
Impaired p53-Mediated DNA Damage Response Contributes to Microcephaly in Nijmegen Breakage Syndrome Patient-Derived Cerebral Organoids

 ,
,  , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. iPSCs Derivation and Culture Methods
2.2. Generation of Cerebral Organoids
2.3. Single Cell Dissociation and 2D Neuronal Culture
2.4. Organoid Sectioning and Immunostainings
2.5. Quantitative Assessment of Cerebral Organoids and Image Analysis of Histological Sections
2.6. Reverse Transcriptase PCR (RT-PCR)
2.7. Microarray Analysis
2.8. Western Blot
2.9. Flow Cytometry
2.10. Karyotyping and Chromosomal Microarray
2.11. Analysis of Mutational Status of TP53: Library Preparation and Massive Parallel Sequencing
2.12. Bisulfite Genomic Sequencing
2.13. Statistical Analysis
3. Results
3.1. iPSCs-Derived from NBS Patients Exhibit Chromosome Instability
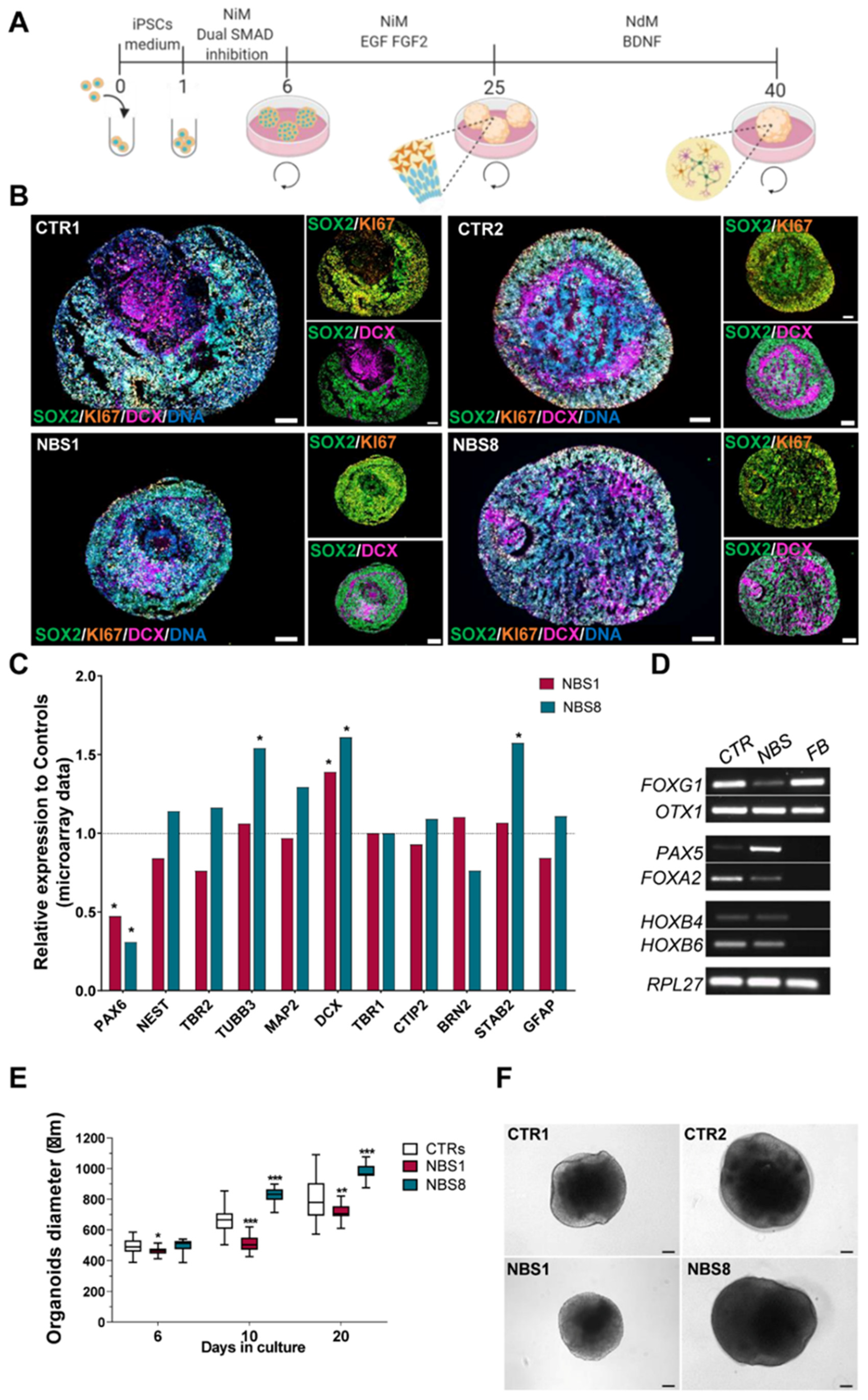
3.2. NBS-iPSCs Efficiently Differentiate into Cerebral Organoids and Recapitulate the Microcephaly Phenotype
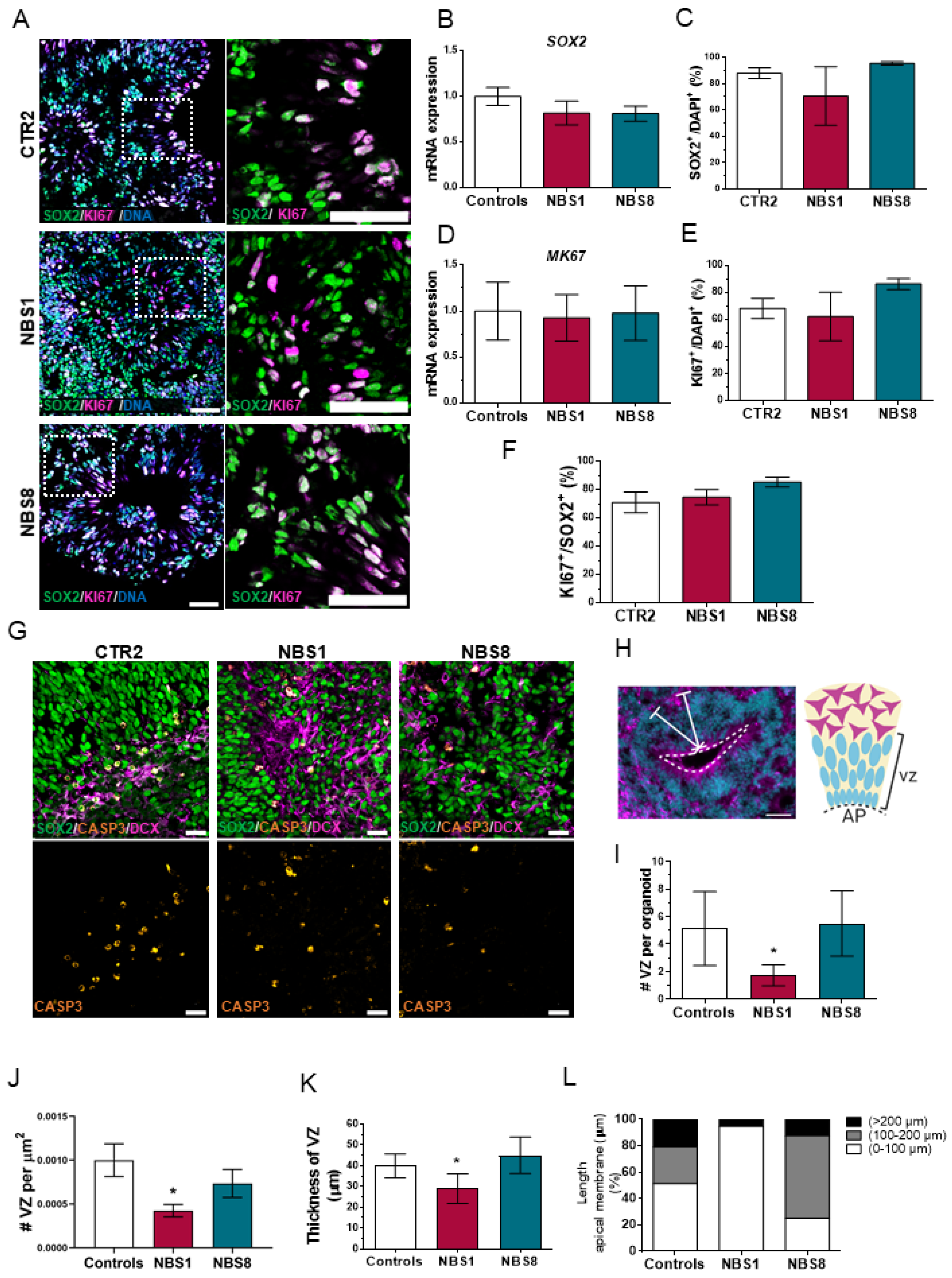
