
Molecular Mechanism of Lipotoxicity as an Interesting Aspect in the Development of Pathological States—Current View of Knowledge


Abstract
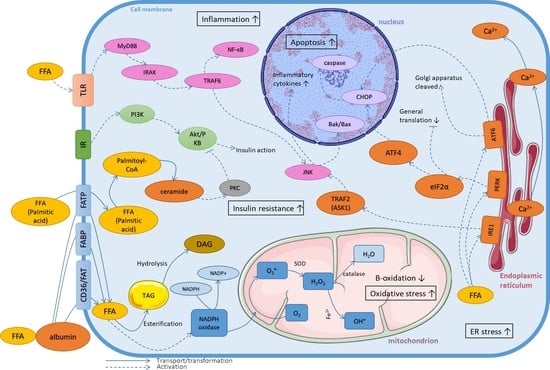
1. Introduction
2. The Role of Homeostasis Disturbances of Fatty Acids in Lipotoxicity
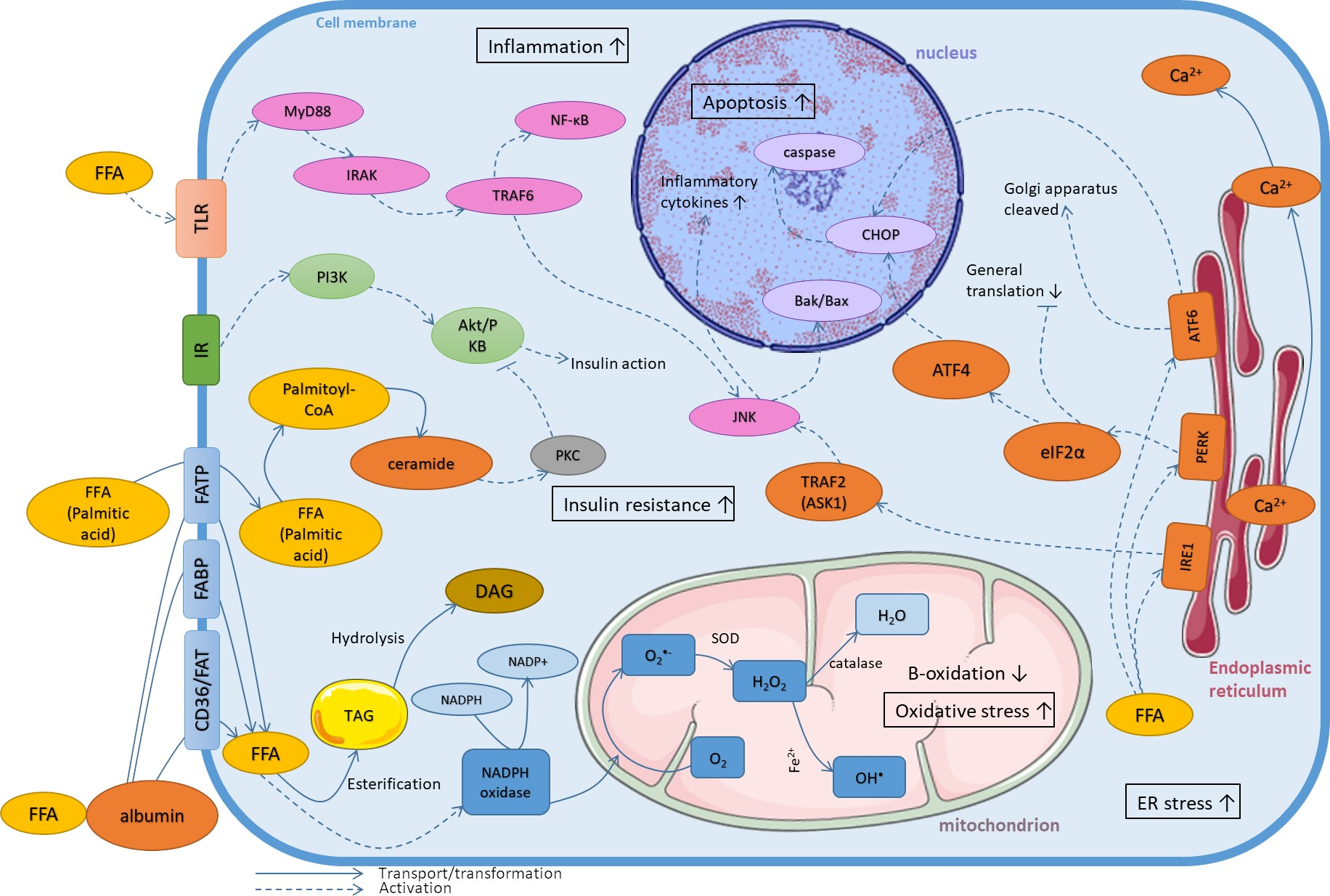
3. Mechanisms of Lipotoxicity on the Cellular Level
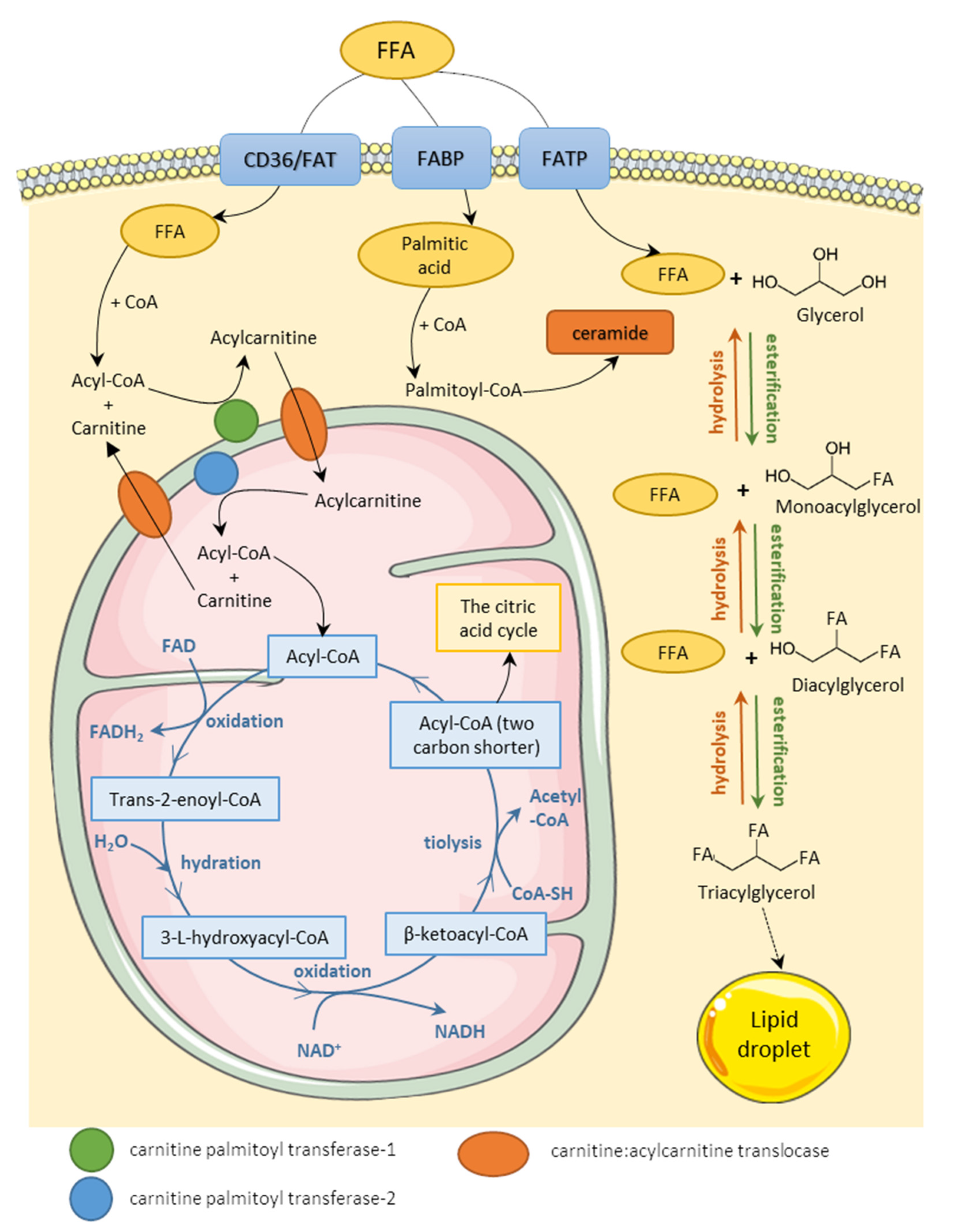
3.1. Fatty Acids Metabolism
3.2. Oxidative Stress
3.3. Endoplasmic Reticulum Stress
3.4. Inflammatory State
3.5. Induction of Insulin Resistance
3.6. Autophagy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lee, Y.; Hirose, H.; Ohneda, M.; Johnson, J.H.; McGarry, J.D.; Unger, R.H. β-Cell Lipotoxicity in the Pathogenesis of Non-Insulin-Dependent Diabetes Mellitus of Obese Rats: Impairment in Adipocyte-β-Cell Relationships. Proc. Natl. Acad. Sci. USA 1994, 91, 10878–10882. [Google Scholar] [CrossRef] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential Effect of Oleic and Palmitic Acid on Lipid Accumulation and Apoptosis in Cultured Hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.A.; LeBrasseur, N.K.; Cote, G.M.; Trucillo, M.P.; Pimentel, D.R.; Ido, Y.; Ruderman, N.B.; Sawyer, D.B. Oleate Prevents Palmitate-Induced Cytotoxic Stress in Cardiac Myocytes. Biochem. Biophys. Res. Commun. 2005, 336, 309–315. [Google Scholar] [CrossRef]
- Peng, G.; Li, L.; Liu, Y.; Pu, J.; Zhang, S.; Yu, J.; Zhao, J.; Liu, P. Oleate Blocks Palmitate-Induced Abnormal Lipid Distribution, Endoplasmic Reticulum Expansion and Stress, and Insulin Resistance in Skeletal Muscle. Endocrinology 2011, 152, 2206–2218. [Google Scholar] [CrossRef] [PubMed]
- Doege, H.; Grimm, D.; Falcon, A.; Tsang, B.; Storm, T.A.; Xu, H.; Ortegon, A.M.; Kazantzis, M.; Kay, M.A.; Stahl, A. Silencing of Hepatic Fatty Acid Transporter Protein 5 in Vivo Reverses Diet-Induced Non-Alcoholic Fatty Liver Disease and Improves Hyperglycemia. J. Biol. Chem. 2008, 283, 22186–22192. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yan, Q.; Lv, M.; Song, K.; Dai, Y.; Huang, Y.; Zhang, L.; Zhang, C.; Gao, H. Involvement of FATP2-Mediated Tubular Lipid Metabolic Reprogramming in Renal Fibrogenesis. Cell Death Dis. 2020, 11, 994. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xiao, Y.; Tang, L.; Zhong, F.; Huang, G.; Xu, J.M.; Xu, A.M.; Dai, R.P.; Zhou, Z.G. Adipocyte Fatty Acid-Binding Protein Promotes Palmitate-Induced Mitochondrial Dysfunction and Apoptosis in Macrophages. Front. Immunol. 2018, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Lee, K.Y.; Jung, S.H.; Kim, H.S.; Shim, G.; Kim, M.G.; Oh, Y.K.; Oh, S.H.; Jun, D.W.; Lee, B.H. Activation of AMPK by Berberine Induces Hepatic Lipid Accumulation by Upregulation of Fatty Acid Translocase CD36 in Mice. Toxicol. Appl. Pharmacol. 2017, 316, 74–82. [Google Scholar] [CrossRef]
- Gao, Q.; Sarkar, A.; Chen, Y.; Xu, B.; Zhu, X.; Yuan, Y.; Guan, T. Overexpression of Heart-Type Fatty Acid Binding Protein Enhances Fatty Acid-Induced Podocyte Injury. Exp. Ther. Med. 2018, 15, 2054–2061. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Gieseler, R.K.; Sowa, J.P.; Kahraman, A.; Erhard, J.; Wedemeyer, I.; Emons, B.; Jochum, C.; Feldkamp, T.; Gerken, G.; et al. Apoptosis Is Associated with CD36/Fatty Acid Translocase Upregulation in Non-Alcoholic Steatohepatitis. Liver Int. 2010, 30, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.E.; Bucarey, J.L.; Espinosa, A. Muscle Lipid Metabolism: Role of Lipid Droplets and Perilipins. J. Diabetes Res. 2017, 2017, 1789395. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sreenevasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a Lipid Droplet-Associated Protein, Provides Physical and Metabolic Linkage to Mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 Improves Hepatic Lipotoxicity by Inhibiting Lipolysis. Hepatology 2015, 61, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Wronska, A.; Kmiec, Z. Structural and Biochemical Characteristics of Various White Adipose Tissue Depots. Acta Physiol. 2012, 205, 194–208. [Google Scholar] [CrossRef]
- Siemińska, L. Adipose Tissue. Pathophysiology, Distribution, Sex Differences and the Role in Inflammation and Cancerogenesis. Endokrynol. Pol. 2007, 58, 330–342. [Google Scholar] [PubMed]
- Burdge, G.C.; Calder, P.C. Introduction to Fatty Acids and Lipids. World Rev. Nutr. Diet. 2015, 112, 1–16. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-Oxidation of Saturated Fatty Acids in Humans TO. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef]
- Rinaldo, P.; Matern, D.; Bennett, M.J. Fatty Acid Oxidation Disorders. Annu. Rev. Physiol. 2002, 64, 477–502. [Google Scholar] [CrossRef]
- Chavez, J.A.; Summers, S.A. Perspective A Ceramide-Centric View of Insulin Resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef]
- Stremmel, W.; Pohl, J.; Ring, A.; Herrmann, T. A New Concept of Cellular Uptake and Intracellular Trafficking of Long-Chain Fatty Acids. Lipids 2001, 36, 981–989. [Google Scholar] [CrossRef]
- Kume, S.; Uzu, T.; Araki, S.I.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Kubota, N.; Terauchi, Y.; Kadowaki, T.; et al. Role of Altered Renal Lipid Metabolism in the Development of Renal Injury Induced by a High-Fat Diet. J. Am. Soc. Nephrol. 2007, 18, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.E.; Jung, I.R.; Lee, Y.J.; Lee, S.J.; Lee, J.H.; Kim, Y.; Jun, H.S.; Lee, K.W.; Park, C.B.; Kang, Y. Stimulation of Lipogenesis as Well as Fatty Acid Oxidation Protects against Palmitate-Induced INS-1 β-Cell Death. Endocrinology 2011, 152, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Kampe, K.; Sieber, J.; Orellana, J.M.; Mundel, P.; Jehle, A.W. Susceptibility of Podocytes to Palmitic Acid Is Regulated by Fatty Acid Oxidation and Inversely Depends on Acetyl-CoA Carboxylases 1 and 2. Am. J. Physiol. Ren. Physiol. 2014, 306, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Mayrhofer, C.; Krieger, S.; Huttary, N.; Chang, M.W.F.; Grillari, J.; Allmaier, G.; Kerjaschki, D. Alterations in Fatty Acid Utilization and an Impaired Antioxidant Defense Mechanism Are Early Events in Podocyte Injury. Am. J. Pathol. 2009, 174, 1191–1202. [Google Scholar] [CrossRef]
- Manukyan, L.; Ubhayasekera, S.J.K.A.; Bergquist, J.; Sargsyan, E.; Bergsten, P. Palmitate-Induced Impairments of Beta-Cell Function Are Linked With Generation of Specific Ceramide Species via Acylation of Sphingosine. Endocrinology 2015, 156, 802–812. [Google Scholar] [CrossRef]
- Akoumi, A.; Haffar, T.; Mousterji, M.; Kiss, R.S.; Bousette, N. Palmitate Mediated Diacylglycerol Accumulation Causes Endoplasmic Reticulum Stress, Plin2 Degradation, and Cell Death in H9C2 Cardiomyoblasts. Exp. Cell Res. 2017, 354, 85–94. [Google Scholar] [CrossRef]
- Mingrone, G.; Rosa, G.; Di Rocco, P.; Manco, M.; Capristo, E.; Castagneto, M.; Vettor, R.; Gasbarrini, G.; Greco, A.V. Skeletal Muscle Triglycerides Lowering Is Associated with Net Improvement of Insulin Sensitivity, TNF-α Reduction and GLUT4 Expression Enhancement. Int. J. Obes. 2002, 26, 1165–1172. [Google Scholar] [CrossRef]
- Hwang, J.H.; Stein, D.T.; Barzilai, N.; Cui, M.H.; Tonelli, J.; Kishore, P.; Hawkins, M. Increased Intrahepatic Triglyceride Is Associated with Peripheral Insulin Resistance: In Vivo MR Imaging and Spectroscopy Studies. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 1663–1669. [Google Scholar] [CrossRef]
- Cheon, H.G.; Cho, Y.S. Protection of Palmitic Acid-Mediated Lipotoxicity by Arachidonic Acid via Channeling of Palmitic Acid into Triglycerides in C2C12. J. Biomed. Sci. 2014, 21, 13. [Google Scholar] [CrossRef]
- Mantzaris, M.D.; Tsianos, E.V.; Galaris, D. Interruption of Triacylglycerol Synthesis in the Endoplasmic Reticulum Is the Initiating Event for Saturated Fatty Acid-Induced Lipotoxicity in Liver Cells. FEBS J. 2011, 278, 519–530. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride Accumulation Protects against Fatty Acid-Induced Lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.B.; Newgard, C.B.; et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef]
- Haffar, T.; Akoumi, A.; Bousette, N. Lipotoxic Palmitate Impairs the Rate of β-Oxidation and Citric Acid Cycle Flux in Rat Neonatal Cardiomyocytes. Cell. Physiol. Biochem. 2016, 40, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, A.S.; Gaidhu, M.P.; Habib, S.; So, M.; Fediuc, S.; Mirpourian, M.; Musheev, M.; Curi, R.; Ceddia, R.B. Prolonged Exposure to Palmitate Impairs Fatty Acid Oxidation despite Activation of AMP-Activated Protein Kinase in Skeletal Muscle Cells. J. Cell. Physiol. 2008, 217, 478–485. [Google Scholar] [CrossRef]
- Tomas, E.; Tsao, T.S.; Saha, A.K.; Murrey, H.E.; Zhang, C.C.; Itani, S.I.; Lodish, H.F.; Ruderman, N.B. Enhanced Muscle Fat Oxidation and Glucose Transport by ACRP30 Globular Domain: Acetyl-CoA Carboxylase Inhibition and AMP-Activated Protein Kinase Activation. Proc. Natl. Acad. Sci. USA 2002, 99, 16309–16313. [Google Scholar] [CrossRef]
- Asem, E.K.; Qin, W.; Rane, S.G. High Carbohydrate Availability Increases LCFA Uptake and Decreases LCFA Oxidation in Perfused Muscle. Am. J. Physiol. Endocrinol. Metab. 2002, 282, 177–183. [Google Scholar] [CrossRef][Green Version]
- Hickson-Bick, D.L.M.; Buja, M.L.; McMillin, J.B. Palmitate-Mediated Alterations in the Fatty Acid Metabolism of Rat Neonatal Cardiac Myocytes. J. Mol. Cell. Cardiol. 2000, 32, 511–519. [Google Scholar] [CrossRef]
