stress Signaling in Cancer Development and Treatment. Nat. Struct. Mol. Biol. 2012, 19, 5–7. [Google Scholar] [CrossRef]
- Tam, W.-L.; Ang, Y.-S.; Lim, B. The Molecular Basis of Ageing in Stem Cells. Mech. Ageing Dev. 2007, 128, 137–148. [Google Scholar] [CrossRef]
- Olovnikov, A.M. Telomeres, Telomerase, and Aging: Origin of the Theory. Exp. Gerontol. 1996, 31, 443–448. [Google Scholar] [CrossRef]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Palacios, J.A.; Herranz, D.; De Bonis, M.L.; Velasco, S.; Serrano, M.; Blasco, M.A. SIRT1 Contributes to Telomere Maintenance and Augments Global Homologous Recombination. J. Cell Biol. 2010, 191, 1299–1313. [Google Scholar] [CrossRef]
- Rijo-Ferreira, F.; Takahashi, J.S. Genomics of Circadian Rhythms in Health and Disease. Genome Med. 2019, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Feillet, C.; van der Horst, G.T.J.; Levi, F.; Rand, D.A.; Delaunay, F. Coupling between the Circadian Clock and Cell Cycle Oscillators: Implication for Healthy Cells and Malignant Growth. Front. Neurol. 2015, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Langmesser, S.; Tallone, T.; Bordon, A.; Rusconi, S.; Albrecht, U. Interaction of Circadian Clock Proteins PER2 and CRY with BMAL1 and CLOCK. BMC Mol. Biol. 2008, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S. Transcriptional Architecture of the Mammalian Circadian Clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, X. A Wheel of Time: The Circadian Clock, Nuclear Receptors, and Physiology. Genes Dev. 2010, 24, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Cusumano, P.; Damulewicz, M.; Carbognin, E.; Caccin, L.; Puricella, A.; Specchia, V.; Bozzetti, M.P.; Costa, R.; Mazzotta, G.M. The RNA Helicase BELLE Is Involved in Circadian Rhythmicity and in Transposons Regulation in Drosophila melanogaster. Front. Physiol. 2019, 10, 133. [Google Scholar] [CrossRef]
- Stavreva, D.A.; Garcia, D.A.; Fettweis, G.; Gudla, P.R.; Zaki, G.F.; Soni, V.; McGowan, A.; Williams, G.; Huynh, A.; Palangat, M.; et al. Transcriptional Bursting and Co-Bursting Regulation by Steroid Hormone Release Pattern and Transcription Factor Mobility. Mol. Cell 2019, 75, 1161–1177.e11. [Google Scholar] [CrossRef]
- Yan, J.; Goldbeter, A. Robust Synchronization of the Cell Cycle and the Circadian Clock through Bidirectional Coupling. J. R. Soc. Interface 2019, 16, 20190376. [Google Scholar] [CrossRef]
- Kowalska, E.; Ripperger, J.A.; Hoegger, D.C.; Bruegger, P.; Buch, T.; Birchler, T.; Mueller, A.; Albrecht, U.; Contaldo, C.; Brown, S.A. NONO Couples the Circadian Clock to the Cell Cycle. Proc. Natl. Acad. Sci. USA 2013, 110, 1592–1599. [Google Scholar] [CrossRef]
- Umemura, Y.; Yagita, K. Development of the Circadian Core Machinery in Mammals. J. Mol. Biol. 2020, 432, 3611–3617. [Google Scholar] [CrossRef]
- Vallone, D.; Lahiri, K.; Dickmeis, T.; Foulkes, N.S. Start the Clock! Circadian Rhythms and Development. Dev. Dyn. 2007, 236, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, J.M.; O’Connell, M.J. The G(2) DNA Damage Checkpoint Targets Both Wee1 and Cdc25. J. Cell Sci. 2000, 113, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Burchett, J.B.; Knudsen-Clark, A.M.; Altman, B.J. MYC Ran Up the Clock: The Complex Interplay between MYC and the Molecular Circadian Clock in Cancer. Int. J. Mol. Sci. 2021, 22, 7761. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Vila-Caballer, M.; Santos, C.S.; Liu, J.; Yang, J.; Finkielstein, C.V. The Circadian Factor Period 2 Modulates p53 Stability and Transcriptional Activity in Unstressed Cells. Mol. Biol. Cell 2014, 25, 3081–3093. [Google Scholar] [CrossRef]
