
The Cardiomyocyte in Heart Failure with Preserved Ejection Fraction—Victim of Its Environment?


{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Dilemma of HFpEF Definition
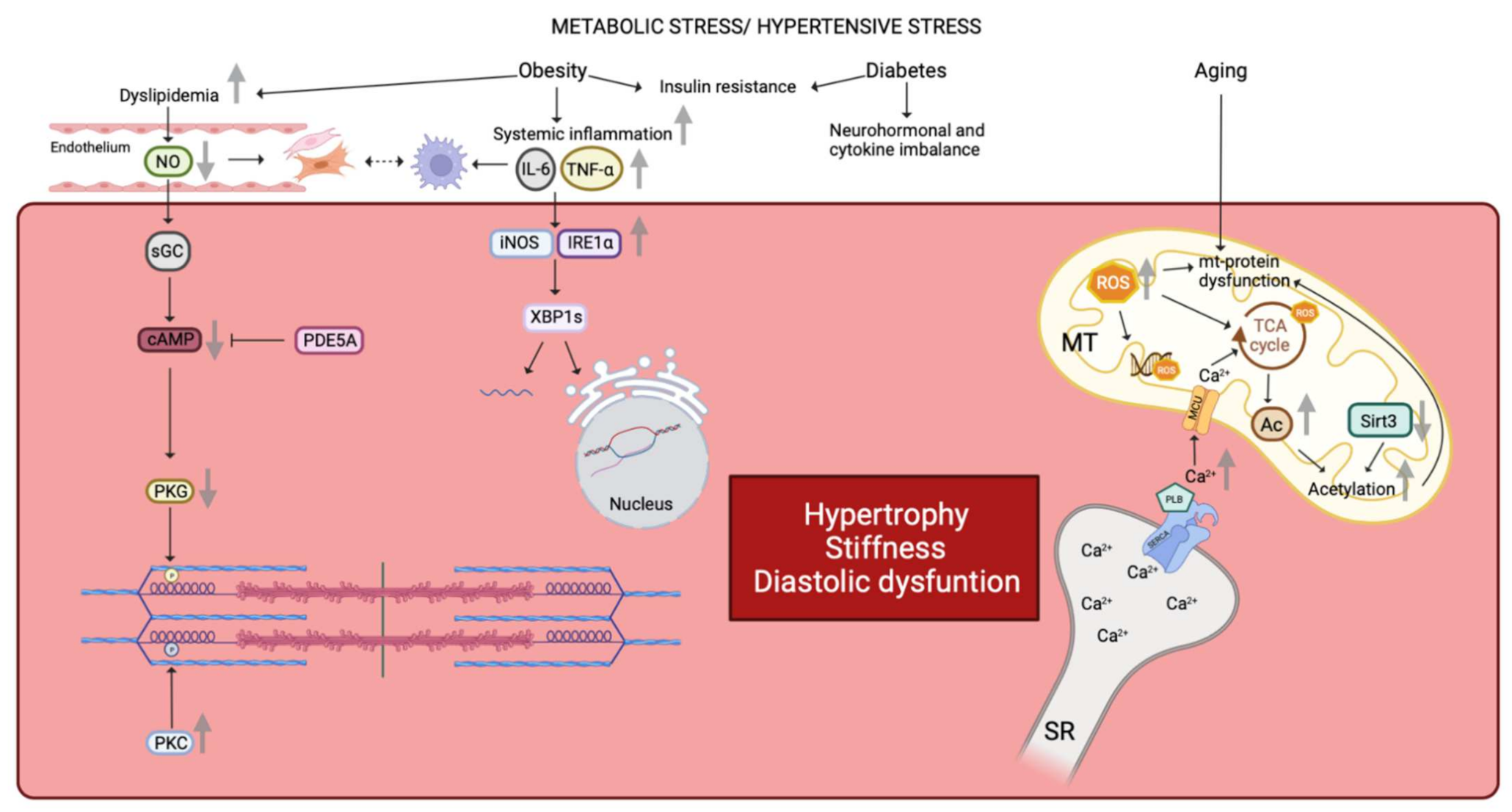
2.1. Structural and Functional Changes of the Cardiomyocyte in HFpEF
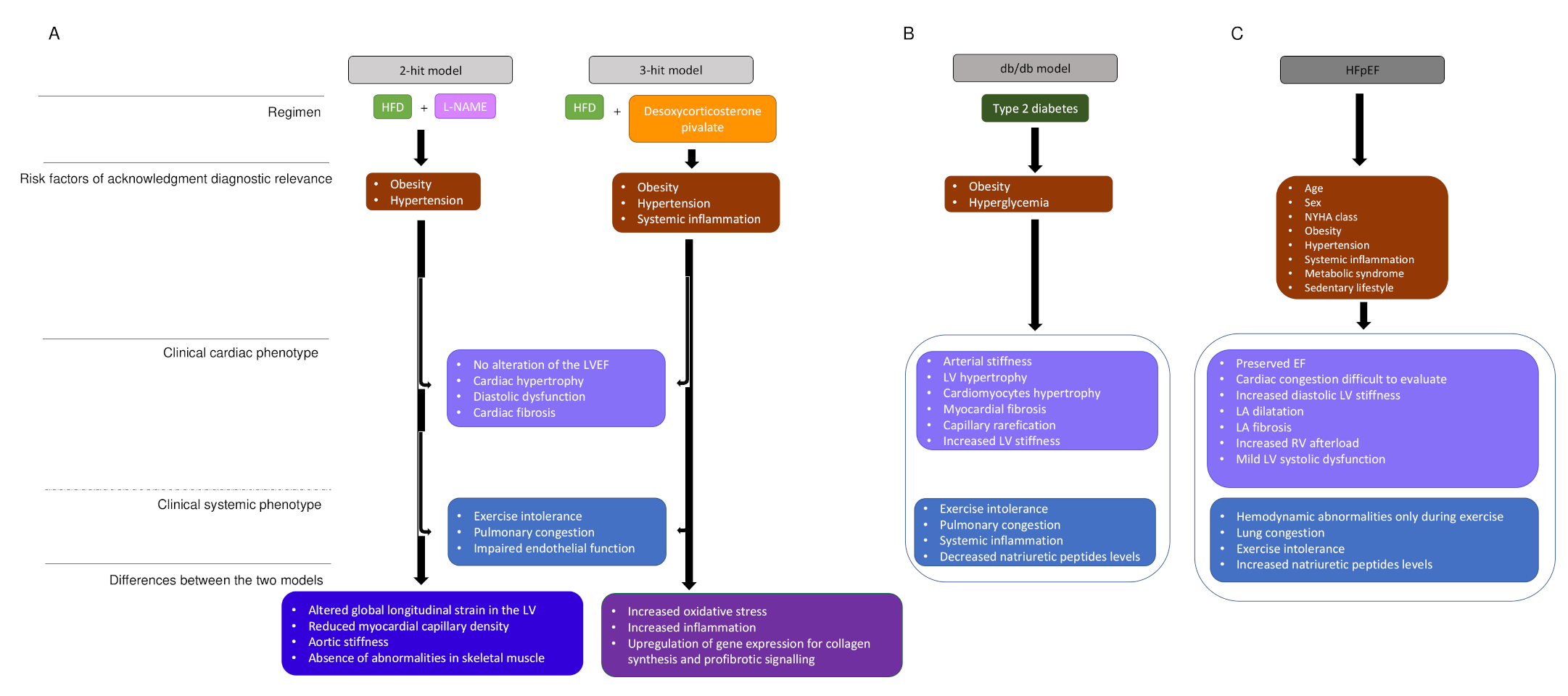
2.2. The Impact of Age, Hypertension, Obesity, and Diabetes in HFpEF
2.3. Impact of Systemic Inflammation on Cardiomyocyte Function, Cardiac Fibrosis, and Vascular Function
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Joyce, E.; Givertz, M.M. Turning Failure into Success: Trials of the Heart Failure Clinical Research Network. Curr. Cardiol. Rep. 2016, 18, 121. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Després, J.-P.; Fullerton, H.J.; et al. Heart Disease and Stroke Statistics—2016 Update. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef] [PubMed]
- Senni, M.; Caravita, S.; Paulus, W.J. Do Existing Definitions Identify Subgroup Phenotypes or Reflect the Natural History of Heart Failure With Preserved Ejection Fraction? Circulation 2019, 140, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Tadic, M.; Cuspidi, C.; Calicchio, F.; Grassi, G.; Mancia, G. Diagnostic algorithm for HFpEF: How much is the recent consensus applicable in clinical practice? Heart Fail. Rev. 2021, 26, 1485–1493. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Shah, A.M.; Borlaug, B.A. Heart Failure With Preserved Ejection Fraction In Perspective. Circ. Res. 2019, 124, 1598–1617. [Google Scholar] [CrossRef]
- Shah, S.J.; Kitzman, D.W.; Borlaug, B.A.; van Heerebeek, L.; Zile, M.R.; Kass, D.A.; Paulus, W.J. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation 2016, 134, 73–90. [Google Scholar] [CrossRef]
- Borlaug, B.A. Evaluation and management of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2020, 17, 559–573. [Google Scholar] [CrossRef]
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2014, 370, 1383–1392. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin–Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Coats, A.J.; Tsutsui, H.; Abdelhamid, M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Böhm, M.; Butler, J.; et al. Universal Definition and Classification of Heart Failure. J. Card. Fail. 2021, 27, 387–413. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Butler, J.; Abboud, F.M.; Armstrong, P.W.; Adamopoulos, S.; Atherton, J.J.; Backs, J.; Bauersachs, J.; Burkhoff, D.; Bonow, R.O.; et al. The continuous heart failure spectrum: Moving beyond an ejection fraction classification. Eur. Heart J. 2019, 40, 2155–2163. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Gorter, T.M.; Obokata, M.; Reddy, Y.N.V.; Melenovsky, V.; Borlaug, B.A. Exercise unmasks distinct pathophysiologic features in heart failure with preserved ejection fraction and pulmonary vascular disease. Eur. Heart J. 2018, 39, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Borlaug, B.A.; Kitzman, D.W.; McCulloch, A.D.; Blaxall, B.C.; Agarwal, R.; Chirinos, J.A.; Collins, S.; Deo, R.C.; Gladwin, M.T.; et al. Research Priorities for Heart Failure With Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation 2020, 141, 1001–1026. [Google Scholar] [CrossRef]
- Shah, K.S.; Xu, H.; Matsouaka, R.A.; Bhatt, D.L.; Heidenreich, P.A.; Hernandez, A.F.; Devore, A.D.; Yancy, C.W.; Fonarow, G.C. Heart Failure With Preserved, Borderline, and Reduced Ejection Fraction. J. Am. Coll. Cardiol. 2017, 70, 2476–2486. [Google Scholar] [CrossRef]
- Lourenço, A.P.; Leite-Moreira, A.F.; Balligand, J.-L.; Bauersachs, J.; Dawson, D.; de Boer, R.A.; de Windt, L.J.; Falcão-Pires, I.; Fontes-Carvalho, R.; Franz, S.; et al. An integrative translational approach to study heart failure with preserved ejection fraction: A position paper from the Working Group on Myocardial Function of the European Society of Cardiology: Translational research in HFpEF. Eur. J. Heart Fail. 2018, 20, 216–227. [Google Scholar] [CrossRef]
- Mishra, S.; Kass, D.A. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2021, 18, 400–423. [Google Scholar] [CrossRef]
- Ho, J.E.; Zern, E.K.; Wooster, L.; Bailey, C.S.; Cunningham, T.; Eisman, A.S.; Hardin, K.M.; Zampierollo, G.A.; Jarolim, P.; Pappagianopoulos, P.P.; et al. Differential Clinical Profiles, Exercise Responses, and Outcomes Associated with Existing HFpEF Definitions. Circulation 2019, 140, 353–365. [Google Scholar] [CrossRef]