- Young, M.E.; Goodwin, G.W.; Ying, J.; Guthrie, P.; Wilson, C.R.; Laws, F.A.; Taegtmeyer, H. Regulation of Cardiac and Skeletal Muscle Malonyl-CoA Decarboxylase by Fatty Acids. Am. J. Physiol. Endocrinol. Metab. 2001, 280, 471–479. [Google Scholar] [CrossRef]
- Bruce, C.R.; Hoy, A.J.; Turner, N.; Watt, M.J.; Allen, T.L.; Carpenter, K.; Cooney, G.J.; Febbraio, M.A.; Kraegen, E.W. Overexpression of Carnitine Palmitoyltransferase-1 in Skeletal Muscle Is Sufficient to Enhance Fatty Acid Oxidation and Improve High-Fat Diet-Induced Insulin Resistance. Diabetes 2009, 58, 550–558. [Google Scholar] [CrossRef]
- Young, M.E.; Guthrie, P.H.; Razeghi, P.; Leighton, B.; Abbasi, S.; Patil, S.; Youker, K.A.; Taegtmeyer, H. Impaired Long-Chain Fatty Acid Oxidation and Contractile Dysfunction in the Obese Zucker Rat Heart. Diabetes 2002, 51, 2587–2595. [Google Scholar] [CrossRef]
- Wensaas, A.J.; Rustan, A.C.; Just, M.; Berge, R.K.; Drevon, C.A.; Gaster, M. Fatty Acid Incubation of Myotubes from Humans with Type 2 Diabetes Leads to Enhanced Release of β-Oxidation Products Because of Impaired Fatty Acid Oxidation: Effects of Tetradecylthioacetic Acid and Eicosapentaenoic Acid. Diabetes 2009, 58, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective Fatty Acid Oxidation in Renal Tubular Epithelial Cells Plays a Key Role in Kidney Fibrosis Development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-Induced Insulin Resistance in Human Muscle Is Associated with Changes in Diacylglycerol, Protein Kinase C, and IκB-α. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Montell, E.; Turini, M.; Marotta, M.; Roberts, M.; Noé, V.; Ciudad, C.J.; Macé, K.; Gómez-Foix, A.M. DAG Accumulation from Saturated Fatty Acids Desensitizes Insulin Stimulation of Glucose Uptake in Muscle Cells. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E229–E237. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. Characterizing the Effects of Saturated Fatty Acids on Insulin Signaling and Ceramide and Diacylglycerol Accumulation in 3T3-L1 Adipocytes and C2C12 Myotubes. Arch. Biochem. Biophys. 2003, 419, 101–109. [Google Scholar] [CrossRef]
- Macrae, K.; Stretton, C.; Lipina, C.; Blachnio-zabielska, A.; Baranowski, M.; Gorski, J.; Marley, A.; Hundal, H.S. Defining the Role of DAG, Mitochondrial Function, and Lipid Deposition in Palmitate-Induced Proinfl Ammatory Signaling and Its Counter-Modulation by Palmitoleate. J. Lipid Res. 2013, 54, 2366–2378. [Google Scholar] [CrossRef]
- Aguer, C.; McCoin, C.S.; Knotts, T.A.; Thrush, A.B.; Ono-Moore, K.; McPherson, R.; Dent, R.; Hwang, D.H.; Adams, S.H.; Harper, M.E. Acylcarnitines: Potential Implications for Skeletal Muscle Insulin Resistance. FASEB J. 2015, 29, 336–345. [Google Scholar] [CrossRef]
- Blackburn, M.L.; Ono-moore, K.; Sobhi, H.F.; Adams, S.H. Carnitine Palmitoyltransferase 2 (CPT2) Knockout Potentiates Palmitate-Induced Insulin Resistance in C2C12 Myotubes. Am. J. Physiol. Endocrinol. Metab. 2020, 319, 265–275. [Google Scholar] [CrossRef]
- Blachnio-Zabielska, A.U.; Chacinska, M.; Vendelbo, M.H.; Zabielski, P. The Crucial Role of C18-Cer in Fat-Induced Skeletal Muscle Insulin Resistance. Cell. Physiol. Biochem. 2016, 40, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Pickersgill, L.; Litherland, G.J.; Greenberg, S.; Walker, M.; Stephen, J. Key Role for Ceramides in Mediating Insulin Resistance in Human Muscle Cells. J. Biol. Chem. 2007, 282, 12583–12589. [Google Scholar] [CrossRef]
- Turpin, S.M.; Lancaster, G.I.; Darby, I.; Febbraio, M.A.; Watt, M.J. Apoptosis in Skeletal Muscle Myotubes Is Induced by Ceramides and Is Positively Related to Insulin Resistance. Am. J. Physiol. Endocrinol. Metab. 2006, 291, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Itami, N.; Shirasuna, K.; Kuwayama, T.; Iwata, H. Palmitic Acid Induces Ceramide Accumulation, Mitochondrial Protein Hyperacetylation, and Mitochondrial Dysfunction in Porcine Oocytes. Biol. Reprod. 2018, 98, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Wehinger, S.; Ortiz, R.; Díaz, M.I.; Aguirre, A.; Valenzuela, M.; Llanos, P.; Mc Master, C.; Leyton, L.; Quest, A.F.G. Phosphorylation of caveolin-1 on tyrosine-14 induced by ROS enhances palmitate-induced death of beta-pancreatic cells. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 693–708. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chavez, J.A.; Knotts, T.A.; Wang, L.P.; Li, G.; Dobrowsky, R.T.; Florant, G.L.; Summers, S.A. A Role for Ceramide, but Not Diacylglycerol, in the Antagonism of Insulin Signal Transduction by Saturated Fatty Acids. J. Biol. Chem. 2003, 278, 10297–10303. [Google Scholar] [CrossRef]
- Schmitz-Peiffer, C.; Craig, D.L.; Biden, T.J. Ceramide Generation Is Sufficient to Account for the Inhibition of the Insulin-Stimulated PKB Pathway in C2C12 Skeletal Muscle Cells Pretreated with Palmitate. J. Biol. Chem. 1999, 274, 24202–24210. [Google Scholar] [CrossRef]
- Powell, D.J.; Turban, S.; Gray, A.; Hajduch, E.; Hundal, H.S. Intracellular Ceramide Synthesis and Protein Kinase Cζ Activation Play an Essential Role in Palmitate-Induced Insulin Resistance in Rat L6 Skeletal Muscle Cells. Biochem. J. 2004, 382, 619–629. [Google Scholar] [CrossRef]
- Sergi, D.; Morris, A.C.; Kahn, D.E.; McLean, F.H.; Hay, E.A.; Kubitz, P.; MacKenzie, A.; Martinoli, M.G.; Drew, J.E.; Williams, L.M. Palmitic Acid Triggers Inflammatory Responses in N42 Cultured Hypothalamic Cells Partially via Ceramide Synthesis but Not via TLR4. Nutr. Neurosci. 2020, 23, 321–334. [Google Scholar] [CrossRef]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic Acid: Physiological Role, Metabolism and Nutritional Implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef]
- Baldwin, A.C.; Green, C.D.; Olson, L.K.; Moxley, M.A.; Corbett, J.A. A Role for Aberrant Protein Palmitoylation in FFA-Induced ER Stress and β-Cell Death. Am. J. Physiol.-Endocrinol. Metab. 2012, 302, E1390–E1399. [Google Scholar] [CrossRef]
- Hsiao, Y.H.; Lin, C.I.; Liao, H.; Chen, Y.H.; Lin, S.H. Palmitic Acid-Induced Neuron Cell Cycle G2/M Arrest and Endoplasmic Reticular Stress through Protein Palmitoylation in SH-SY5Y Human Neuroblastoma Cells. Int. J. Mol. Sci. 2014, 15, 20876–20899. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J.; Krause, K.H. Reactive Oxygen Species: From Health to Disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef] [PubMed]
- Alnahdi, A.; John, A.; Raza, H. Augmentation of Glucotoxicity, Oxidative Stress, Apoptosis and Mitochondrial Dysfunction in Hepg2 Cells by Palmitic Acid. Nutrients 2019, 11, 1979. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Silvestri, S.; Orlando, P.; Mazibuko-Mbeje, S.E.; Johnson, R.; Marcheggiani, F.; Cirilli, I.; Muller, C.J.F.; Louw, J.; Chellan, N.; et al. Palmitate-Induced Toxicity Is Associated with Impaired Mitochondrial Respiration and Accelerated Oxidative Stress in Cultured Cardiomyocytes: The Critical Role of Coenzyme Q9/10. Toxicol. In Vitro 2020, 68, 104948. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S.V. Protection of Mitochondria Prevents High-Fat Diet–Induced Glomerulopathy and Proximal Tubular Injury. Kidney Int. 2016, 90, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, I.; Solís-Muñoz, P.; Fernández-Moreira, D.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. In Vitro Treatment of HepG2 Cells with Saturated Fatty Acids Reproduces Mitochondrial Dysfunction Found in Nonalcoholic Steatohepatitis. DMM Dis. Models Mech. 2015, 8, 183–191. [Google Scholar] [CrossRef]
- Samartsev, V.N.; Kozhina, O.V. Oxidative stress as regulatory factor for fatty-acid-induced uncoupling involving liver mitochondrial ADP/ATP and aspartate/glutamate antiporters of old rats. Biochem. Biokhim. 2008, 73, 783–790. [Google Scholar] [CrossRef]
- Rial, E.; Rodríguez-Sánchez, L.; Gallardo-Vara, E.; Zaragoza, P.; Moyano, E.; González-Barroso, M. Lipotoxicity, fatty acid uncoupling and mitochondrial carrier function. Biochim. Biophys. Acta-Bioenerg. 2010, 1797, 800–806. [Google Scholar] [CrossRef]
- Ma, S.; Yang, D.; Li, D.; Tan, Y.; Tang, B.; Yang, Y. Inhibition of uncoupling protein 2 with genipin exacerbates palmitate-induced hepatic steatosis. Lipids Health Dis. 2012, 11, 154. [Google Scholar] [CrossRef]
- Patanè, G.; Anello, M.; Piro, S.; Vigneri, R.; Purrello, F.; Rabuazzo, A. Role of ATP Production and Uncoupling Protein-2 in the Insulin Secretory Defect Induced by Chronic Exposure to High Glucose or Free Fatty Acids and Effects of Peroxisome Proliferator-Activated Receptor-γ Inhibition. Diabetes 2002, 51, 2749–2756. [Google Scholar] [CrossRef]
- Lou, J.; Wang, Y.; Wang, X.; Jiang, Y. Uncoupling Protein 2 Regulates Palmitic Acid-Induced Hepatoma Cell Autophagy. BioMed Res. Int. 2014, 2014, 810401. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Kowluru, A.; Kowluru, R.A. Lipotoxicity Augments Glucotoxicity-Induced Mitochondrial Damage in the Development of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2985–2992. [Google Scholar] [CrossRef]
- Rachek, L.I.; Musiyenko, S.I.; LeDoux, S.P.; Wilson, G.L. Palmitate Induced Mitochondrial Deoxyribonucleic Acid Damage and Apoptosis in L6 Rat Skeletal Muscle Cells. Endocrinology 2007, 148, 293–299. [Google Scholar] [CrossRef]