3.3. NBS Organoids Present a Disrupted Cytoarchitecture with Normal Proliferation of the NPCs
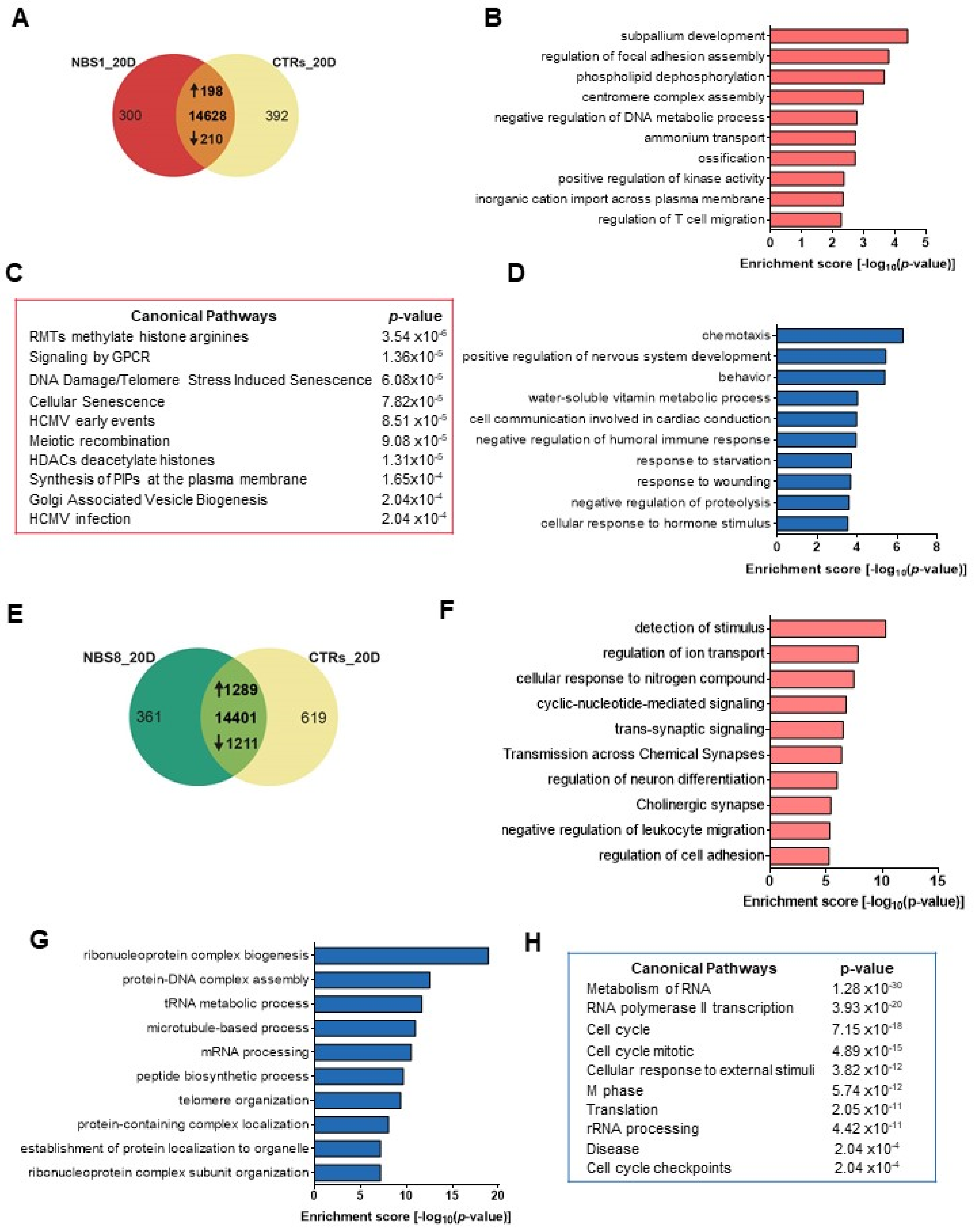
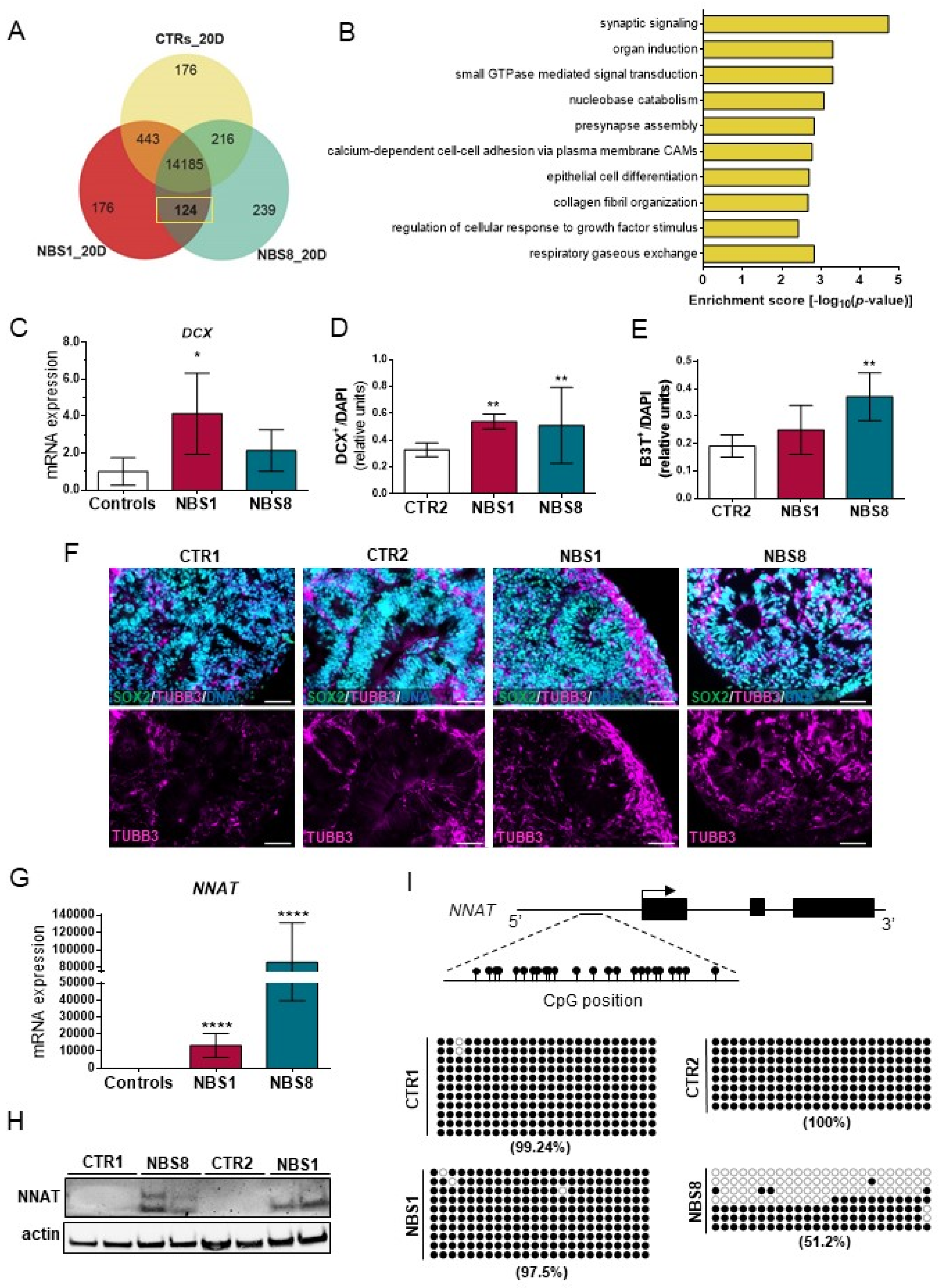
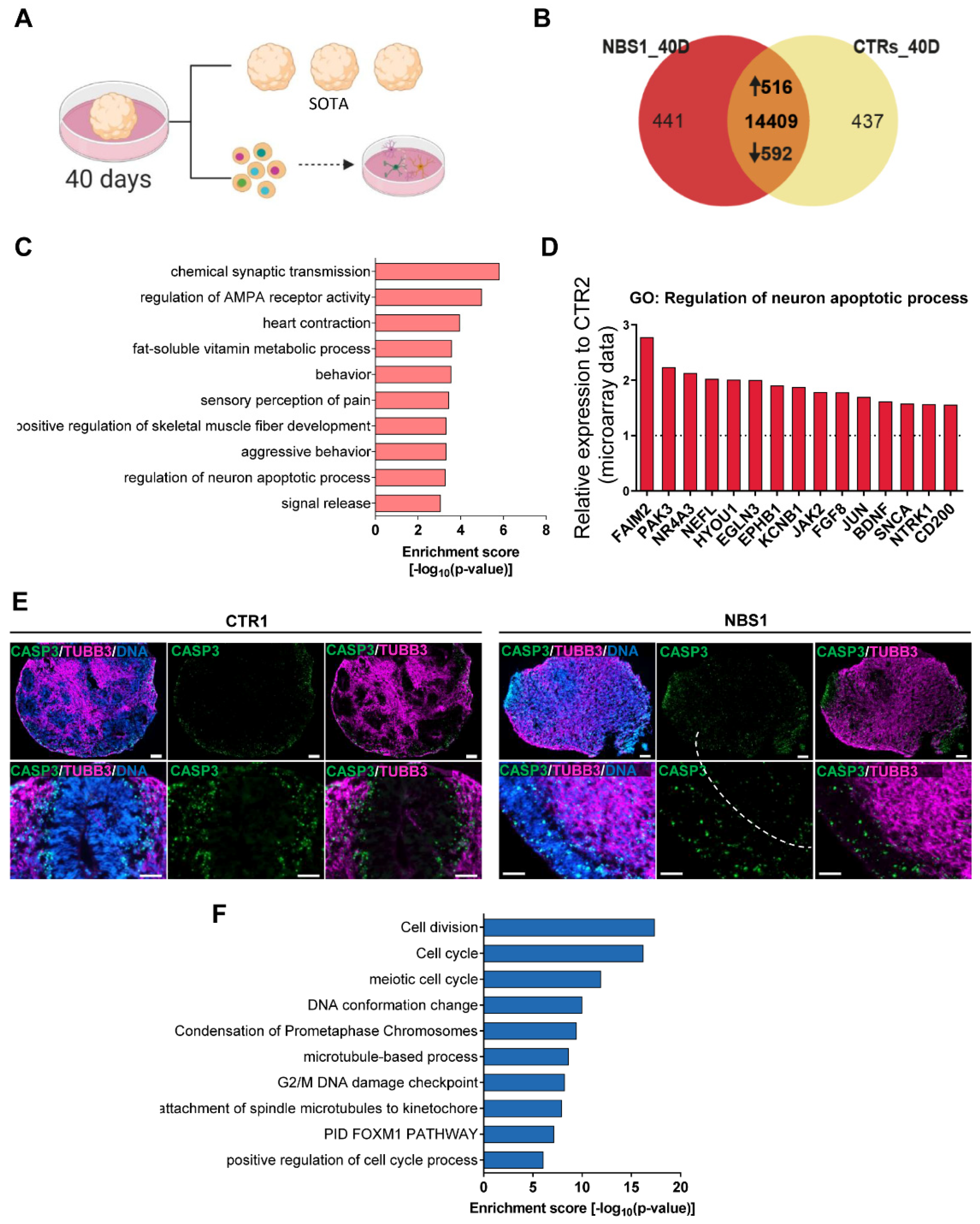
3.4. NBS1 and NBS8 Organoids Show a Distinct Transcriptomic Profile