- Zile, M.R.; Baicu, C.F.; SIkonomidis, J.; Stroud, R.E.; Nietert, P.J.; Bradshaw, A.D.; Slater, R.; Palmer, B.M.; Van Buren, P.; Meyer, M.; et al. Myocardial Stiffness in Patients with Heart Failure and a Preserved Ejection Fraction: Contributions of Collagen and Titin. Circulation 2015, 131, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Granzier, H.L.; Labeit, S. The Giant Protein Titin: A Major Player in Myocardial Mechanics, Signaling, and Disease. Circ. Res. 2004, 94, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, M.; Vicenzi, M.; Arena, R.; Guazzi, M.D. Pulmonary Hypertension in Heart Failure with Preserved Ejection Fraction: A target of phosphodiesterase-5 inhibition in a 1-year study. Circulation 2011, 124, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Valero-Muñoz, M.; Backman, W.; Sam, F. Murine Models of Heart Failure With Preserved Ejection Fraction. JACC Basic Transl. Sci. 2017, 2, 770–789. [Google Scholar] [CrossRef]
- Freiburg, A.; Trombitas, K.; Hell, W.; Cazorla, O.; Fougerousse, F.; Centner, T.; Kolmerer, B.; Witt, C.; Beckmann, J.S.; Gregorio, C.C.; et al. Series of Exon-Skipping Events in the Elastic Spring Region of Titin as the Structural Basis for Myofibrillar Elastic Diversity. Circ. Res. 2000, 86, 1114–1121. [Google Scholar] [CrossRef]
- Neagoe, C.; Kulke, M.; del Monte, F.; Gwathmey, J.K.; de Tombe, P.P.; Hajjar, R.J.; Linke, W.A. Titin Isoform Switch in Ischemic Human Heart Disease. Circulation 2002, 106, 1333–1341. [Google Scholar] [CrossRef]
- Nagueh, S.F.; Shah, G.; Wu, Y.; Torre-Amione, G.; King, N.M.P.; Lahmers, S.; Witt, C.C.; Becker, K.; Labeit, S.; Granzier, H.L. Altered Titin Expression, Myocardial Stiffness, and Left Ventricular Function in Patients with Dilated Cardiomyopathy. Circulation 2004, 110, 155–162. [Google Scholar] [CrossRef]
- Makarenko, I.; Opitz, C.A.; Leake, M.C.; Neagoe, C.; Kulke, M.; Gwathmey, J.K.; del Monte, F.; Hajjar, R.J.; Linke, W.A. Passive Stiffness Changes Caused by Upregulation of Compliant Titin Isoforms in Human Dilated Cardiomyopathy Hearts. Circ. Res. 2004, 95, 708–716. [Google Scholar] [CrossRef]
- Valero-Munoz, M.; Li, S.; Wilson, R.M.; Boldbaatar, B.; Iglarz, M.; Sam, F. Dual Endothelin-A/Endothelin-B Receptor Blockade and Cardiac Remodeling in Heart Failure With Preserved Ejection Fraction. Circ. Heart Fail. 2016, 9, e003381. [Google Scholar] [CrossRef]
- Hamdani, N.; Bishu, K.G.; von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2013, 97, 464–471. [Google Scholar] [CrossRef]
- LeWinter, M.M.; Granzier, H. Cardiac Titin: A Multifunctional Giant. Circulation 2010, 121, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure With Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Borbély, A.; Falcao-Pires, I.; van Heerebeek, L.; Hamdani, N.; Édes, I.; Gavina, C.; Leite-Moreira, A.F.; Bronzwaer, J.G.F.; Papp, Z.; van der Velden, J.; et al. Hypophosphorylation of the Stiff N2B Titin Isoform Raises Cardiomyocyte Resting Tension in Failing Human Myocardium. Circ. Res. 2009, 104, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Hudson, B.; Bogomolovas, J.; Zhu, Y.; Anderson, B.; Greaser, M.; Labeit, S.; Granzier, H. PKC Phosphorylation of Titin’s PEVK Element: A Novel and Conserved Pathway for Modulating Myocardial Stiffness. Circ. Res. 2009, 105, 631–638. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.V.; Bronzwaer, J.G.F.; van der Velden, J.; Stienen, G.J.M.; Laarman, G.J.; Somsen, A.; et al. Low Myocardial Protein Kinase G Activity in Heart Failure with Preserved Ejection Fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef]
- Belin, R.J.; Sumandea, M.P.; Allen, E.J.; Schoenfelt, K.; Wang, H.; Solaro, R.J.; de Tombe, P.P. Augmented Protein Kinase C-α–Induced Myofilament Protein Phosphorylation Contributes to Myofilament Dysfunction in Experimental Congestive Heart Failure. Circ. Res. 2007, 101, 195–204. [Google Scholar] [CrossRef]
- Hudson, B.; Hidalgo, C.; Saripalli, C.; Granzier, H. Hyperphosphorylation of Mouse Cardiac Titin Contributes to Transverse Aortic Constriction-Induced Diastolic Dysfunction. Circ. Res. 2011, 109, 858–866. [Google Scholar] [CrossRef]
- Takimoto, E.; Champion, H.C.; Li, M.; Belardi, D.; Ren, S.; Rodriguez, E.R.; Bedja, D.; Gabrielson, K.L.; Wang, Y.; Kass, D.A. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 2005, 11, 214–222. [Google Scholar] [CrossRef]