- Yuzefovych, L.V.; LeDoux, S.P.; Wilson, G.L.; Rachek, L.I. Mitochondrial DNA Damage via Augmented Oxidative Stress Regulates Endoplasmic Reticulum Stress and Autophagy: Crosstalk, Links and Signaling. PLoS ONE 2013, 8, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Sparagna, G.C.; Hickson-Bick, D.L.; Buja, L.M.; Mcmillin, J.B. A Metabolic Role for Mitochondria in Palmitate-Induced Cardiac Myocyte Apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Berk, M.; Mcintyre, T.M.; Gores, G.J.; Feldstein, A.E. The Lysosomal-Mitochondrial Axis in Free Fatty Acid–Induced Hepatic Lipotoxicity. Hepatology 2008, 47, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, L.; Gurley, E.; Studer, E.; Shang, J.; Wang, T.; Wang, C.; Yan, M.; Jiang, Z.; Hylemon, P.B.; et al. Prevention of Free Fatty Acid-Induced Hepatic Lipotoxicity by 18β-Glycyrrhetinic Acid through Lysosomal and Mitochondrial Pathways. Hepatology 2008, 47, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Lambertucci, R.H.; Hirabara, S.M.; Dos, L.; Silveira, R.; Levada-pires, A.C.; Curi, R.U.I.; Pithon-curi, T.C. Palmitate Increases Superoxide Production Through Mitochondrial Electron Transport Chain and NADPH Oxidase Activity in Skeletal Muscle Cells. J. Cell. Physiol. 2008, 796–804. [Google Scholar] [CrossRef]
- Chinen, I.; Shimabukuro, M.; Yamakawa, K.; Higa, N.; Matsuzaki, T.; Noguchi, K.; Ueda, S.; Sakanashi, M.; Takasu, N. Vascular Lipotoxicity: Endothelial Dysfunction via Fatty-Acid-Induced Reactive Oxygen Species Overproduction in Obese Zucker Diabetic Fatty Rats. Endocrinology 2007, 148, 160–165. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High Glucose Level and Free Fatty Acid Stimulate Protein Kinase C—Dependent Activation of NAD (P) H Oxidase in Cultured Vascular Cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef]
- Gao, D.; Nong, S.; Huang, X.; Lu, Y.; Zhao, H.; Lin, Y.; Man, Y.; Wang, S.; Yang, J.; Li, J. The Effects of Palmitate on Hepatic Insulin Resistance Are Mediated by NADPH Oxidase 3-Derived Reactive Oxygen Species through JNK and P38 MAPK Pathways. J. Biol. Chem. 2010, 285, 29965–29973. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Lu, J.; Yang, S. Oleic/Palmitate Induces Apoptosis in Human Articular Chondrocytes via Upregulation of Nox4 Expression and Ros Production. Ann. Clin. Lab. Sci. 2016, 46, 353–359. [Google Scholar]
- Joseph, L.C.; Barca, E.; Subramanyam, P.; Komrowski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of Napdh Oxidase 2 (Nox2) Prevents Oxidative Stress and Mitochondrial Abnormalities Caused by Saturated Fat in Cardiomyocytes. PLoS ONE 2016, 11, e0145750. [Google Scholar] [CrossRef]
- Yang, L.; Guan, G.; Lei, L.; Liu, J.; Cao, L.; Wang, X. Oxidative and Endoplasmic Reticulum Stresses Are Involved in Palmitic Acid-Induced H9c2 Cell Apoptosis. Biosci. Rep. 2019, 39, BSR20190225. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.J.; Zhao, L.M.; Pang, Z.D.; She, G.; Song, Z.; Cheng, X.; Du, X.J.; Deng, X.L. Oxidative Stress Induced by Palmitic Acid Modulates KCa2.3 Channels in Vascular Endothelium. Exp. Cell Res. 2019, 383, 111552. [Google Scholar] [CrossRef] [PubMed]
- Yuzefovych, L.; Wilson, G.; Rachek, L. Different Effects of Oleate vs. Palmitate on Mitochondrial Function, Apoptosis, and Insulin Signaling in L6 Skeletal Muscle Cells: Role of Oxidative Stress. Am. J. Physiol. Endocrinol. Metab. 2010, 299. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-González, S.; Marín-Royo, G.; Jurado-López, R.; Bartolomé, M.V.; Romero-Miranda, A.; Luaces, M.; Islas, F.; Nieto, M.L.; Martínez-Martínez, E.; Cachofeiro, V. The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells 2020, 9, 451. [Google Scholar] [CrossRef] [PubMed]
- Nyunt, T.; Britton, M.; Wanichthanarak, K.; Budamagunta, M. Mitochondrial Oxidative Stress-Induced Transcript Variants of ATF3 Mediate Lipotoxic Brain Microvascular Injury. Free Radic. Biol. Med. 2020, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Shi, W.; Gou, Y.; Zhou, X.; Tak, Y.A.; Zhou, Y.; Liu, Z. Fatty Acid-Mediated Intracellular Iron Translocation: A Synergistic Mechanism of Oxidative Injury. Free Radic. Biol. Med. 2005, 39, 1385–1398. [Google Scholar] [CrossRef]
- Hua, W.; Huang, H.Z.; Tan, L.T.; Wan, J.M.; Gui, H.B.; Zhao, L.; Ruan, X.Z.; Chen, X.M.; Du, X.G. CD36 Mediated Fatty Acid-Induced Podocyte Apoptosis via Oxidative Stress. PLoS ONE 2015, 10, e0127507. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Chen, Q.; Ma, K.; Ju, Y.; Ji, T.; Wang, Z.; Li, W.; Li, W. Astragaloside IV Inhibits Palmitate-Mediated Oxidative Stress and Fibrosis in Human Glomerular Mesangial Cells via Downregulation of CD36 Expression. Pharmacol. Rep. 2019, 71, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ren, C.; Zhang, M.; Zhong, Y. Perilipin 5 Reduces Oxidative Damage Associated With Lipotoxicity by Activating the PI3K/ERK-Mediated Nrf2-ARE Signaling Pathway in INS-1 Pancreatic β-Cells. Front. Endocrinol. 2020, 11, 166. [Google Scholar] [CrossRef]
- Xu, S.; Nam, S.M.; Kim, J.; Das, R.; Choi, S.; Nguyen, T.T.; Quan, X.; Choi, S.J.; Chung, C.H.; Lee, E.Y.; et al. Palmitate Induces ER Calcium Depletion and Apoptosis in Mouse Podocytes Subsequent to Mitochondrial Oxidative Stress. Cell Death Dis. 2015, 6, e1976. [Google Scholar] [CrossRef] [PubMed]
- Aung, H.H.; Altman, R.; Nyunt, T.; Kim, J.; Nuthikattu, S.; Voss, J.C.; Wilson, D.; Rutledge, J.C.; Villablanca, A.C. Lipotoxic Brain Microvascular Injury Is Mediated by Activating Transcription Factor 3-Dependent Inflammatory and Oxidative Stress Pathways. J. Lipid Res. 2016, 57, 955–968. [Google Scholar] [CrossRef]
- Li, P.; Li, L.; Zhang, C.; Cheng, X.; Zhang, Y.; Guo, Y.; Long, M.; Yang, S.; He, J. Palmitic Acid and β-Hydroxybutyrate Induce Inflammatory Responses in Bovine Endometrial Cells by Activating Oxidative Stress-Mediated NF-ΚB Signaling. Molecules 2019, 24, 2421. [Google Scholar] [CrossRef]
- Barazzoni, R.; Zanetti, M.; Cappellari, G.G.; Semolic, A.; Boschelle, M.; Codarin, E.; Pirulli, A.; Cattin, L.; Guarnieri, G. Fatty Acids Acutely Enhance Insulin-Induced Oxidative Stress and Cause Insulin Resistance by Increasing Mitochondrial Reactive Oxygen Species (ROS) Generation and Nuclear Factor-ΚB Inhibitor (IκB)-Nuclear Factor-ΚB (NFκB) Activation in Rat Muscle, in the Absence of Mitochondrial Dysfunction. Diabetologia 2012, 55, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.C.; Ugas, M.A.; Hagman, D.K.; Parazzoli, S.D.; Poitout, V. Evidence Against the Involvement of Oxidative Stress in Fatty Acid Inhibition of Insulin Secretion. Diabetes 2004, 53, 2610–2616. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. ER Stress and the Unfolded Protein Response. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. Protein Folding in the Endoplasmic Reticulum and the Unfolded Protein Response. Handb. Exp. Pharmacol. 2006, 172, 69–91. [Google Scholar] [CrossRef]
- Nivala, A.M.; Reese, L.; Frye, M.; Gentile, C.L.; Pagliassotti, M.J. Fatty Acid-Mediated Endoplasmic Reticulum Stressin Vivo: Differential Response to the Infusion of Soybean and Lard Oil in Rats. Metabolism 2014, 62, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Pardo, V.; González-Rodríguez, Á.; Muntané, J.; Kozma, S.C.; Valverde, Á.M. Role of Hepatocyte S6K1 in Palmitic Acid-Induced Endoplasmic Reticulum Stress, Lipotoxicity, Insulin Resistance and in Oleic Acid-Induced Protection. Food Chem. Toxicol. 2015, 80, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Sieber, J.; Lindenmeyer, M.T.; Kampe, K.; Campbell, K.N.; Cohen, C.D.; Hopfer, H.; Mundel, P.; Jehle, A.W. Regulation of Podocyte Survival and Endoplasmic Reticulum Stress by Fatty Acids. Am. J. Physiol. Ren. Physiol. 2010, 299. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-K.; Lee, J.; Jang, Y.; Kwon, Y.H. Involvement of Endoplasmic Reticulum Stress in Palmitate-Induced Apoptosis in HepG2 Cells. Toxicol. Res. 2008, 24, 129–135. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Park, M.J.; Han, H.J.; Kim, D. Il Lipotoxicity-Induced PRMT1 Exacerbates Mesangial Cell Apoptosis via Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2017, 18, 1421. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Li, X.; Wu, N.; Jia, P.; Liu, C.; Jia, D. Palmitate Induces Myocardial Lipotoxic Injury via the Endoplasmic Reticulum Stress-Mediated Apoptosis Pathway. Mol. Med. Rep. 2017, 16, 6934–6939. [Google Scholar] [CrossRef]
- Tan, L.; Yammani, R.R. Nupr1 Regulates Palmitate-Induced Apoptosis in Human Articular Chondrocytes. Biosci. Rep. 2019, 39, BSR20181473. [Google Scholar] [CrossRef]
- Martinez, S.C.; Tanabe, K.; Cras-me, C.; Abumrad, N.A.; Bernal-mizrachi, E.; Permutt, M.A. Inhibition of Foxo1 Protects Pancreatic Islet β-Cells Stress—Induced Apoptosis. Diabetes 2008, 57, 846–859. [Google Scholar] [CrossRef]
- Lim, J.C.; Lim, S.K.; Han, H.J.; Park, S.H. Cannabinoid Receptor 1 Mediates Palmitic Acid-Induced Apoptosis via Endoplasmic Reticulum Stress in Human Renal Proximal Tubular Cells. J. Cell. Physiol. 2010, 225, 654–663. [Google Scholar] [CrossRef]
- Lhoták, Š.; Sood, S.; Brimble, E.; Carlisle, R.E.; Colgan, S.M.; Mazzetti, A.; Dickhout, J.G.; Ingram, A.J.; Austin, R.C. ER Stress Contributes to Renal Proximal Tubule Injury by Increasing SREBP-2- Mediated Lipid Accumulation and Apoptotic Cell Death. Am. J. Physiol. Ren. Physiol. 2012, 303, 266–278. [Google Scholar] [CrossRef]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic Palmitate But Not Oleate Exposure Induces Endoplasmic Reticulum Stress, Which May Contribute to INS-1 Pancreatic β-Cell Apoptosis. Endocrinology 2015, 147, 3398–3407. [Google Scholar] [CrossRef]