- Stokes, K.; Nunes, M.; Trombley, C.; Flôres, D.E.F.L.; Wu, G.; Taleb, Z.; Alkhateeb, A.; Banskota, S.; Harris, C.; Love, O.P.; et al. The Circadian Clock Gene, Bmal1, Regulates Intestinal Stem Cell Signaling and Represses Tumor Initiation. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1847–1872.e0. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, F.; Fan, W.; Zheng, M.; Kang, J.; Huang, F.; He, H. Expression of Circadian Clock Genes during Differentiation of Rat Dental Papilla Cells in Vitro. Biol. Rhythm Res. 2020. [Google Scholar] [CrossRef]
- Morse, D.; Cermakian, N.; Brancorsini, S.; Parvinen, M.; Sassone-Corsi, P. No Circadian Rhythms in Testis: Period 1 Expression Is Clock Independent and Developmentally Regulated in the Mouse. Mol. Endocrinol. 2003, 17, 141–151. [Google Scholar] [CrossRef]
- Beaver, L.M.; Rush, B.L.; Gvakharia, B.O.; Giebultowicz, J.M. Noncircadian Regulation and Function of Clock Genes Period and Timeless in Oogenesis of Drosophila melanogaster. J. Biol. Rhythm. 2003, 18, 463–472. [Google Scholar] [CrossRef]
- Bittman, E.L. Timing in the Testis. J. Biol. Rhythm. 2016, 31, 12–36. [Google Scholar] [CrossRef]
- Lu, Y.; Zheng, X.; Hu, W.; Bian, S.; Zhang, Z.; Tao, D.; Liu, Y.; Ma, Y. Cancer/testis Antigen PIWIL2 Suppresses Circadian Rhythms by Regulating the Stability and Activity of BMAL1 and CLOCK. Oncotarget 2017, 8, 54913–54924. [Google Scholar] [CrossRef]
- Solov’eva, L.; Svetlova, M.; Bodinski, D.; Zalensky, A.O. Nature of Telomere Dimers and Chromosome Looping in Human Spermatozoa. Chromosome Res. 2004, 12, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; Cragg, M.S.; Erenpreisa, J. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Mayer, W.; Smith, A.; Fundele, R.; Haaf, T. Spatial Separation of Parental Genomes in Preimplantation Mouse Embryos. J. Cell Biol. 2000, 148, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Curran, K.L.; Allen, L.; Porter, B.B.; Dodge, J.; Lope, C.; Willadsen, G.; Fisher, R.; Johnson, N.; Campbell, E.; VonBergen, B.; et al. Circadian Genes, xBmal1 and xNocturnin, Modulate the Timing and Differentiation of Somites in Xenopus Laevis. PLoS ONE 2014, 9, e108266. [Google Scholar] [CrossRef]
- Bellet, M.M.; Orozco-Solis, R.; Sahar, S.; Eckel-Mahan, K.; Sassone-Corsi, P. The Time of Metabolism: NAD+, SIRT1, and the Circadian Clock. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 31–38. [Google Scholar] [CrossRef]
- Wang, R.-H.; Zhao, T.; Cui, K.; Hu, G.; Chen, Q.; Chen, W.; Wang, X.-W.; Soto-Gutierrez, A.; Zhao, K.; Deng, C.-X. Negative Reciprocal Regulation between Sirt1 and Per2 Modulates the Circadian Clock and Aging. Sci. Rep. 2016, 6, 28633. [Google Scholar] [CrossRef]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 Regulates Circadian Clock Gene Expression through PER2 Deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef]
- Chao, H.-W.; Doi, M.; Fustin, J.-M.; Chen, H.; Murase, K.; Maeda, Y.; Hayashi, H.; Tanaka, R.; Sugawa, M.; Mizukuchi, N.; et al. Circadian Clock Regulates Hepatic Polyploidy by Modulating Mkp1-Erk1/2 Signaling Pathway. Nat. Commun. 2017, 8, 2238. [Google Scholar] [CrossRef]
- Wu, Y.; Tao, B.; Zhang, T.; Fan, Y.; Mao, R. Pan-Cancer Analysis Reveals Disrupted Circadian Clock Associates with T Cell Exhaustion. Front. Immunol. 2019, 10, 2451. [Google Scholar] [CrossRef]
- Shilts, J.; Chen, G.; Hughey, J.J. Evidence for Widespread Dysregulation of Circadian Clock Progression in Human Cancer. PeerJ 2018, 6, e4327. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Fung-Uceda, J.; Lee, K.; Seo, P.J.; Polyn, S.; De Veylder, L.; Mas, P. The Circadian Clock Sets the Time of DNA Replication Licensing to Regulate Growth in Arabidopsis. Dev. Cell 2018, 45, 101–113.e4. [Google Scholar] [CrossRef] [PubMed]