- Giannetta, E.; Isidori, A.M.; Galea, N.; Carbone, I.; Mandosi, E.; Vizza, C.D.; Naro, F.; Morano, S.; Fedele, F.; Lenzi, A. Chronic Inhibition of cGMP Phosphodiesterase 5A Improves Diabetic Cardiomyopathy: A Randomized, Controlled Clinical Trial Using Magnetic Resonance Imaging With Myocardial Tagging. Circulation 2012, 125, 2323–2333. [Google Scholar] [CrossRef]
- Redfield, M.M.; Chen, H.H.; Borlaug, B.A.; Semigran, M.J.; Lee, K.L.; Lewis, G.; LeWinter, M.M.; Rouleau, J.L.; Bull, D.A.; Mann, D.L.; et al. Effect of Phosphodiesterase-5 Inhibition on Exercise Capacity and Clinical Status in Heart Failure with Preserved Ejection Fraction: A Randomized Clinical Trial. JAMA 2013, 309, 1268. [Google Scholar] [CrossRef]
- Redfield, M.M.; Anstrom, K.J.; Levine, J.A.; Koepp, G.A.; Borlaug, B.A.; Chen, H.H.; LeWinter, M.M.; Joseph, S.M.; Shah, S.J.; Semigran, M.J.; et al. Isosorbide Mononitrate in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2015, 373, 2314–2324. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Kagiyama, N.; Yanamala, N.; Segar, M.W.; Cho, J.S.; Tokodi, M.; Sengupta, P.P. Deep-Learning Models for the Echocardiographic Assessment of Diastolic Dysfunction. JACC Cardiovasc. Imaging 2021, 14, 1887–1900. [Google Scholar] [CrossRef] [PubMed]
- Goldspink, D.F.; Burniston, J.G.; Tan, L.-B. Cardiomyocyte Death and the Ageing and Failing Heart. Exp. Physiol. 2003, 88, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Jin, L.; de Tombe, P.P. Cardiac thin filament regulation. Pflugers Arch. 2008, 10, 37–46. [Google Scholar] [CrossRef]
- Hamdani, N.; Kooij, V.; van Dijk, S.; Merkus, D.; Paulus, W.J.; Remedios, C.D.; Duncker, D.J.; Stienen, G.J.M.; van der Velden, J. Sarcomeric dysfunction in heart failure. Cardiovasc. Res. 2007, 77, 649–658. [Google Scholar] [CrossRef]
- Solaro, R.J.; Kobayashi, T. Protein Phosphorylation and Signal Transduction in Cardiac Thin Filaments. J. Biol. Chem. 2011, 286, 9935–9940. [Google Scholar] [CrossRef]
- Roh, J.; Houstis, N.; Rosenzweig, A. Why Don’t We Have Proven Treatments for HFpEF? Circ. Res. 2017, 120, 1243–1245. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part II: The Aging Heart in Health: Links to Heart Disease. Circulation 2003, 107, 346–354. [Google Scholar] [CrossRef]
- Shah, A.M.; Claggett, B.; Loehr, L.R.; Chang, P.P.; Matsushita, K.; Kitzman, D.; Konety, S.; Kucharska-Newton, A.; Sueta, C.A.; Mosley, T.H.; et al. Heart Failure Stages Among Older Adults in the Community: The Atherosclerosis Risk in Communities Study. Circulation 2017, 135, 224–240. [Google Scholar] [CrossRef]
- Olivetti, G.; Melissari, M.; Capasso, J.M.; Anversa, P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ. Res. 1991, 68, 1560–1568. [Google Scholar] [CrossRef]
- Loffredo, F.S.; Nikolova, A.P.; Pancoast, J.R.; Lee, R.T. Heart Failure With Preserved Ejection Fraction: Molecular Pathways of the Aging Myocardium. Circ. Res. 2014, 115, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.-F.; Rabinovitch, P.S. Cardiac Aging in Mice and Humans: The Role of Mitochondrial Oxidative Stress. Trends Cardiovasc. Med. 2009, 19, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part III: Cellular and Molecular Clues to Heart and Arterial Aging. Circulation 2003, 107, 490–497. [Google Scholar] [CrossRef]
- Uijl, A.; Savarese, G.; Vaartjes, I.; Dahlström, U.; Brugts, J.J.; Linssen, G.C.M.; Empel, V.; Brunner-La Rocca, H.; Asselbergs, F.W.; Lund, L.H.; et al. Identification of distinct phenotypic clusters in heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2021, 23, 973–982. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA Guideline for the Management of Heart Failure. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Hardy, R.J.; Francis, D.P.; Chaturvedi, N.; Pellerin, D.; Deanfield, J.; Kuh, D.; Mayet, J.; Hughes, A.D. Midlife blood pressure change and left ventricular mass and remodelling in older age in the 1946 British birth cohort study. Eur. Heart J. 2014, 35, 3287–3295. [Google Scholar] [CrossRef]
- Samson, R.; Jaiswal, A.; Ennezat, P.V.; Cassidy, M.; Le Jemtel, T.H. Clinical Phenotypes in Heart Failure With Preserved Ejection Fraction. JAHA 2016, 5, e002477. [Google Scholar] [CrossRef]