- Haywood, J.; Yammani, R.R. Free Fatty Acid Palmitate Activates Unfolded Protein Response Pathway and Promotes Apoptosis in Meniscus Cells. Osteoarthr. Cartil. 2016, 24, 942–945. [Google Scholar] [CrossRef]
- Yamamoto, T.; Endo, J.; Kataoka, M.; Matsuhashi, T.; Katsumata, Y.; Shirakawa, K.; Isobe, S.; Moriyama, H.; Goto, S.; Shimanaka, Y.; et al. Palmitate Induces Cardiomyocyte Death via Inositol Requiring Enzyme-1 (IRE1)-Mediated Signaling Independent of X-Box Binding Protein 1 (XBP1). Biochem. Biophys. Res. Commun. 2020, 526, 122–127. [Google Scholar] [CrossRef]
- Robblee, M.M.; Kim, C.C.; Abate, J.P.; Valdearcos, M.; Sandlund, K.L.M.; Shenoy, M.; Volmer, R.; Iwawaki, T.; Koliwad, S.K. Saturated Fatty Acids Engage an IRE1α-Dependent Pathway to Activate the NLRP3 Inflammasome in Myeloid Cells. Cell Rep. 2016, 14, 2611–2623. [Google Scholar] [CrossRef]
- Wang, J.; Chen, Y.; Song, Q.; Griffiths, A.; Song, Z. MTORC1-IRE1α Pathway Activation Contributes to Palmitate-Elicited Triglyceride Secretion and Cell Death in Hepatocytes. Exp. Biol. Med. 2020, 245, 1268–1279. [Google Scholar] [CrossRef]
- Chen, Y.; Griffiths, A.; Wang, J.; Zhang, T.; Song, Q.; Song, Z. Inositol Requiring Enzyme 1 Alpha (IRE1α) Links Palmitate-Induced MTOR Activation and Lipotoxicity in Hepatocytes. Am. J. Physiol. Cell Physiol. 2020, 319, 1130–1140. [Google Scholar] [CrossRef]
- Deldicque, L.; Cani, P.D.; Philp, A.; Raymackers, J.M.; Meakin, P.J.; Ashford, M.L.J.; Delzenne, N.M.; Francaux, M.; Baar, K. The Unfolded Protein Response Is Activated in Skeletal Muscle by High-Fat Feeding: Potential Role in the Downregulation of Protein Synthesis. Am. J. Physiol. Endocrinol. Metab. 2010, 299, 695–705. [Google Scholar] [CrossRef]
- Guo, W.; Wong, S.; Xie, W.; Lei, T.; Luo, Z. Palmitate Modulates Intracellular Signaling, Induces Endoplasmic Reticulum Stress, and Causes Apoptosis in Mouse 3T3-L1 and Rat Primary Preadipocytes. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 576–586. [Google Scholar] [CrossRef]
- Kim, S.K.; Oh, E.; Yun, M.; Lee, S.B.; Chae, G.T. Palmitate Induces Cisternal ER Expansion via the Activation of XBP-1/CCTα-Mediated Phospholipid Accumulation in RAW 264.7 Cells. Lipids Health Dis. 2015, 14, 73. [Google Scholar] [CrossRef]
- Diakogiannaki, E.; Welters, H.J.; Morgan, N.G. Differential Regulation of the Endoplasmic Reticulum Stress Response in Pancreatic b -Cells Exposed to Long-Chain Saturated and Monounsaturated Fatty Acids. J. Endocrinol. 2007, 553–563. [Google Scholar] [CrossRef]
- Rennert, C.; Heil, T.; Schicht, G.; Stilkerich, A.; Seidemann, L.; Kegel-Hübner, V.; Seehofer, D.; Damm, G. Prolonged Lipid Accumulation in Cultured Primary Human Hepatocytes Rather Leads to Er Stress than Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 7097. [Google Scholar] [CrossRef]
- Kuo, T.F.; Tatsukawa, H.; Matsuura, T.; Nagatsuma, K.; Hirose, S.; Kojima, S. Free Fatty Acids Induce Transglutaminase 2-Dependent Apoptosis in Hepatocytes via ER Stress-Stimulated PERK Pathways. J. Cell. Physiol. 2012, 227, 1130–1137. [Google Scholar] [CrossRef]
- Cunha, D.A.; Igoillo-esteve, M.; Gurzov, E.N.; Germano, C.M.; Naamane, N.; Marhfour, I.; Fukaya, M.; Vanderwinden, J.; Gysemans, C.; Mathieu, C.; et al. Death Protein 5 and P53-Upregulated Modulator of Apoptosis Mediate the Endoplasmic Reticulum Stress—Mitochondrial Dialog Triggering Lipotoxic Rodent and Human b -Cell Apoptosis. Diabetes 2012, 61, 2763–2775. [Google Scholar] [CrossRef] [PubMed]
- Ladrière, L.; Igoillo-esteve, M.; Cunha, D.A.; Brion, J.; Bugliani, M.; Marchetti, P.; Eizirik, D.L.; Cnop, M. Enhanced Signaling Downstream of Ribonucleic Acid-Activated Protein Kinase-Like Endoplasmic Reticulum Kinase Potentiates Lipotoxic Endoplasmic Reticulum Stress in Human Islets. J. Clin. Endocrinol. Metab. 2010, 95, 1442–1449. [Google Scholar] [CrossRef]
- Anusornvongchai, T.; Nangaku, M.; Jao, T.M.; Wu, C.H.; Ishimoto, Y.; Maekawa, H.; Tanaka, T.; Shimizu, A.; Yamamoto, M.; Suzuki, N.; et al. Palmitate Deranges Erythropoietin Production via Transcription Factor ATF4 Activation of Unfolded Protein Response. Kidney Int. 2018, 94, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Dai, D.L.; Yao, L.; Yu, H.H.; Ning, B.; Zhang, Q.; Chen, J.; Cheng, W.H.; Shen, W.; Yang, Z.X. Saturated Fatty Acid Induction of Endoplasmic Reticulum Stress and Apoptosis in Human Liver Cells via the PERK/ATF4/CHOP Signaling Pathway. Mol. Cell. Biochem. 2012, 364, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Pirot, P.; Ortis, F.; Cnop, M.; Ma, Y.; Hendershot, L.M.; Eizirik, L. Transcriptional Regulation of the Endoplasmic Reticulum Stress Gene Chop in Pancreatic. Diabetes 2007, 56, 1069–1077. [Google Scholar] [CrossRef]
- Marwarha, G.; Claycomb, K.; Schommer, J.; Collins, D.; Sciences, H.; Forks, G.; Forks, G.; Nutrition, H.; Forks, G. Palmitate-Induced Endoplasmic Reticulum Stress and Subsequent C/EBPα Homologous Protein Activation Attenuates Leptin and Insulin-like Growth Factor 1 Expression in the Brain. Cell. Signal. 2017, 28, 1789–1805. [Google Scholar] [CrossRef]
- Sharmin, M.M.; Mizusawa, M.; Hayashi, S.; Arai, W.; Sakata, S.; Yonekura, S. Effects of Fatty Acids on Inducing Endoplasmic Reticulum Stress in Bovine Mammary Epithelial Cells. J. Dairy Sci. 2020, 103, 8643–8654. [Google Scholar] [CrossRef]
- Boslem, E.; Weir, J.M.; Macintosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of Endoplasmic Reticulum Lipid Rafts Contributes to Lipotoxicity in Pancreatic β-Cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef]
- Preston, A.M.; Gurisik, E.; Bartley, C.; Laybutt, D.R. Reduced Endoplasmic Reticulum (ER)-to-Golgi Protein Trafficking Contributes to ER Stress in Lipotoxic Mouse Beta Cells by Promoting Protein Overload. Diabetologia 2009, 52, 2369–2373. [Google Scholar] [CrossRef]
- Cunha, D.A.; Hekerman, P.; Ladrière, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and Execution of Lipotoxic ER Stress in Pancreatic β-Cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef]
- Gwiazda, K.S.; Yang, T.B.; Lin, Y.; Johnson, J.D. Effects of Palmitate on ER and Cytosolic Ca2+ Homeostasis in β-Cells. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E690–E701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xue, R.; Zhang, Z.; Yang, X.; Shi, H. Palmitic and Linoleic Acids Induce ER Stress and Apoptosis in Hepatoma Cells. Lipids Health Dis. 2012, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Egnatchik, R.A.; Leamy, A.K.; Jacobson, D.A.; Shiota, M.; Young, J.D. ER Calcium Release Promotes Mitochondrial Dysfunction and Hepatic Cell Lipotoxicity in Response to Palmitate Overload. Mol. Metab. 2014, 3, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.D.; Ly, D.D.; Nguyen, N.T.; Kim, J.H.; Yoo, H.; Chung, J.; Lee, M.S.; Cha, S.K.; Park, K.S. Mitochondrial Ca2+ Uptake Relieves Palmitate-Induced Cytosolic Ca2+ Overload in MIN6 Cells. Mol. Cells 2020, 43, 66–75. [Google Scholar] [CrossRef]
- Choi, S.E.; Kim, H.E.; Shin, H.C.; Jang, H.J.; Lee, K.W.; Kim, Y.; Kang, S.S.; Chun, J.; Kang, Y. Involvement of Ca2+-Mediated Apoptotic Signals in Palmitate-Induced MIN6N8a Beta Cell Death. Mol. Cell. Endocrinol. 2007, 272, 50–62. [Google Scholar] [CrossRef]
- Hara, T.; Mahadevan, J.; Kanekura, K.; Hara, M.; Lu, S.; Urano, F. Calcium Efflux from the Endoplasmic Reticulum Leads to β-Cell Death. Endocrinology 2014, 155, 758–768. [Google Scholar] [CrossRef]
- Kim, S.; Joe, Y.; Jeong, S.O.; Zheng, M.; Back, S.H.; Park, S.W.; Ryter, S.W.; Chung, H.T. Endoplasmic Reticulum Stress Is Sufficient for the Induction of IL-1β Production via Activation of the NF-ΚB and Inflammasome Pathways. Innate Immun. 2014, 20, 799–815. [Google Scholar] [CrossRef]
- Kim, J.A.; Jang, H.J.; Hwang, D.H. Toll-like Receptor 4-Induced Endoplasmic Reticulum Stress Contributes to Impairment of Vasodilator Action of Insulin. Am. J. Physiol. Endocrinol. Metab. 2015, 309, 767–776. [Google Scholar] [CrossRef]
- Yang, L.; Guan, G.; Lei, L.; Lv, Q.; Liu, S.; Zhan, X.; Jiang, Z.; Gu, X. Palmitic Acid Induces Human Osteoblast-like Saos-2 Cell Apoptosis via Endoplasmic Reticulum Stress and Autophagy. Cell Stress Chaperones 2018, 23, 1283–1294. [Google Scholar] [CrossRef]
- Katsoulieris, E.; Mabley, J.G.; Samai, M.; Sharpe, M.A.; Green, I.C.; Chatterjee, P.K. Lipotoxicity in Renal Proximal Tubular Cells: Relationship between Endoplasmic Reticulum Stress and Oxidative Stress Pathways. Free Radic. Biol. Med. 2010, 48, 1654–1662. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic Reticulum Stress Links Obesity, Insulin Action and Type 2 Diabetes. Metab. Clin. Exp. 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Wang, D.; Wei, Y.; Pagliassotti, M.J. Saturated Fatty Acids Promote Endoplasmic Reticulum Stress and Liver Injury in Rats with Hepatic Steatosis. Endocrinology 2006, 147, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, J.; Li, J.S.; Chen, H.; Huang, K.; Zheng, L. Accumulation of Endoplasmic Reticulum Stress and Lipogenesis in the Liver through Generational Effects of High Fat Diets. J. Hepatol. 2012, 56, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Ebersbach-Silva, P.; Poletto, A.C.; David-Silva, A.; Seraphim, P.M.; Anhê, G.F.; Passarelli, M.; Furuya, D.T.; Machado, U.F. Palmitate-Induced Slc2a4/GLUT4 Downregulation in L6 Muscle Cells: Evidence of Inflammatory and Endoplasmic Reticulum Stress Involvement. Lipids Health Dis. 2018, 17, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hu, G.; Lu, Y.; Zheng, J.; Chen, J.; Wang, X.; Zeng, Y. Palmitic Acid Aggravates Inflammation of Pancreatic Acinar Cells by Enhancing Unfolded Protein Response Induced CCAAT-Enhancer-Binding Protein β–CCAAT-Enhancer-Binding Protein α Activation. Int. J. Biochem. Cell Biol. 2016, 79, 181–193. [Google Scholar] [CrossRef]