- Dakup, P.P.; Porter, K.I.; Gajula, R.P.; Goel, P.N.; Cheng, Z.; Gaddameedhi, S. The Circadian Clock Protects against Ionizing Radiation-Induced Cardiotoxicity. FASEB J. 2020, 34, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Dakup, P.P.; Porter, K.I.; Gaddameedhi, S. The Circadian Clock Protects against Acute Radiation-Induced Dermatitis. Toxicol. Appl. Pharmacol. 2020, 399, 115040. [Google Scholar] [CrossRef] [PubMed]
- Relógio, A.; Thomas, P.; Medina-Pérez, P.; Reischl, S.; Bervoets, S.; Gloc, E.; Riemer, P.; Mang-Fatehi, S.; Maier, B.; Schäfer, R.; et al. Ras-Mediated Deregulation of the Circadian Clock in Cancer. PLoS Genet. 2014, 10, e1004338. [Google Scholar] [CrossRef]
- Van Dycke, K.C.G.; Rodenburg, W.; van Oostrom, C.T.M.; van Kerkhof, L.W.M.; Pennings, J.L.A.; Roenneberg, T.; van Steeg, H.; van der Horst, G.T.J. Chronically Alternating Light Cycles Increase Breast Cancer Risk in Mice. Curr. Biol. 2015, 25, 1932–1937. [Google Scholar] [CrossRef] [PubMed]
- Papagiannakopoulos, T.; Bauer, M.R.; Davidson, S.M.; Heimann, M.; Subbaraj, L.; Bhutkar, A.; Bartlebaugh, J.; Vander Heiden, M.G.; Jacks, T. Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab. 2016, 24, 324–331. [Google Scholar] [CrossRef]
- Zhang, J.; Lv, H.; Ji, M.; Wang, Z.; Wu, W. Low Circadian Clock Genes Expression in Cancers: A Meta-Analysis of Its Association with Clinicopathological Features and Prognosis. PLoS ONE 2020, 15, e0233508. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An Immeasurable Source of Knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Rahman, M.; Jackson, L.K.; Johnson, W.E.; Li, D.Y.; Bild, A.H.; Piccolo, S.R. Alternative Preprocessing of RNA-Sequencing Data in The Cancer Genome Atlas Leads to Improved Analysis Results. Bioinformatics 2015, 31, 3666–3672. [Google Scholar] [CrossRef]
- Lu, C.; Yang, Y.; Zhao, R.; Hua, B.; Xu, C.; Yan, Z.; Sun, N.; Qian, R. Role of Circadian Gene Clock during Differentiation of Mouse Pluripotent Stem Cells. Protein Cell 2016, 7, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute Quantification of Somatic DNA Alterations in Human Cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e3. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, J.W.; Irie, N.; Lodder, P.; van Pelt, A.M.M.; Koster, J.; Hamer, G. Tumors Widely Express Hundreds of Embryonic Germline Genes. Cancers 2020, 12, 3812. [Google Scholar] [CrossRef] [PubMed]
- Kalejs, M.; Ivanov, A.; Plakhins, G.; Cragg, M.S.; Emzinsh, D.; Illidge, T.M.; Erenpreisa, J. Upregulation of Meiosis-Specific Genes in Lymphoma Cell Lines Following Genotoxic Insult and Induction of Mitotic Catastrophe. BMC Cancer 2006, 6, 6. [Google Scholar] [CrossRef]
- Lange, U.C.; Saitou, M.; Western, P.S.; Barton, S.C.; Surani, M.A. The Fragilis Interferon-Inducible Gene Family of Transmembrane Proteins Is Associated with Germ Cell Specifica tion in Mice. BMC Dev. Biol. 2003, 3, 1. [Google Scholar] [CrossRef]
- Sheng, X.; Tian, C.; Liu, L.; Wang, L.; Ye, X.; Li, J.; Zeng, M.; Liu, L. Characterization of Oogonia Stem Cells in Mice by Fragilis. Protein Cell 2019, 10, 825–831. [Google Scholar] [CrossRef]
- Grundmann, E. The concept of Julius Cohnheim on tumor formation and metastasis from the viewpoint of new research results. Zentralbl. Allg. Pathol. 1985, 130, 323–331. [Google Scholar]
- Vinnitsky, V. The Development of a Malignant Tumor Is due to a Desperate Asexual Self-Cloning Process in Which Cancer Stem Cells Develop the Ability to Mimic the Genetic Program of Germline Cells. Intrinsically Disord. Proteins 2014, 2, e29997. [Google Scholar] [CrossRef][Green Version]
- Rajaraman, R.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, S.R. Neosis-A Parasexual Somatic Reduction Division in Cancer. Int. J. Hum. Genet. 2007, 7, 29–48. [Google Scholar] [CrossRef]
- Prigogine, I. Time, Structure and Fluctuations: Nobel Lecture, 8 December 1977. Available online: https://www.nobelprize.org/uploads/2018/06/prigogine-lecture.pdf (accessed on 5 November 2021).