- Tanaka, K.; Valero-Muñoz, M.; Wilson, R.M.; Essick, E.E.; Fowler, C.T.; Nakamura, K.; van den Hoff, M.; Ouchi, N.; Sam, F. Follistatin-Like 1 Regulates Hypertrophy in Heart Failure With Preserved Ejection Fraction. JACC Basic Transl. Sci. 2016, 1, 207–221. [Google Scholar] [CrossRef]
- Pandey, A.; Shah, S.J.; Butler, J.; Kellogg, D.L.; Lewis, G.D.; Forman, D.E.; Mentz, R.J.; Borlaug, B.A.; Simon, M.A.; Chirinos, J.A.; et al. Exercise Intolerance in Older Adults With Heart Failure With Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2021, 78, 1166–1187. [Google Scholar] [CrossRef]
- Kitzman, D.W.; Shah, S.J. The HFpEF Obesity Phenotype. J. Am. Coll. Cardiol. 2016, 68, 200–203. [Google Scholar] [CrossRef]
- de las Fuentes, L.; Waggoner, A.D.; Mohammed, B.S.; Stein, R.I.; Miller, B.V.; Foster, G.D.; Wyatt, H.R.; Klein, S.; Davila-Roman, V.G. Effect of Moderate Diet-Induced Weight Loss and Weight Regain on Cardiovascular Structure and Function. J. Am. Coll. Cardiol. 2009, 54, 2376–2381. [Google Scholar] [CrossRef]
- Kitzman, D.W.; Brubaker, P.; Morgan, T.; Haykowsky, M.; Hundley, G.; Kraus, W.E.; Eggebeen, J.; Nicklas, B.J. Effect of Caloric Restriction or Aerobic Exercise Training on Peak Oxygen Consumption and Quality of Life in Obese Older Patients With Heart Failure With Preserved Ejection Fraction: A Randomized Clinical Trial. JAMA 2016, 315, 36. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.I.; Hahn, V.S.; Jani, V.; Hsu, S.; Sharma, K.; Kass, D.A. Reduced Right Ventricular Sarcomere Contractility in Heart Failure With Preserved Ejection Fraction and Severe Obesity. Circulation 2021, 143, 965–967. [Google Scholar] [CrossRef] [PubMed]
- Obokata, M.; Kane, G.C.; Reddy, Y.N.V.; Olson, T.P.; Melenovsky, V.; Borlaug, B.A. Role of Diastolic Stress Testing in the Evaluation for Heart Failure With Preserved Ejection Fraction: A Simultaneous Invasive-Echocardiographic Study. Circulation 2017, 135, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Hahn, V.S.; Knutsdottir, H.; Luo, X.; Bedi, K.; Margulies, K.B.; Haldar, S.M.; Stolina, M.; Yin, J.; Khakoo, A.Y.; Vaishnav, J.; et al. Myocardial Gene Expression Signatures in Human Heart Failure With Preserved Ejection Fraction. Circulation 2021, 143, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.-H.; Vogel, M.W.; Chen, H.H. Pre-Clinical Diastolic Dysfunction. J. Am. Coll. Cardiol. 2014, 63, 407–416. [Google Scholar] [CrossRef]
- Kindermann, M.; Bohm, M. Sweet hearts die earlier--lessons from CHARM. Eur. Heart J. 2008, 29, 1342–1343. [Google Scholar] [CrossRef]
- Barouch, L.A.; Berkowitz, D.E.; Harrison, R.W.; O’Donnell, C.P.; Hare, J.M. Disruption of Leptin Signaling Contributes to Cardiac Hypertrophy Independently of Body Weight in Mice. Circulation 2003, 108, 754–759. [Google Scholar] [CrossRef]
- Reil, J.-C.; Hohl, M.; Reil, G.-H.; Granzier, H.L.; Kratz, M.T.; Kazakov, A.; Fries, P.; Müller, A.; Lenski, M.; Custodis, F.; et al. Heart rate reduction by If-inhibition improves vascular stiffness and left ventricular systolic and diastolic function in a mouse model of heart failure with preserved ejection fraction. Eur. Heart J. 2013, 34, 2839–2849. [Google Scholar] [CrossRef]
- Komajda, M.; Isnard, R.; Cohen-Solal, A.; Metra, M.; Pieske, B.; Ponikowski, P.; Voors, A.A.; Dominjon, F.; Henon-Goburdhun, C.; Pannaux, M.; et al. Effect of ivabradine in patients with heart failure with preserved ejection fraction: The EDIFY randomized placebo-controlled trial: Ivabradine in HFpEF. Eur. J. Heart Fail. 2017, 19, 1495–1503. [Google Scholar] [CrossRef]
- Griffin, K.A.; Abu-Naser, M.; Abu-Amarah, I.; Picken, M.; Williamson, G.A.; Bidani, A.K. Dynamic blood pressure load and nephropathy in the ZSF1 (fa/fa cp) model of type 2 diabetes. Am. J. Physiol.-Ren. Physiol. 2007, 293, F1605–F1613. [Google Scholar] [CrossRef]
- Leite, S.; Moreira-Costa, L.; Cerqueira, R.; Sousa-Mendes, C.; Angélico-Gonçalves, A.; Fontoura, D.; Vasques-Nóvoa, F.; Leite-Moreira, A.F.; Lourenço, A.P. Chronic Sildenafil Therapy in the ZSF1 Obese Rat Model of Metabolic Syndrome and Heart Failure With Preserved Ejection Fraction. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Chirinos, J.A.; Zamani, P. The Nitrate-Nitrite-NO Pathway and Its Implications for Heart Failure and Preserved Ejection Fraction. Curr. Heart Fail. Rep. 2016, 13, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Gould, N.; Doulias, P.-T.; Tenopoulou, M.; Raju, K.; Ischiropoulos, H. Regulation of Protein Function and Signaling by Reversible Cysteine S-Nitrosylation. J. Biol. Chem. 2013, 288, 26473–26479. [Google Scholar] [CrossRef]