- Gu, X.; Li, K.; Laybutt, D.R.; He, M.L.; Zhao, H.L.; Chan, J.C.N.; Xu, G. Bip Overexpression, but Not CHOP Inhibition, Attenuates Fatty-Acid-Induced Endoplasmic Reticulum Stress and Apoptosis in HepG2 Liver Cells. Life Sci. 2010, 87, 724–732. [Google Scholar] [CrossRef]
- Girona, J.; Rosales, R.; Saavedra, P.; Masana, L.; Vallve, J.-C. Palmitate Decreases Migration and Proliferation and Increases Oxidative Stress and Inflammation in Smooth Muscle Cells. Role of the Nrf2 Signaling Pathway. Am. J. Physiol. Cell Physiol. 2019, 316, 888–897. [Google Scholar] [CrossRef]
- Krogmann, A.; Staiger, K.; Haas, C.; Gommer, N.; Peter, A.; Heni, M.; Machicao, F.; Häring, H.; Staiger, H. Inflammatory Response of Human Coronary Artery Endothelial Cells to Saturated Long-Chain Fatty Acids. Microvasc. Res. 2011, 81, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Lipina, C.; Macrae, K.; Suhm, T.; Weigert, C.; Blachnio-Zabielska, A.; Baranowski, M.; Gorski, J.; Burgess, K.; Hundal, H.S. Mitochondrial Substrate Availability and Its Role in Lipid-Induced Insulin Resistance and Proinflammatory Signaling in Skeletal Muscle. Diabetes 2013, 62, 3426–3436. [Google Scholar] [CrossRef][Green Version]
- Mugabo, Y.; Mukaneza, Y.; Renier, G. Palmitate Induces C-Reactive Protein Expression in Human Aortic Endothelial Cells. Relevance to Fatty Acid-Induced Endothelial Dysfunction. Metab. Clin. Exp. 2011, 60, 640–648. [Google Scholar] [CrossRef]
- Weisberg, S.P.; Mccann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity Is Associated with Macrophage Accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Delproposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic Switching of Adipose Tissue Macrophages With Obesity Is Generated by Spatiotemporal Differences in Macrophage Subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [PubMed]
- Kitade, H.; Sawamoto, K.; Nagashimada, M.; Inoue, H.; Yamamoto, Y.; Sai, Y.; Takamura, T.; Yamamoto, H.; Miyamoto, K.; Ginsberg, H.N.; et al. CCR5 Plays a Critical Role in Obesity-Induced Adipose Tissue In Fl Ammation and Insulin Resistance by Regulating Both Macrophage Recruitment and M1/M2 Status. Diabetes 2012, 61, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Patsouris, D.; Cao, J.; Vial, G.; Bravard, A.; Lefai, E.; Durand, A.; Laugerette, F.; Debard, C.; Durand, C. Insulin Resistance Is Associated with MCP1-Mediated Macrophage Accumulation in Skeletal Muscle in Mice and Humans. PLoS ONE 2014, 9, e110653. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Xue, J.; Huang, Q.; Dai, Z.; Xu, Y. Involvement of Receptor-Interacting Protein 140 in Palmitate-Stimulated Macrophage Infiltration of Pancreatic Beta Cells. Exp. Ther. Med. 2017, 14, 483–494. [Google Scholar] [CrossRef][Green Version]
- Takahashi, K.; Yamaguchi, S.; Shimoyama, T.; Seki, H.; Miyokawa, K.; Katsuta, H.; Tanaka, T.; Yoshimoto, K.; Ohno, H.; Nagamatsu, S.; et al. JNK- and IκB-Dependent Pathways Regulate MCP-1 but Not Adiponectin Release from Artificially Hypertrophied 3T3-L1 Adipocytes Preloaded with Palmitate in Vitro. Am. J. Physiol. Endocrinol. Metab. 2008, 294, 898–909. [Google Scholar] [CrossRef]
- Dai, L.; Bhargava, P.; Stanya, K.J.; Alexander, R.K.; Liou, Y.; Jacobi, D.; Knudsen, N.H.; Hyde, A.; Gangl, M.R.; Liu, S.; et al. Macrophage Alternative Activation Confers Protection against Lipotoxicity-Induced Cell Death. Mol. Metab. 2017, 6, 1186–1197. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in Inflammation and Metabolic Disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Xiu, F.; Diao, L.; Qi, P.; Catapano, M.; Jeschke, M.G. Palmitate Differentially Regulates the Polarization of Differentiating and Differentiated Macrophages. Immunology 2016, 147, 82–96. [Google Scholar] [CrossRef]
- Samokhvalov, V.; Bilan, P.J.; Schertzer, J.D.; Antonescu, C.N.; Klip, A. Palmitate- And Lipopolysaccharide-Activated Macrophages Evoke Contrasting Insulin Responses in Muscle Cells. Am. J. Physiol. Endocrinol. Metab. 2009, 296. [Google Scholar] [CrossRef]
- Tang, T.; Sui, Y.; Lian, M.; Li, Z.; Hua, J. Pro-Inflammatory Activated Kupffer Cells by Lipids Induce Hepatic NKT Cells Deficiency through Activation-Induced Cell Death. PLoS ONE 2013, 8, e81949. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic Recruitment of Macrophages Promotes Nonalcoholic Steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Sepehri, Z.; Kiani, Z.; Nasiri, A.A.; Kohan, F. Toll-like Receptor 2 and Type 2 Diabetes. Cell. Mol. Biol. Lett. 2016, 21, 2. [Google Scholar] [CrossRef] [PubMed]
- Schilling, J.D.; Machkovech, H.M.; He, L.; Diwan, A.; Schaffer, J.E. TLR4 Activation Under Lipotoxic Conditions Leads to Synergistic Macrophage Cell Death Through a TRIF-Dependent Pathway. J. Immunol. 2014, 190, 1285–1296. [Google Scholar] [CrossRef]
- Lee, J.Y.; Zhao, L.; Hwang, D.H. Modulation of Pattern Recognition Receptor-Mediated Inflammation and Risk of Chronic Diseases by Dietary Fatty Acids. Nutr. Rev. 2010, 68, 38–61. [Google Scholar] [CrossRef]
- Lee, S.M.; Choi, S.E.; Lee, J.H.; Lee, J.J.; Jung, I.R.; Lee, S.J.; Lee, K.W.; Kang, Y. Involvement of the TLR4 (Toll-like Receptor4) Signaling Pathway in Palmitate-Induced INS-1 Beta Cell Death. Mol. Cell. Biochem. 2011, 354, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated Fatty Acids Activate TLR-Mediated Proinflammatory Signaling Pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef]
- Snodgrass, R.G.; Huang, S.; Choi, I.-W.; Rutledge, J.C.; Hwang, D.H. Inflammasome-Mediated Secretion of IL-1β in Human Monocytes through TLR2 Activation; Modulation by Dietary Fatty Acids. J. Immunol. 2013, 191, 4337–4347. [Google Scholar] [CrossRef]
- Cazanave, S.C.; Mott, J.L.; Elmi, N.A.; Bronk, S.F.; Werneburg, N.W.; Akazawa, Y.; Kahraman, A.; Garrison, S.P.; Zambetti, G.P.; Charlton, M.R.; et al. JNK1-Dependent PUMA Expression Contributes to Hepatocyte Lipoapoptosis. J. Biol. Chem. 2009, 284, 26591–26602. [Google Scholar] [CrossRef]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free Fatty Acids Induce JNK-Dependent Hepatocyte Lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef]
- Wang, Y.; Ausman, L.M.; Russell, R.M.; Greenberg, A.S.; Wang, X.-D. Increased Apoptosis in High-Fat Diet–Induced Nonalcoholic Steatohepatitis in Rats Is Associated with c-Jun NH2 -Terminal Kinase Activation and Elevated Proapoptotic Bax1,2. J. Nutr. 2008, 138, 1866–1871. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, Y.; Cazanave, S.; Mott, J.L.; Elmi, N.A.; Bronk, S.F.; Kohno, S.; Charlton, M.R.; Gores, G.J. Palmitoleate Attenuates Palmitate-Induced Bim and PUMA up- Regulation and Hepatocyte Lipoapoptosis. J. Hepatol. 2010, 52, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Litwak, S.A.; Wali, J.A.; Pappas, E.G.; Saadi, H.; Stanley, W.J.; Varanasi, L.C.; Kay, T.W.H.; Thomas, H.E.; Gurzov, E.N. Lipotoxic Stress Induces Pancreatic β-Cell Apoptosis through Modulation of Bcl-2 Proteins by the Ubiquitin-Proteasome System. J. Diabetes Res. 2015, 2015, 280615. [Google Scholar] [CrossRef]
- Qinan, W.; Xiaguang, G.; Xiaotian, L.; Wuquan, D.; Ling, Z.; Bing, C. Par-4/NF-B Mediates the Apoptosis of Islet β Cells Induced by Glucolipotoxicity. J. Diabetes Res. 2016, 2016, 4692478. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.K.; Cambiaghi, T.D.; Luchessi, A.D.; Hirabara, S.M.; Vinolo, M.A.R.; Newsholme, P.; Curi, R. Activation of Survival and Apoptotic Signaling Pathways in Lymphocytes Exposed to Palmitic Acid. J. Cell. Physiol. 2012, 227, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.A.; Favelyukis, S.; Nguyen, A.K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A Subpopulation of Macrophages Infiltrates Hypertrophic Adipose Tissue and Is Activated by Free Fatty Acids via Toll-like Receptors 2 and 4 and JNK-Dependent Pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef]
- Senn, J.J. Toll-like Receptor-2 Is Essential for the Development of Palmitate-Induced Insulin Resistance in Myotubes. J. Biol. Chem. 2006, 281, 26865–26875. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Wang, F.; Liu, S.; Zhao, S.; Ling, E.A.; Hao, A. Saturated Fatty Acids Activate Microglia via Toll-like Receptor 4/NF-ΚB Signalling. Br. J. Nutr. 2012, 107, 229–241. [Google Scholar] [CrossRef]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-Like Receptor 9 Promotes Steatohepatitis by Induction of Interleukin-1β in Mice. Gastroenterology 2010, 139, 323–340. [Google Scholar] [CrossRef]