- Noble, R.; Tasaki, K.; Noble, P.J.; Noble, D. Biological Relativity Requires Circular Causality but Not Symmetry of Causation: So, Where, What and When Are the Boundaries? Front. Physiol. 2019, 10, 827. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. On the Intrinsic Inevitability of Cancer: From Foetal to Fatal Attraction. Semin. Cancer Biol. 2011, 21, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F. Is Cancer Cell Reversion to Normalcy Possible? Un Update. In Novel Approaches in Cancer Study; Crimson Publishers: New York, NY, USA, 2020; Volume 5. [Google Scholar]
- Lotem, J.; Sachs, L. Epigenetics Wins over Genetics: Induction of Differentiation in Tumor Cells. Semin. Cancer Biol. 2002, 12, 339–346. [Google Scholar] [CrossRef]
- Telerman, A.; Amson, R. The Molecular Programme of Tumour Reversion: The Steps beyond Malignant Transformation. Nat. Rev. Cancer 2009, 9, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Krigerts, J.; Salmina, K.; Freivalds, T.; Zayakin, P.; Rumnieks, F.; Inashkina, I.; Giuliani, A.; Hausmann, M.; Erenpreisa, J. Differentiating Cancer Cells Reveal Early Large-Scale Genome Regulation by Pericentric Domains. Biophys. J. 2021, 120, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, M.; Giuliani, A.; Cucina, A.; Minini, M. Redifferentiation Therapeutic Strategies in Cancer. Drug Discov. Today 2020, 25, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.M.; Bissell, M.J. Of Extracellular Matrix, Scaffolds, and Signaling: Tissue Architecture Regulates Development, Homeostasis, and Cancer. Annu. Rev. Cell Dev. Biol. 2006, 22, 287–309. [Google Scholar] [CrossRef]
- Lévi, F. Circadian Chronotherapy for Human Cancers. Lancet Oncol. 2001, 2, 307–315. [Google Scholar] [CrossRef]
- Battaglin, F.; Chan, P.; Pan, Y.; Soni, S.; Qu, M.; Spiller, E.R.; Castanon, S.; Roussos Torres, E.T.; Mumenthaler, S.M.; Kay, S.A.; et al. Clocking Cancer: The Circadian Clock as a Target in Cancer Therapy. Oncogene 2021, 40, 3187–3200. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vainshelbaum, N.M.; Salmina, K.; Gerashchenko, B.I.; Lazovska, M.; Zayakin, P.; Cragg, M.S.; Pjanova, D.; Erenpreisa, J. Role of the Circadian Clock “Death-Loop” in the DNA Damage Response Underpinning Cancer Treatment Resistance. Cells 2022, 11, 880. https://doi.org/10.3390/cells11050880
Vainshelbaum NM, Salmina K, Gerashchenko BI, Lazovska M, Zayakin P, Cragg MS, Pjanova D, Erenpreisa J. Role of the Circadian Clock “Death-Loop” in the DNA Damage Response Underpinning Cancer Treatment Resistance. Cells. 2022; 11(5):880. https://doi.org/10.3390/cells11050880
Chicago/Turabian StyleVainshelbaum, Ninel Miriam, Kristine Salmina, Bogdan I. Gerashchenko, Marija Lazovska, Pawel Zayakin, Mark Steven Cragg, Dace Pjanova, and Jekaterina Erenpreisa. 2022. "Role of the Circadian Clock “Death-Loop” in the DNA Damage Response Underpinning Cancer Treatment Resistance" Cells 11, no. 5: 880. https://doi.org/10.3390/cells11050880
APA StyleVainshelbaum, N. M., Salmina, K., Gerashchenko, B. I., Lazovska, M., Zayakin, P., Cragg, M. S., Pjanova, D., & Erenpreisa, J. (2022). Role of the Circadian Clock “Death-Loop” in the DNA Damage Response Underpinning Cancer Treatment Resistance. Cells, 11(5), 880. https://doi.org/10.3390/cells11050880