- Tian, N.; Moore, R.S.; Braddy, S.; Rose, R.A.; Gu, J.-W.; Hughson, M.D.; Manning, R.D. Interactions between oxidative stress and inflammation in salt-sensitive hypertension. Am. J. Physiol.-Heart Circ. Physiol. 2007, 293, H3388–H3395. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Deng, Y.; Yang, L.; Ou, W.; Xie, M.; Ding, L.; Jiang, C.; Yu, H.; Li, Q.; et al. Mitochondrial protein hyperacetylation underpins heart failure with preserved ejection fraction in mice. J. Mol. Cell. Cardiol. 2022, 165, 76–85. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef]
- Lee, C.F.; Chavez, J.D.; Garcia-Menendez, L.; Choi, Y.; Roe, N.D.; Chiao, Y.A.; Edgar, J.S.; Goo, Y.A.; Goodlett, D.R.; Bruce, J.E.; et al. Normalization of NAD + Redox Balance as a Therapy for Heart Failure. Circulation 2016, 134, 883–894. [Google Scholar] [CrossRef]
- Horton, J.L.; Martin, O.J.; Lai, L.; Riley, N.M.; Richards, A.L.; Vega, R.B.; Leone, T.C.; Pagliarini, D.J.; Muoio, D.M.; Bedi, K.C.; et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2016, 1, e84897. [Google Scholar] [CrossRef] [PubMed]
- Bayes-Genis, A.; Liu, P.P.; Lanfear, D.E.; de Boer, R.A.; González, A.; Thum, T.; Emdin, M.; Januzzi, J.L. Omics phenotyping in heart failure: The next frontier. Eur. Heart J. 2020, 41, 3477–3484. [Google Scholar] [CrossRef] [PubMed]
- van den Brom, C.E.; Huisman, M.C.; Vlasblom, R.; Boontje, N.M.; Duijst, S.; Lubberink, M.; Molthoff, C.F.; Lammertsma, A.A.; van der Velden, J.; Boer, C.; et al. Altered myocardial substrate metabolism is associated with myocardial dysfunction in early diabetic cardiomyopathy in rats: Studies using positron emission tomography. Cardiovasc. Diabetol. 2009, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Franssen, C.; Lourenço, A.; Falcão-Pires, I.; Fontoura, D.; Leite, S.; Plettig, L.; López, B.; Ottenheijm, C.A.; Becher, P.M.; et al. Myocardial Titin Hypophosphorylation Importantly Contributes to Heart Failure with Preserved Ejection Fraction in a Rat Metabolic Risk Model. Circ. Heart Fail. 2013, 6, 1239–1249. [Google Scholar] [CrossRef]
- Hullin, R.; Biel, M.; Flockerzi, V.; Hofmann, F. Tissue-specific expression of calcium channels. Trends Cardiovasc. Med. 1993, 3, 48–53. [Google Scholar] [CrossRef]
- Tian, R.; Ingwall, J.S. Energetic basis for reduced contractile reserve in isolated rat hearts. Am. J. Physiol.-Heart Circ. Physiol. 1996, 270, H1207–H1216. [Google Scholar] [CrossRef]
- Flarsheim, C.E.; Grupp, I.L.; Matlib, M.A. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am. J. Physiol.-Heart Circ. Physiol. 1996, 271, H192–H202. [Google Scholar] [CrossRef]
- Stüdeli, R.; Jung, S.; Mohacsi, P.; Perruchoud, S.; Castiglioni, P.; Wenaweser, P.; Heimbeck, G.; Feller, M.; Hullin, R. Diastolic Dysfunction in Human Cardiac Allografts is Related with Reduced SERCA2a Gene Expression: Reduced SERCA2a Expression in Diastolic Failure. Am. J. Transplant. 2006, 6, 775–782. [Google Scholar] [CrossRef]
- Hullin, R.; Asmus, F.; Ludwig, A.; Hersel, J.; Boekstegers, P. Subunit Expression of the Cardiac L-Type Calcium Channel Is Differentially Regulated in Diastolic Heart Failure of the Cardiac Allograft. Circulation 1999, 100, 155–163. [Google Scholar] [CrossRef][Green Version]
- Miranda-Silva, D.; Wüst, R.C.I.; Conceição, G.; Gonçalves-Rodrigues, P.; Gonçalves, N.; Gonçalves, A.; Kuster, D.W.D.; Leite-Moreira, A.F.; Velden, J.; Sousa Beleza, J.M.; et al. Disturbed cardiac mitochondrial and cytosolic calcium handling in a metabolic risk-related rat model of heart failure with preserved ejection fraction. Acta Physiol. 2020, 228, e13378. [Google Scholar] [CrossRef]
- Maack, C.; O’Rourke, B. Excitation-contraction coupling and mitochondrial energetics. Basic Res. Cardiol. 2007, 102, 369–392. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; O’Rourke, B. Enhancing Mitochondrial Ca2+ Uptake in Myocytes From Failing Hearts Restores Energy Supply and Demand Matching. Circ. Res. 2008, 103, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W.; Maack, C. SR and mitochondria: Calcium cross-talk between kissing cousins. J. Mol. Cell. Cardiol. 2013, 55, 42–49. [Google Scholar] [CrossRef]