- Jové, M.; Planavila, A.; Laguna, J.C.; Vázquez-Carrera, M. Palmitate-Induced Interleukin 6 Production Is Mediated by Protein Kinase C and Nuclear-Factor B Activation and Leads to Glucose Transporter 4 down-Regulation in Skeletal Muscle Cells. Endocrinology 2005, 146, 3087–3095. [Google Scholar] [CrossRef]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-Induced Insulin Resistance Mediated by the Proinflammatory Receptor TLR4 Requires Saturated Fatty Acid-Induced Ceramide Biosynthesis in Mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated Fatty Acids, but Not Unsaturated Fatty Acids, Induce the Expression of Cyclooxygenase-2 Mediated through Toll-like Receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Zheng, Z.; Chang, L.; Zhao, Y.; Tan, C.; Dandekar, A.; Zhang, Z.; Lin, Z.; Gui, M.; Li, X.; et al. Toll-like Receptor-Mediated IRE1 a Activation as a Therapeutic Target for Inflammatory Arthritis. EMBO J. 2013, 32, 2477–2490. [Google Scholar] [CrossRef]
- Martinon, F.; Chen, X.; Lee, A.-H.; Glimcher, L.H. Toll-like Receptor Activation of XBP1 Regulates Innate Immune Responses in Macrophages. Nat. Immunol. 2011, 11, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Joe, Y.; Kim, H.J.; Kim, Y.-S.; Jeong, S.O.; Pae, H.-O.; Ryter, S.W.; Surh, Y.-J.; Chung, H.T. Endoplasmic Reticulum Stress–Induced IRE1α Activation Mediates Cross-Talk of GSK-3β and XBP-1 To Regulate Inflammatory Cytokine Production. J. Immunol. 2015, 194, 4498–4506. [Google Scholar] [CrossRef]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty Acid and Endotoxin Activate Inflammasomes in Mouse Hepatocytes That Release Danger Signals to Stimulate Immune Cells. Hepatology 2011, 51, 133–144. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Pan, J.; Ou, Z.; Cai, C.; Li, P.; Gong, J.; Ruan, X.Z.; He, K. Fatty Acid Activates NLRP3 Inflammasomes in Mouse Kupffer Cells through Mitochondrial DNA Release. Cell. Immunol. 2018, 332, 111–120. [Google Scholar] [CrossRef]
- Weber, K.; Schilling, J.D. Lysosomes Integrate Metabolic-Inflammatory Cross-Talk in Primary Macrophage Inflammasome Activation. J. Biol. Chem. 2014, 289, 9158–9171. [Google Scholar] [CrossRef]
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High Fat Induces Acute and Chronic Inflammation in the Hypothalamus: Effect of High-Fat Diet, Palmitate and TNF-α on Appetite-Regulating NPY Neurons. Int. J. Obes. 2017, 41, 149–158. [Google Scholar] [CrossRef]
- Jové, M.; Planavila, A.; Sánchez, R.M.; Merlos, M.; Laguna, J.C.; Vázquez-Carrera, M. Palmitate Induces Tumor Necrosis Factor-α Expression in C2C12 Skeletal Muscle Cells by a Mechanism Involving Protein Kinase C and Nuclear Factor-ΚB Activation. Endocrinology 2006, 147, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free Fatty Acids Promote Hepatic Lipotoxicity by Stimulating TNF-α Expression via a Lysosomal Pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Barreyro, F.J.; Isomoto, H.; Bronk, S.F.; Gores, G.J. Free Fatty Acids Sensitise Hepatocytes to TRAIL Mediated Cytotoxicity. Gut 2007, 56, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Boller, S.; Debray, S.; Bouzakri, K.; Meier, D.T.; Prazak, R.; Kerr-conte, J.; Pattou, F.; Ehses, J.A.; Schuit, F.C.; Donath, M.Y. Free Fatty Acids Induce a Proinflammatory Response in Islets via the Abundantly Expressed Interleukin-1 Receptor I. Endocrinology 2009, 150, 5218–5229. [Google Scholar] [CrossRef]
- Joshi-Barve, S.; Barve, S.S.; Amancherla, K.; Gobejishvili, L.; Hill, D.; Cave, M.; Hote, P.; McClain, C.J. Palmitic Acid Induces Production of Proinflammatory Cytokine Interleukin-8 from Hepatocytes. Hepatology 2007, 46, 823–830. [Google Scholar] [CrossRef]
- Hoeks, J.; Mensink, M.; Hesselink, M.K.C.; Ekroos, K.; Schrauwen, P. Long- and Medium-Chain Fatty Acids Induce Insulin Resistance to a Similar Extent in Humans despite Marked Differences in Muscle Fat Accumulation. J. Clin. Endocrinol. Metab. 2012, 97, 208–216. [Google Scholar] [CrossRef]
- Delarue, J.; Magnan, C. Free Fatty Acids and Insulin Resistance. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 142–148. [Google Scholar] [CrossRef]
- Boden, G.; Cheung, P.; Peter Stein, T.; Kresge, K.; Mozzoli, M. FFA Cause Hepatic Insulin Resistance by Inhibiting Insulin Suppression of Glycogenolysis. Am. J. Physiol. Endocrinol. Metab. 2002, 283, 12–19. [Google Scholar] [CrossRef]
- Hirabara, S.M.; Curi, R.; Maechler, P. Saturated Fatty Acid-Induced Insulin Resistance Is Associated with Mitochondrial Dysfunction in Skeletal Muscle Cells. J. Cell. Physiol. 2010, 222, 187–194. [Google Scholar] [CrossRef]
- Shinjo, S.; Jiang, S.; Nameta, M.; Suzuki, T.; Kanai, M.; Nomura, Y.; Goda, N. Disruption of the Mitochondria-Associated ER Membrane (MAM) Plays a Central Role in Palmitic Acid–Induced Insulin Resistance. Exp. Cell Res. 2017, 359, 86–93. [Google Scholar] [CrossRef]
- Sebastián, D.; Herrero, L.; Serra, D.; Asins, G.; Hegardt, F.G. CPT I Overexpression Protects L6E9 Muscle Cells from Fatty Acid-Induced Insulin Resistance. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E677–E686. [Google Scholar] [CrossRef] [PubMed]
- Capel, F.; Cheraiti, N.; Acquaviva, C.; Hénique, C.; Bertrand-Michel, J.; Vianey-Saban, C.; Prip-Buus, C.; Morio, B. Oleate Dose-Dependently Regulates Palmitate Metabolism and Insulin Signaling in C2C12 Myotubes. Biochim. Et Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 2000–2010. [Google Scholar] [CrossRef] [PubMed]
- Coll, T.; Eyre, E.; Rodríguez-Calvo, R.; Palomer, X.; Sánchez, R.M.; Merlos, M.; Laguna, J.C.; Vázquez-Carrera, M. Oleate Reverses Palmitate-Induced Insulin Resistance and Inflammation in Skeletal Muscle Cells. J. Biol. Chem. 2008, 283, 11107–11116. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical Nodes in Signalling Pathways: Insights into Insulin Action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Anderwald, C.; Brunmair, B.; Stadlbauer, K.; Krebs, M.; Fürnsinn, C.; Roden, M. Effects of Free Fatty Acids on Carbohydrate Metabolism and Insulin Signalling in Perfused Rat Liver. Eur. J. Clin. Investig. 2007, 37, 774–782. [Google Scholar] [CrossRef]
- Lennon, R.; Pons, D.; Sabin, M.A.; Wei, C.; Shield, J.P.; Coward, R.J.; Tavaré, J.M.; Mathieson, P.W.; Saleem, M.A.; Welsh, G.I. Saturated Fatty Acids Induce Insulin Resistance in Human Podocytes: Implications for Diabetic Nephropathy. Nephrol. Dial. Transplant. 2009, 24, 3288–3296. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, S.; Nguyen, Y.H.; Skrobuk, P.; Koistinen, H.A. Palmitate and Oleate Exert Differential Effects on Insulin Signalling and Glucose Uptake in Human Skeletal Muscle Cells. Endocr. Connect. 2017, 6, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Mandarino, L.; Kashyap, S.; Wirfel, K.; Pratipanawatr, T.; Berria, R.; DeFronzo, R.A.; Cusi, K. Dose-Response Effect of Elevated Plasma Free Fatty Acid on Insulin Signaling. Diabetes 2005, 54, 1640–1648. [Google Scholar] [CrossRef]
- Frangioudakis, G.; Cooney, G.J. Acute Elevation of Circulating Fatty Acids Impairs Downstream Insulin Signalling in Rat Skeletal Muscle in Vivo Independent of Effects on Stress Signalling. J. Endocrinol. 2008, 197, 277–285. [Google Scholar] [CrossRef][Green Version]
- Ruddock, M.W.; Stein, A.; Landaker, E.; Park, J.; Cooksey, R.C.; McClain, D.; Patti, M.E. Saturated Fatty Acids Inhibit Hepatic Insulin Action by Modulating Insulin Receptor Expression and Post-Receptor Signalling. J. Biochem. 2008, 144, 599–607. [Google Scholar] [CrossRef]
- Lee, J.S.; Pinnamaneni, S.K.; Su, J.E.; In, H.C.; Jae, H.P.; Chang, K.K.; Sinclair, A.J.; Febbraio, M.A.; Watt, M.J. Saturated, but Not n-6 Polyunsaturated, Fatty Acids Induce Insulin Resistance: Role of Intramuscular Accumulation of Lipid Metabolites. J. Appl. Physiol. 2006, 100, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Badin, P.M.; Louche, K.; Mairal, A.; Liebisch, G.; Schmitz, G.; Rustan, A.C.; Smith, S.R.; Langin, D.; Moro, C. Altered Skeletal Muscle Lipase Expression and Activity Contribute to Insulin Resistance in Humans. Diabetes 2011, 60, 1734–1742. [Google Scholar] [CrossRef]
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of Protein Kinase Cε Prevents Hepatic Insulin Resistance in Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.L.; Coghlan, M.; Hundal, H.S. Modulating Serine Palmitoyl Transferase (SPT)Expression and Activity Unveils a Crucial Role in Lipid-Induced Insulin Resistance in Rat Skeletal Muscle Cells. Biochem. J. 2009, 417, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by Which Fatty Acids Inhibit Insulin Activation of Insulin Receptor Substrate-1 (IRS-1)-Associated Phosphatidylinositol 3-Kinase Activity in Muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.C.; Kemp, C.J.; Elias, C.F.; Abplanalp, W.; Herman, J.P.; Migrenne, S.; Lefevre, A.L.; Cruciani-Guglielmacci, C.; Magnan, C.; Yu, F.; et al. Palmitic Acid Mediates Hypothalamic Insulin Resistance by Altering PKC-θ Subcellular Localization in Rodents. J. Clin. Investig. 2009, 119, 2577–2589. [Google Scholar] [CrossRef]
- Kewalramani, G.; Fink, L.N.; Asadi, F.; Klip, A. Palmitate-Activated Macrophages Confer Insulin Resistance to Muscle Cells by a Mechanism Involving Protein Kinase C θ and ε. PLoS ONE 2011, 6, e26947. [Google Scholar] [CrossRef]
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.D.; Bain, J.R.; Newgard, C.B.; Farese, R.V.; et al. Dissociation of Hepatic Steatosis and Insulin Resistance in Mice Overexpressing DGAT in the Liver. Cell Metab. 2007, 6, 69–78. [Google Scholar] [CrossRef]
- Pardo, V.; González-Rodríguez, Á.; Guijas, C.; Balsinde, J.; Valverde, Á.M. Opposite Cross-Talk by Oleate and Palmitate on Insulin Signaling in Hepatocytes through Macrophage Activation. J. Biol. Chem. 2015, 290, 11663–11677. [Google Scholar] [CrossRef]