- Wüst, R.C.I.; de Vries, H.J.; Wintjes, L.T.; Rodenburg, R.J.; Niessen, H.W.M.; Stienen, G.J.M. Mitochondrial complex I dysfunction and altered NAD(P)H kinetics in rat myocardium in cardiac right ventricular hypertrophy and failure. Cardiovasc. Res. 2016, 111, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Ritterhoff, J.; Tian, R. Metabolism in cardiomyopathy: Every substrate matters. Cardiovasc. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef]
- Chirinos, J.A.; Orlenko, A.; Zhao, L.; Basso, M.D.; Cvijic, M.E.; Li, Z.; Spires, T.E.; Yarde, M.; Wang, Z.; Seiffert, D.A.; et al. Multiple Plasma Biomarkers for Risk Stratification in Patients With Heart Failure and Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2020, 75, 1281–1295. [Google Scholar] [CrossRef]
- Hage, C.; Michaëlsson, E.; Linde, C.; Donal, E.; Daubert, J.-C.; Gan, L.-M.; Lund, L.H. Inflammatory Biomarkers Predict Heart Failure Severity and Prognosis in Patients With Heart Failure with Preserved Ejection Fraction: A Holistic Proteomic Approach. Circ. Cardiovasc. Genet. 2017, 10, e001633. [Google Scholar] [CrossRef]
- Hahn, V.S.; Yanek, L.R.; Vaishnav, J.; Ying, W.; Vaidya, D.; Lee, Y.Z.J.; Riley, S.J.; Subramanya, V.; Brown, E.E.; Hopkins, C.D.; et al. Endomyocardial Biopsy Characterization of Heart Failure with Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020, 8, 712–724. [Google Scholar] [CrossRef]
- Yamamoto, K. Myocardial stiffness is determined by ventricular fibrosis, but not by compensatory or excessive hypertrophy in hypertensive heart. Cardiovasc. Res. 2002, 55, 76–82. [Google Scholar] [CrossRef]
- Graham, H.K.; Trafford, A.W. Spatial disruption and enhanced degradation of collagen with the transition from compensated ventricular hypertrophy to symptomatic congestive heart failure. Am. J. Physiol.-Heart Circ. Physiol. 2007, 292, H1364–H1372. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Handoko, M.L.; Falcao-Pires, I.; Musters, R.J.; Kupreishvili, K.; Ijsselmuiden, A.J.J.; Schalkwijk, C.G.; Bronzwaer, J.G.F.; Diamant, M.; et al. Diastolic Stiffness of the Failing Diabetic Heart: Importance of Fibrosis, Advanced Glycation End Products, and Myocyte Resting Tension. Circulation 2008, 117, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Alex, L.; Russo, I.; Holoborodko, V.; Frangogiannis, N.G. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am. J. Physiol.-Heart Circ. Physiol. 2018, 315, H934–H949. [Google Scholar] [CrossRef] [PubMed]
- Kurose, H. Cardiac Fibrosis and Fibroblasts. Cells 2021, 10, 1716. [Google Scholar] [CrossRef]
- López, B.; González, A.; Díez, J. Circulating Biomarkers of Collagen Metabolism in Cardiac Diseases. Circulation 2010, 121, 1645–1654. [Google Scholar] [CrossRef]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac Inflammation Contributes to Changes in the Extracellular Matrix in Patients with Heart Failure and Normal Ejection Fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef]
- Cavalera, M.; Frangogiannis, N. Targeting the Chemokines in Cardiac Repair. CPD 2014, 20, 1971–1979. [Google Scholar] [CrossRef]
- Johns, R.A.; Takimoto, E.; Meuchel, L.W.; Elsaigh, E.; Zhang, A.; Heller, N.M.; Semenza, G.L.; Yamaji-Kegan, K. Hypoxia-Inducible Factor 1α Is a Critical Downstream Mediator for Hypoxia-Induced Mitogenic Factor (FIZZ1/RELMα)–Induced Pulmonary Hypertension. ATVB 2016, 36, 134–144. [Google Scholar] [CrossRef]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef]
- Meléndez, G.C.; McLarty, J.L.; Levick, S.P.; Du, Y.; Janicki, J.S.; Brower, G.L. Interleukin 6 Mediates Myocardial Fibrosis, Concentric Hypertrophy, and Diastolic Dysfunction in Rats. Hypertension 2010, 56, 225–231. [Google Scholar] [CrossRef]
- Meier, H.; Bullinger, J.; Marx, G.; Deten, A.; Horn, L.-C.; Raßler, B.; Zimmer, H.-G.; Briest, W. Crucial Role of Interleukin-6 in the Development of Norepinephrine-induced Left Ventricular Remodeling in Mice. Cell Physiol. Biochem. 2009, 23, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Coles, B.; Fielding, C.A.; Rose-John, S.; Scheller, J.; Jones, S.A.; O’Donnell, V.B. Classic Interleukin-6 Receptor Signaling and Interleukin-6 trans-Signaling Differentially Control Angiotensin II-Dependent Hypertension, Cardiac Signal Transducer and Activator of Transcription-3 Activation, and Vascular Hypertrophy in Vivo. Am. J. Pathol. 2007, 171, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Wang, G.; Zheng, N.; Cheng, W.; Ouyang, K.; Lin, H.; Liao, Y.; Liu, J. HIMF (Hypoxia-Induced Mitogenic Factor)-IL (Interleukin)-6 Signaling Mediates Cardiomyocyte-Fibroblast Crosstalk to Promote Cardiac Hypertrophy and Fibrosis. Hypertension 2019, 73, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Kalogeropoulos, A.; Georgiopoulou, V.; Psaty, B.M.; Rodondi, N.; Smith, A.L.; Harrison, D.G.; Liu, Y.; Hoffmann, U.; Bauer, D.C.; Newman, A.B.; et al. Inflammatory Markers and Incident Heart Failure Risk in Older Adults. J. Am. Coll. Cardiol. 2010, 55, 2129–2137. [Google Scholar] [CrossRef]
- Greulich, S.; Maxhera, B.; Vandenplas, G.; de Wiza, D.H.; Smiris, K.; Mueller, H.; Heinrichs, J.; Blumensatt, M.; Cuvelier, C.; Akhyari, P.; et al. Secretory Products From Epicardial Adipose Tissue of Patients With Type 2 Diabetes Mellitus Induce Cardiomyocyte Dysfunction. Circulation 2012, 126, 2324–2334. [Google Scholar] [CrossRef]
- Mazurek, T.; Zhang, L.; Zalewski, A.; Mannion, J.D.; Diehl, J.T.; Arafat, H.; Sarov-Blat, L.; O’Brien, S.; Keiper, E.A.; Johnson, A.G.; et al. Human Epicardial Adipose Tissue Is a Source of Inflammatory Mediators. Circulation 2003, 108, 2460–2466. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hausegger, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef]
- Ather, S.; Chan, W.; Bozkurt, B.; Aguilar, D.; Ramasubbu, K.; Zachariah, A.A.; Wehrens, X.H.T.; Deswal, A. Impact of Noncardiac Comorbidities on Morbidity and Mortality in a Predominantly Male Population With Heart Failure and Preserved Versus Reduced Ejection Fraction. J. Am. Coll. Cardiol. 2012, 59, 998–1005. [Google Scholar] [CrossRef]
- Lam, C.S.P.; Lyass, A.; Kraigher-Krainer, E.; Massaro, J.M.; Lee, D.S.; Ho, J.E.; Levy, D.; Redfield, M.M.; Pieske, B.M.; Benjamin, E.J.; et al. Cardiac Dysfunction and Noncardiac Dysfunction as Precursors of Heart Failure with Reduced and Preserved Ejection Fraction in the Community. Circulation 2011, 124, 24–30. [Google Scholar] [CrossRef]
- Taube, A.; Schlich, R.; Sell, H.; Eckardt, K.; Eckel, J. Inflammation and metabolic dysfunction: Links to cardiovascular diseases. Am. J. Physiol.-Heart Circ. Physiol. 2012, 302, H2148–H2165. [Google Scholar] [CrossRef] [PubMed]
- Jelic, S.; Lederer, D.J.; Adams, T.; Padeletti, M.; Colombo, P.C.; Factor, P.H.; Le Jemtel, T.H. Vascular Inflammation in Obesity and Sleep Apnea. Circulation 2010, 121, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Haass, M.; Kitzman, D.W.; Anand, I.S.; Miller, A.; Zile, M.R.; Massie, B.M.; Carson, P.E. Body Mass Index and Adverse Cardiovascular Outcomes in Heart Failure Patients With Preserved Ejection Fraction: Results From the Irbesartan in Heart Failure With Preserved Ejection Fraction (I-PRESERVE) Trial. Circ. Heart Fail. 2011, 4, 324–331. [Google Scholar] [CrossRef]
- Hummel, S.L.; Seymour, E.M.; Brook, R.D.; Kolias, T.J.; Sheth, S.S.; Rosenblum, H.R.; Wells, J.M.; Weder, A.B. Low-Sodium Dietary Approaches to Stop Hypertension Diet Reduces Blood Pressure, Arterial Stiffness, and Oxidative Stress in Hypertensive Heart Failure With Preserved Ejection Fraction. Hypertension 2012, 60, 1200–1206. [Google Scholar] [CrossRef]
- Macdougall, I.C.; Canaud, B.; de Francisco, A.L.M.; Filippatos, G.; Ponikowski, P.; Silverberg, D.; van Veldhuisen, D.J.; Anker, S.D. Beyond the cardiorenal anaemia syndrome: Recognizing the role of iron deficiency. Eur. J. Heart Fail. 2012, 14, 882–886. [Google Scholar] [CrossRef]
- Shah, K.B.; Kop, W.J.; Christenson, R.H.; Diercks, D.B.; Henderson, S.; Hanson, K.; Li, S.-Y.; deFilippi, C.R. Prognostic Utility of ST2 in Patients with Acute Dyspnea and Preserved Left Ventricular Ejection Fraction. Clin. Chem. 2011, 57, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, J.; Sugiyama, S.; Nozaki, T.; Sugamura, K.; Konishi, M.; Ohba, K.; Matsuzawa, Y.; Akiyama, E.; Yamamoto, E.; Sakamoto, K.; et al. Pentraxin 3 Is a New Inflammatory Marker Correlated with Left Ventricular Diastolic Dysfunction and Heart Failure With Normal Ejection Fraction. J. Am. Coll. Cardiol. 2011, 57, 861–869. [Google Scholar] [CrossRef]
- Kallikourdis, M.; Martini, E.; Carullo, P.; Sardi, C.; Roselli, G.; Greco, C.M.; Vignali, D.; Riva, F.; Ormbostad Berre, A.M.; Stølen, T.O.; et al. T cell costimulation blockade blunts pressure overload-induced heart failure. Nat. Commun. 2017, 8, 14680. [Google Scholar] [CrossRef]