- Ragheb, R.; Shanab, G.M.L.; Medhat, A.M.; Seoudi, D.M.; Adeli, K.; Fantus, I.G. Free Fatty Acid-Induced Muscle Insulin Resistance and Glucose Uptake Dysfunction: Evidence for PKC Activation and Oxidative Stress-Activated Signaling Pathways. Biochem. Biophys. Res. Commun. 2009, 389, 211–216. [Google Scholar] [CrossRef]
- Nakamura, S.; Takamura, T.; Matsuzawa-nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate Induces Insulin Resistance in H4IIEC3 Hepatocytes through Reactive Oxygen Species Produced. J. Biol. Chem. 2009, 284, 14809–14818. [Google Scholar] [CrossRef]
- De Figueiredo, A.S.P.; Salmon, A.B.; Bruno, F.; Jimenez, F.; Martinez, H.G.; Halade, G.V.; Ahuja, S.S.; Clark, R.A.; DeFronzo, R.A.; Abboud, H.E.; et al. Nox2 Mediates Skeletal Muscle Insulin Resistance Induced by a High Fat Diet. J. Biol. Chem. 2015, 290, 13427–13439. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Han, T.T.; Liu, Y.; Zheng, S.; Zhang, Y.; Liu, W.; Hu, Y.M. Insulin Resistance Caused by Lipotoxicity Is Related to Oxidative Stress and Endoplasmic Reticulum Stress in LPL Gene Knockout Heterozygous Mice. Atherosclerosis 2015, 239, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Hage Hassan, R.; Hainault, I.; Vilquin, J.T.; Sama, C.; Lasnier, F.; Ferré, P.; Foufelle, F.; Hajduch, E. Endoplasmic Reticulum Stress Does Not Mediate Palmitate-Induced Insulin Resistance in Mouse and Human Muscle Cells. Diabetologia 2012, 55, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.J.; Kim, H.S.; Hwang, D.H.; Quon, M.J.; Kim, J.A. Toll-like Receptor 2 Mediates High-Fat Diet-Induced Impairment of Vasodilator Actions of Insulin. Am. J. Physiol. Endocrinol. Metab. 2013, 304, 1077–1088. [Google Scholar] [CrossRef]
- Davis, J.E.; Gabler, N.K.; Walker-Daniels, J.; Spurlock, M.E. The C-Jun N-Terminal Kinase Mediates the Induction of Oxidative Stress and Insulin Resistance by Palmitate and Toll-like Receptor 2 and 4 Ligands in 3T3-L1 Adipocytes. Horm. Metab. Res. 2009, 41, 523–530. [Google Scholar] [CrossRef]
- Carpentier, A.; Mittelman, S.D.; Lamarche, B.; Bergman, R.N.; Giacca, A.; Lewis, G.F. Acute enhancement of insulin secretion by FFA in humans is lost with prolonged FFA elevation. Am. J. Physiol. Cell Physiol. 2019, 276, 1055–1066. [Google Scholar] [CrossRef]
- Bollheimer, L.C.; Kemptner, D.M.; Kagerbauer, S.M.; Kestler, T.M.; Wrede, C.E.; Buettner, R. Intracellular Depletion of Insulin: A Comparative Study with Palmitate, Oleate and Elaidate in INS-1 Cells. Eur. J. Endocrinol. 2003, 148, 481–486. [Google Scholar] [CrossRef]
- Cousin, S.P.; Hu, S.R.; Myers, M.G.; Rhodes, C.J. Free Fatty Acid-Induced Inhibition of Glucose and Acid Synthesis in the Pancreatic β-Cell Line INS-1. Endocrinology 2015, 142, 229–240. [Google Scholar] [CrossRef]
- Jacqueminet, S.; Briaud, I.; Rouault, C.; Reach, G.; Poitout, V. Inhibition of Insulin Gene Expression by Long-Term Exposure of Pancreatic /3 Cells to Palmitate Is Dependent on the Presence of a Stimulatory Glucose Concentration. Metabolism 2000, 49, 532–536. [Google Scholar] [CrossRef]
- Briaud, I.; Harmon, J.S.; Kelpe, C.L.; Segu, V.B.G.; Poitout, V. Lipotoxicity of the Pancreatic β-Cell Is Associated with Glucose-Dependent Esterification of Fatty Acids into Neutral Lipids. Diabetes 2001, 50, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Naugler, W.; Galimi, F.; Lee, M.; Karin, M. Saturated Fatty Acids Inhibit Induction of Insulin Gene Transcription by JNK-Mediated Phosphorylation of Insulin-Receptor Substrates. Proc. Natl. Acad. Sci. USA 2006, 103, 16454–16459. [Google Scholar] [CrossRef] [PubMed]
- Kelpe, C.L.; Moore, P.C.; Parazzoli, S.D.; Wicksteed, B.; Rhodes, C.J.; Poitout, V. Palmitate Inhibition of Insulin Gene Expression Is Mediated at the Transcriptional Level via Ceramide Synthesis. J. Biol. Chem. 2003, 278, 30015–30021. [Google Scholar] [CrossRef] [PubMed]
- Hagman, D.K.; Hays, L.B.; Parazzoli, S.D.; Poitout, V. Palmitate Inhibits Insulin Gene Expression by Altering Pdx-1 Nuclear Localization and Reducing Mafa Expression in Isolated Rat Islets of Langerhans. J. Biol. Chem. 2006, 280, 32413–32418. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Bender, E.; Schamberger, J.; Eitner, F. Pharmacology of Free Fatty Acid Receptors and Their Allosteric Modulators. Int. J. Mol. Sci. 2021, 22, 1763. [Google Scholar] [CrossRef]
- Marafie, S.K.; Al-Shawaf, E.M.; Abubaker, J.; Arefanian, H. Palmitic Acid-Induced Lipotoxicity Promotes a Novel Interplay between Akt-MTOR, IRS-1, and FFAR1 Signaling in Pancreatic β-Cells. Biol. Res. 2019, 52, 44. [Google Scholar] [CrossRef]
- Kristinsson, H.; Smith, D.M.; Bergsten, P.; Sargsyan, E. FFAR1 Is Involved in Both the Acute and Chronic Effects of Palmitate on Insulin Secretion. Endocrinology 2013, 154, 4078–4088. [Google Scholar] [CrossRef]
- Graciano, M.F.; Valle, M.M.; Curi, R.; Carpinelli, A.R.; Graciano, M.F.; Valle, M.M.; Curi, R.; Carpinelli, A.R. Evidence for the Involvement of GPR40 and NADPH Oxidase in Palmitic Acid-Induced Superoxide Production and Insulin Secretion. Islets 2013, 5, 139–148. [Google Scholar] [CrossRef]
- Shen, X.; Yang, L.; Yan, S.; Wei, W.; Liang, L.; Zheng, H.; Cai, X. The Effect of FFAR1 on Pioglitazone-Mediated Attenuation of Palmitic Acid-Induced Oxidative Stress and Apoptosis in ΒTC6 Cells. Metabolism 2014, 63, 335–351. [Google Scholar] [CrossRef]
- Natalicchio, A.; Labarbuta, R.; Tortosa, F.; Biondi, G.; Marrano, N.; Peschechera, A.; Carchia, E.; Orlando, M.R.; Leonardini, A.; Cignarelli, A.; et al. Exendin-4 Protects Pancreatic Beta Cells from Palmitate-Induced Apoptosis by Interfering with GPR40 and the MKK4 / 7 Stress Kinase Signalling Pathway. Diabetologia 2013, 56, 2456–2466. [Google Scholar] [CrossRef]
- Somesh, B.P.; Verma, M.K.; Sadasivuni, M.K.; Mammen-Oommen, A. Chronic Glucolipotoxic Conditions in Pancreatic Islets Impair Insulin Secretion Due to Dysregulated Calcium Dynamics, Glucose Responsiveness and Mitochondrial Activity. BMC Cell Biol. 2013, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.; Affourtit, C. Novel Insights into Pancreatic β-Cell Glucolipotoxicity from Real-Time Functional Analysis of Mitochondrial Energy Metabolism in INS-1E Insulinoma Cells. Biochem. J. 2013, 426, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Lee, S.; Lee, Y.; Li, L.; Lee, S.; Lee, J.; Kim, Y.; Jun, H.; Lee, K.; Kang, Y. Protective Role of Autophagy in Palmitate-Induced INS-1 β-Cell Death. Endocrinology 2009, 150, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Wang, Y.; Gu, L.; Fan, N.; Ma, Y.; Peng, Y. Palmitate Induces Endoplasmic Reticulum Stress and Autophagy in Mature Adipocytes: Implications for Apoptosis and Inflammation. Int. J. Mol. Med. 2015, 35, 932–940. [Google Scholar] [CrossRef]
- Reginato, A.; Siqueira, B.P.; Miyamoto, J.É.; Portovedo, M.; Costa, S.D.; de Fante, T.; Rodrigues, H.G.; Ignácio-Souza, L.M.; Torsoni, M.A.; Torsoni, A.S.; et al. Acute Effects of Fatty Acids on Autophagy in NPY Neurons. J. Neuroendocrinol. 2020, 32, e12900. [Google Scholar] [CrossRef]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy Is Important in Islet Homeostasis and Compensatory Increase of Beta Cell Mass in Response to High-Fat Diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef]
- Martino, L.; Masini, M.; Novelli, M.; Beffy, P.; Bugliani, M.; Marselli, L.; Masiello, P.; Marchetti, P.; Tata, V. De Palmitate Activates Autophagy in INS-1E b -Cells and in Isolated Rat and Human Pancreatic Islets. PLoS ONE 2012, 7, e36188. [Google Scholar] [CrossRef]
- Komiya, K.; Uchida, T.; Ueno, T.; Koike, M.; Abe, H.; Hirose, T.; Kawamori, R.; Uchiyama, Y.; Kominami, E.; Fujitani, Y.; et al. Biochemical and Biophysical Research Communications Free Fatty Acids Stimulate Autophagy in Pancreatic b -Cells via JNK Pathway. Biochem. Biophys. Res. Commun. 2010, 401, 561–567. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, L.; Wang, B.; Zou, X.; Mu, Y.; Lu, J. Palmitate Induces Autophagy in Pancreatic β -Cells via Endoplasmic Reticulum Stress and Its Downstream JNK Pathway. Int. J. Mol. Med. 2013, 32, 1401–1406. [Google Scholar] [CrossRef]
- Russo, S.B.; Baicu, C.F.; Van Laer, A.; Geng, T.; Kasiganesan, H.; Zile, M.R.; Cowart, L.A. Ceramide Synthase 5 Mediates Lipid-Induced Autophagy and Hypertrophy in Cardiomyocytes. J. Clin. Investig. 2012, 122, 3919–3930. [Google Scholar] [CrossRef]
- Tan, S.H.; Shui, G.; Zhou, J.; Li, J.J.E.; Bay, B.H.; Wenk, M.R.; Shen, H.M. Induction of Autophagy by Palmitic Acid via Protein Kinase C-Mediated Signaling Pathway Independent of MTOR (Mammalian Target of Rapamycin). J. Biol. Chem. 2012, 287, 14364–14376. [Google Scholar] [CrossRef]
- Quan, W.; Hur, K.Y.; Lim, Y.; Oh, S.H.; Lee, J.; Kim, K.H.; Kim, G.H. Autophagy Deficiency in Beta Cells Leads to Compromised Unfolded Protein Response and Progression from Obesity to Diabetes in Mice. Diabetologia 2012, 55, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Liu, H.; Xiang, H.; Zhou, J.; Zeng, Z.; Chen, R.; Zhao, S.; Xiao, J.; Shu, Z.; Chen, S.; et al. Palmitic Acid-induced Autophagy Increases Reactive Oxygen Species via the Ca2+/PKCα/NOX4 Pathway and Impairs Endothelial Function in Human Umbilical Vein Endothelial Cells. Exp. Ther. Med. 2019, 2425–2432. [Google Scholar] [CrossRef] [PubMed]