- Cuijpers, I.; Simmonds, S.J.; van Bilsen, M.; Czarnowska, E.; González Miqueo, A.; Heymans, S.; Kuhn, A.R.; Mulder, P.; Ratajska, A.; Jones, E.A.V.; et al. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. Basic Res. Cardiol. 2020, 115, 39. [Google Scholar] [CrossRef]
- Falkenham, A.; de Antueno, R.; Rosin, N.; Betsch, D.; Lee, T.D.G.; Duncan, R.; Légaré, J. Nonclassical Resident Macrophages Are Important Determinants in the Development of Myocardial Fibrosis. Am. J. Pathol. 2015, 185, 927–942. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H Oxidase: Role in Cardiovascular Biology and Disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered Mitochondrial Dynamics Contributes to Endothelial Dysfunction in Diabetes Mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef]
- Rajapakse, A.G.; Yepuri, G.; Carvas, J.M.; Stein, S.; Matter, C.M.; Scerri, I.; Ruffieux, J.; Montani, J.-P.; Ming, X.-F.; Yang, Z. Hyperactive S6K1 Mediates Oxidative Stress and Endothelial Dysfunction in Aging: Inhibition by Resveratrol. PLoS ONE 2011, 6, e19237. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; Radauceanu, A. Effect of MR Blockade on Collagen Formation and Cardiovascular Disease with a Specific Emphasis on Heart Failure. Heart Fail. Rev. 2005, 10, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Duprez, D.A.; Gross, M.D.; Kizer, J.R.; Ix, J.H.; Hundley, W.G.; Jacobs, D.R. Predictive Value of Collagen Biomarkers for Heart Failure With and Without Preserved Ejection Fraction: MESA (Multi-Ethnic Study of Atherosclerosis). JAHA 2018, 7, e007885. [Google Scholar] [CrossRef] [PubMed]
- Tschöpe, C.; Bock, C.-T.; Kasner, M.; Noutsias, M.; Westermann, D.; Schwimmbeck, P.-L.; Pauschinger, M.; Poller, W.-C.; Kühl, U.; Kandolf, R.; et al. High Prevalence of Cardiac Parvovirus B19 Infection in Patients with Isolated Left Ventricular Diastolic Dysfunction. Circulation 2005, 111, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Olson, T.P.; Lam, C.S.P.; Flood, K.S.; Lerman, A.; Johnson, B.D.; Redfield, M.M. Global Cardiovascular Reserve Dysfunction in Heart Failure With Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2010, 56, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Haykowsky, M.J.; Brubaker, P.H.; Stewart, K.P.; Morgan, T.M.; Eggebeen, J.; Kitzman, D.W. Effect of Endurance Training on the Determinants of Peak Exercise Oxygen Consumption in Elderly Patients with Stable Compensated Heart Failure and Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2012, 60, 120–128. [Google Scholar] [CrossRef]
- Edelmann, F.; Gelbrich, G.; Düngen, H.-D.; Fröhling, S.; Wachter, R.; Stahrenberg, R.; Binder, L.; Töpper, A.; Lashki, D.J.; Schwarz, S.; et al. Exercise Training Improves Exercise Capacity and Diastolic Function in Patients with Heart Failure with Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2011, 58, 1780–1791. [Google Scholar] [CrossRef]
- Chamsi-Pasha, M.A.; Zhan, Y.; Debs, D.; Shah, D.J. CMR in the Evaluation of Diastolic Dysfunction and Phenotyping of HFpEF. JACC Cardiovasc. Imaging 2020, 13, 283–296. [Google Scholar] [CrossRef]
- Backhaus, S.J.; Lange, T.; George, E.F.; Hellenkamp, K.; Gertz, R.J.; Billing, M.; Wachter, R.; Steinmetz, M.; Kutty, S.; Raaz, U.; et al. Exercise Stress Real-Time Cardiac Magnetic Resonance Imaging for Noninvasive Characterization of Heart Failure with Preserved Ejection Fraction: The HFpEF-Stress Trial. Circulation 2021, 143, 1484–1498. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocca, A.; Heeswijk, R.B.v.; Richiardi, J.; Meyer, P.; Hullin, R. The Cardiomyocyte in Heart Failure with Preserved Ejection Fraction—Victim of Its Environment? Cells 2022, 11, 867. https://doi.org/10.3390/cells11050867
Rocca A, Heeswijk RBv, Richiardi J, Meyer P, Hullin R. The Cardiomyocyte in Heart Failure with Preserved Ejection Fraction—Victim of Its Environment? Cells. 2022; 11(5):867. https://doi.org/10.3390/cells11050867
Chicago/Turabian StyleRocca, Angela, Ruud B. van Heeswijk, Jonas Richiardi, Philippe Meyer, and Roger Hullin. 2022. "The Cardiomyocyte in Heart Failure with Preserved Ejection Fraction—Victim of Its Environment?" Cells 11, no. 5: 867. https://doi.org/10.3390/cells11050867
APA StyleRocca, A., Heeswijk, R. B. v., Richiardi, J., Meyer, P., & Hullin, R. (2022). The Cardiomyocyte in Heart Failure with Preserved Ejection Fraction—Victim of Its Environment? Cells, 11(5), 867. https://doi.org/10.3390/cells11050867