- Mir, S.U.R.; George, N.M.; Zahoor, L.; Harms, R.; Guinn, Z.; Sarvetnick, N.E. Inhibition of Autophagic Turnover in β-Cells by Fatty Acids and Glucose Leads to Apoptotic Cell Death. J. Biol. Chem. 2015, 290, 6071–6085. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Rodriguez, A.; Acaz-Fonseca, E.; Boya, P.; Arevalo, M.A.; Garcia-Segura, L.M. Lipotoxic Effects of Palmitic Acid on Astrocytes Are Associated with Autophagy Impairment. Mol. Neurobiol. 2019, 56, 1665–1680. [Google Scholar] [CrossRef]
- Sadeghi, A.; Shabani, M.; Alizadeh, S.; Meshkani, R. Interplay between Oxidative Stress and Autophagy Function and Its Role in Inflammatory Cytokine Expression Induced by Palmitate in Skeletal Muscle Cells. Cytokine 2020, 125, 154835. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Ni, H.M.; Manley, S.; Bockus, A.; Kassel, K.M.; Luyendyk, J.P.; Copple, B.L.; Ding, W.X. Differential Roles of Unsaturated and Saturated Fatty Acids on Autophagy and Apoptosis in Hepatocytes. J. Pharmacol. Exp. Ther. 2011, 339, 487–498. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Molecular Mechanism of Oxidative Stress | Organism | Cell Culture | Organ/Cell Type | Reference |
|---|---|---|---|---|
| Increased general ROS production | human | isolated chondrocytes | cartilage | [80] |
| cell line Hep2G | liver | [81] | ||
| rat | isolated myocytes | muscle | [77] | |
| cell line H9c2 | heart | [82] | ||
| cell line INS-1 | pancreas | [91] | ||
| mouse | isolated cardiomyocytes | heart | [82] | |
| isolated podocytes | kidney | [89] | ||
| Increased mitochondrial ROS production | rat | cell line L6 | muscle | [85] |
| cell line H9c2 | heart | [63] | ||
| mouse | isolated cardiomyocytes | heart | [82] | |
| isolated podocytes | kidney | [92] | ||
| NOX activation | human | cell line Hep2G | liver | [81] |
| isolated chondrocytes | cartilage | [80] | ||
| rat | isolated myocytes | muscle | [77] | |
| cell line H9c2 | heart | [83] | ||
| mouse | isolated cardiomyocytes | heart | [82] | |
| Reduction in ETC | rat | isolated myocytes | muscle | [77] |
| mouse | isolated cardiomyocytes | heart | [82] | |
| Reduction in MtMP * | human | HUVEC | endothelium | [84] |
| rat | cell line H9c2 | heart | [82] | |
| mouse | isolated podocytes | kidney | [92] | |
| Reduced ATP generation | rat | cell line H9c2 | heart | [63] |
| cell line L6 | muscle | [85] | ||
| mouse | isolated podocytes | kidney | [92] | |
| Iron-mediated toxicity | human | HUVEC | endothelium | [88] |
| increase in mitochondrial Ca2+ | rat | cell line H9c2 | heart | [82] |
| Molecular Mechanism of ER Stress | Organism | Cell Culture | Organ/Cell Type | Reference |
|---|---|---|---|---|
| Activation/phosphorylation of PERK | human | isolated hepatocytes | liver | [119] |
| cell line L02 | liver | [124] | ||
| cell line Hep2G | liver | [124] | ||
| cell line SH-SY5Y | neuroblastoma | [125] | ||
| rat | isolated adipocytes | adipose tissue | [99] | |
| isolated hepatocytes | liver | [99] | ||
| cell line INS-1 | pancreas | [110,130] | ||
| isolated neonatal rat cardiomyocytes | heart | [111] | ||
| mouse | cell line RAW 264.7 | macrophage | [116,117] | |
| cell line N2a | neuroblastoma | [125] | ||
| cell line MIN-6 | pancreas | [131,134] | ||
| Activation/phosphorylation of eIF2α | human | isolated hepatocytes | liver | [100] |
| cell line Hep2G | liver | [146] | ||
| rat | isolated adipocytes | adipose tissue | [99] | |
| isolated hepatocytes | liver | [99] | ||
| cell line INS-1 | pancreas | [130] | ||
| cell line BRIN-BD11 | pancreas | [118] | ||
| mouse | cell line MIN-6 | pancreas | [134] | |
| XBP1 splicing/XBP1s increased expression or activation | human | cell line Hep2G | liver | [123,124] |
| isolated β-cells | pancreas | [131] | ||
| cell line SH-SY5Y | neuroblastoma | [125] | ||
| rat | isolated adipocytes | adipose tissue | [99] | |
| isolated hepatocytes | liver | [99] | ||
| cell line INS-1 | pancreas | [110] | ||
| mouse | isolated podocytes | kidney | [101] | |
| cell line AML12 | liver | [113] | ||
| cell line 3T3-L1 | preadipocytes | [116] | ||
| cell line RAW 264.7 | macrophages | [116,117] | ||
| cell line C2C12 | muscle | [115] | ||
| cell line N2a | neuroblastoma | [125] | ||
| cell line MIN-6 | β-cell | [131] | ||
| CHOP expression | human | isolated hepatocytes | liver | [100] |
| cell line Hep2G | liver | [123,124] | ||
| cell line L02 | liver | [124] | ||
| cell line SH-SY5Y | neuroblastoma | [125] | ||
| isolated β-cells | pancreas | [131] | ||
| rat | cell line INS-1 | pancreas | [110,130] | |
| cell line BRIN-BD11 | pancreas | [118] | ||
| isolated cardiomyocytes | heart | [111] | ||
| mouse | cell line 3T3-L1 | preadipocytes | [116] | |
| cell line RAW 264.7 | macrophages | [116,117] | ||
| cell line C2C12 | muscle | [115] | ||
| cell line N2a | neuroblastoma | [125] | ||
| cell line MIN-6 | pancreas | [131,134] | ||
| Activation/phosphorylation of IRE1 | human | cell line Hep2G | liver | [146] |
| isolated hepatocytes | liver | [119] | ||
| cell line SH-SY5Y | neuroblastoma | [125] | ||
| rat | isolated cardiomyocytes | heart | [111] | |
| cell line INS-1 | pancreas | [130] | ||
| mouse | cell line AML12 | liver | [113] | |
| cell line C2C12 | muscle | [115] | ||
| cell line N2a | neuroblastoma | [125] | ||
| Activation/phosphorylation of ATF4 | human | cell line Hep2G | liver | [123,124] |
| cell line L02 | liver | [124] | ||
| cell line SH-SY5Y | neuroblastoma | [125] | ||
| rat | cell line INS-1 | pancreas | [110] | |
| cell line BRIN-BD11 | pancreas | [118] | ||
| mouse | cell line C2C12 | muscle | [115] | |
| cell line N2a | neuroblastoma | [125] | ||
| Activation/phosphorylation of ATF3 | human | cell line SH-SY5Y | neuroblastoma | [125] |
| rat | cell line INS-1 | pancreas | [130] | |
| mouse | cell line N2a | neuroblastoma | [125] | |
| mTORC1 activation | mouse | cell line AML12 | liver | [113] |
| Perturbation of protein trafficking | mouse | cell line MIN-6 | pancreas | [128,129] |
| ER Ca2+ depletion | human | isolated β-cells | pancreas | [131] |
| rat | cell line INS-1 | pancreas | [130] | |
| cell line H4IIEC3 | liver | [133] | ||
| mouse | cell line MIN-6 | pancreas | [131,134] |
| Molecular Mechanism of Inflammation | Organism | Cell Culture | Organ/Cell Type | Reference |
|---|---|---|---|---|
| Increased IL-6 | human | HCASMC | smooth muscle | [147] |
| hCAEC | endothelium | [148] | ||
| rat | cell line L6 | muscle | [149] | |
| mouse | cell line RAW 264.7 | macrophage | [155] | |
| cell line BV-2 | microglia | [178] | ||
| cell line C2C12 | muscle | [177] | ||
| Increased IL-1beta | human | HCASMC | smooth muscle | [147] |
| cell line THP-1 | monocyte | [168] | ||
| mouse | cell line RAW 264.7 | macrophage | [176] | |
| cell line BV-2 | microglia | [178] | ||
| BMDC | dendritic cells | [112] | ||
| isolated Kupffer Cells | macrophages | [188] | ||
| Increased IL-8 | human | HCASMC | smooth muscle | [147] |
| hCAEC | endothelium | [148] | ||
| mouse | cell line RAW 264.7 | macrophage | [167] | |
| Increased TNF-alfa | human | HCASMC | smooth muscle | [147] |
| cell line Hep2G | liver | [195] | ||
| mouse | cell line RAW 264.7 | macrophage | [155] | |
| cell line 3T3-L1 | preadipocyte | [156] | ||
| cell line BV-2 | microglia | [178] | ||
| cell line C2C12 | muscle | [191] | ||
| Activation of NF-kB | human | primary HAEC | endothelium | [150] |
| cell line THP-1 | monocyte | [168] | ||
| cell line Hep2G | liver | [192] | ||
| rat | cell line L6 | muscle | [149] | |
| cell line INS-1 | pancreas | [166] | ||
| mouse | cell line C2C12 | muscle | [177] | |
| Activation of JNK | human | hCAEC | endothelium | [148] |
| cell line THP-1 | monocyte | [168] | ||
| rat | cell line INS-1 | pancreas | [166] | |
| mouse | cell line RAW 264.7 | macrophage | [176] | |
| cell line C2C12 | muscle | [177] | ||
| Involvement of TLRs | human | cell line THP-1 | monocyte | [168] |
| rat | cell line INS-1 | pancreas | [166] | |
| mouse | cell line RAW 264.7 | macrophage | [176] | |
| cell line BV-2 | microglia | [178] | ||
| cell line C2C12 | muscle | [177] | ||
| Inflammasome activation | mouse | BMDC | dendritic cells | [112] |
| cell line Hepa1–6 | hepatoma | [186] | ||
| cell line RAW 264.7 | macrophage | [186] | ||
| isolated Kupffer Cells | macrophage | [188] | ||
| Induced COX-2 | mouse | cell line RAW 264.7 | macrophage | [182] |
| Increased MCP-1 | human | HCASMC | smooth muscle | [147] |
| hCAEC | endothelium | [148] | ||
| mouse | cell line RAW 264.7 | macrophage | [167] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipke, K.; Kubis-Kubiak, A.; Piwowar, A. Molecular Mechanism of Lipotoxicity as an Interesting Aspect in the Development of Pathological States—Current View of Knowledge. Cells 2022, 11, 844. https://doi.org/10.3390/cells11050844
Lipke K, Kubis-Kubiak A, Piwowar A. Molecular Mechanism of Lipotoxicity as an Interesting Aspect in the Development of Pathological States—Current View of Knowledge. Cells. 2022; 11(5):844. https://doi.org/10.3390/cells11050844
Chicago/Turabian StyleLipke, Katarzyna, Adriana Kubis-Kubiak, and Agnieszka Piwowar. 2022. "Molecular Mechanism of Lipotoxicity as an Interesting Aspect in the Development of Pathological States—Current View of Knowledge" Cells 11, no. 5: 844. https://doi.org/10.3390/cells11050844
APA StyleLipke, K., Kubis-Kubiak, A., & Piwowar, A. (2022). Molecular Mechanism of Lipotoxicity as an Interesting Aspect in the Development of Pathological States—Current View of Knowledge. Cells, 11(5), 844. https://doi.org/10.3390/cells11050844