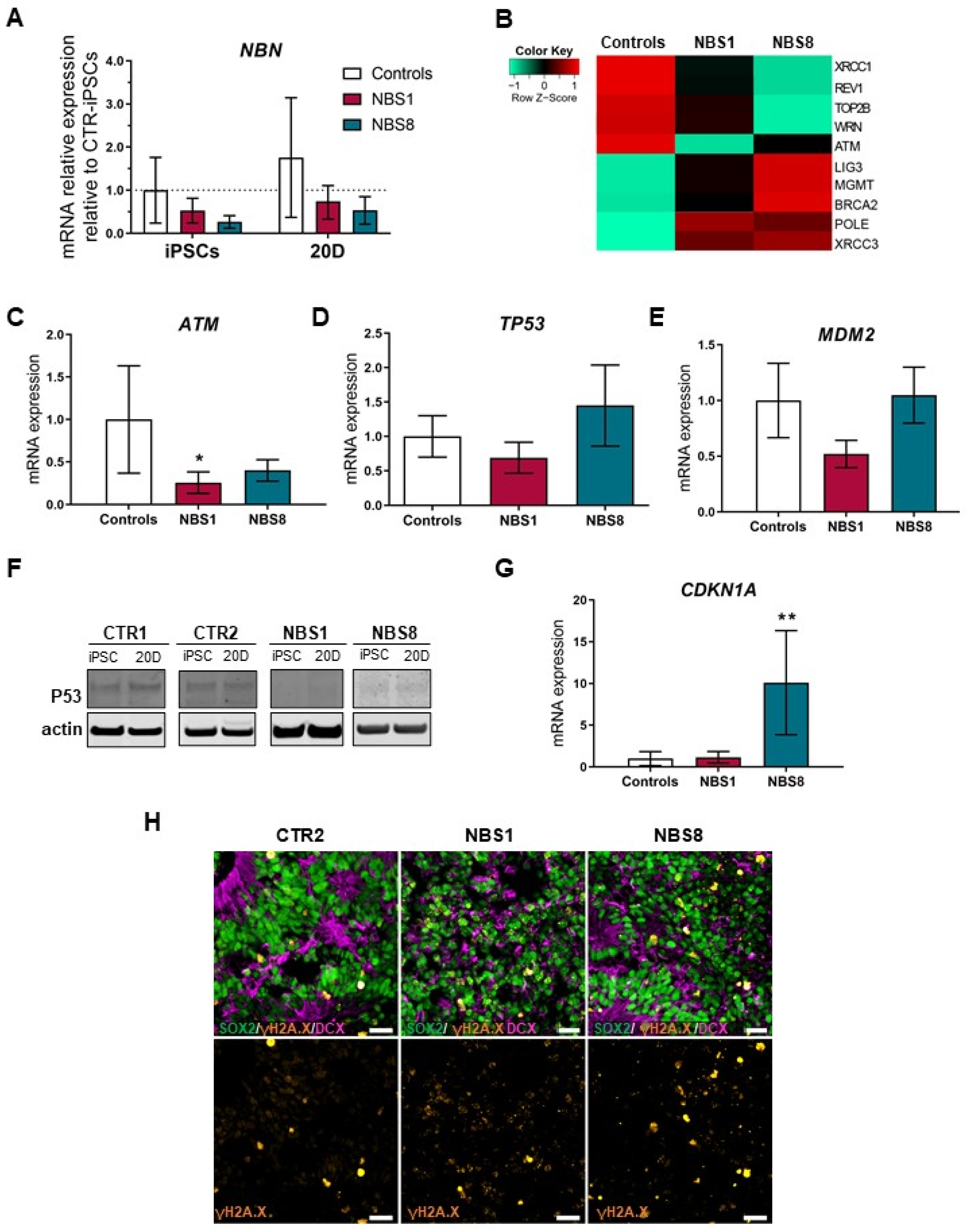
3.5. NBS Organoids Show Accumulation of DNA Damage
3.6. NBS Organoids Exhibit Premature Differentiation Accompanied by NNAT Over-Expression
3.7. NBS Organoids at Day 40 Acquire an Abnormal Regulation of Cell Cycle
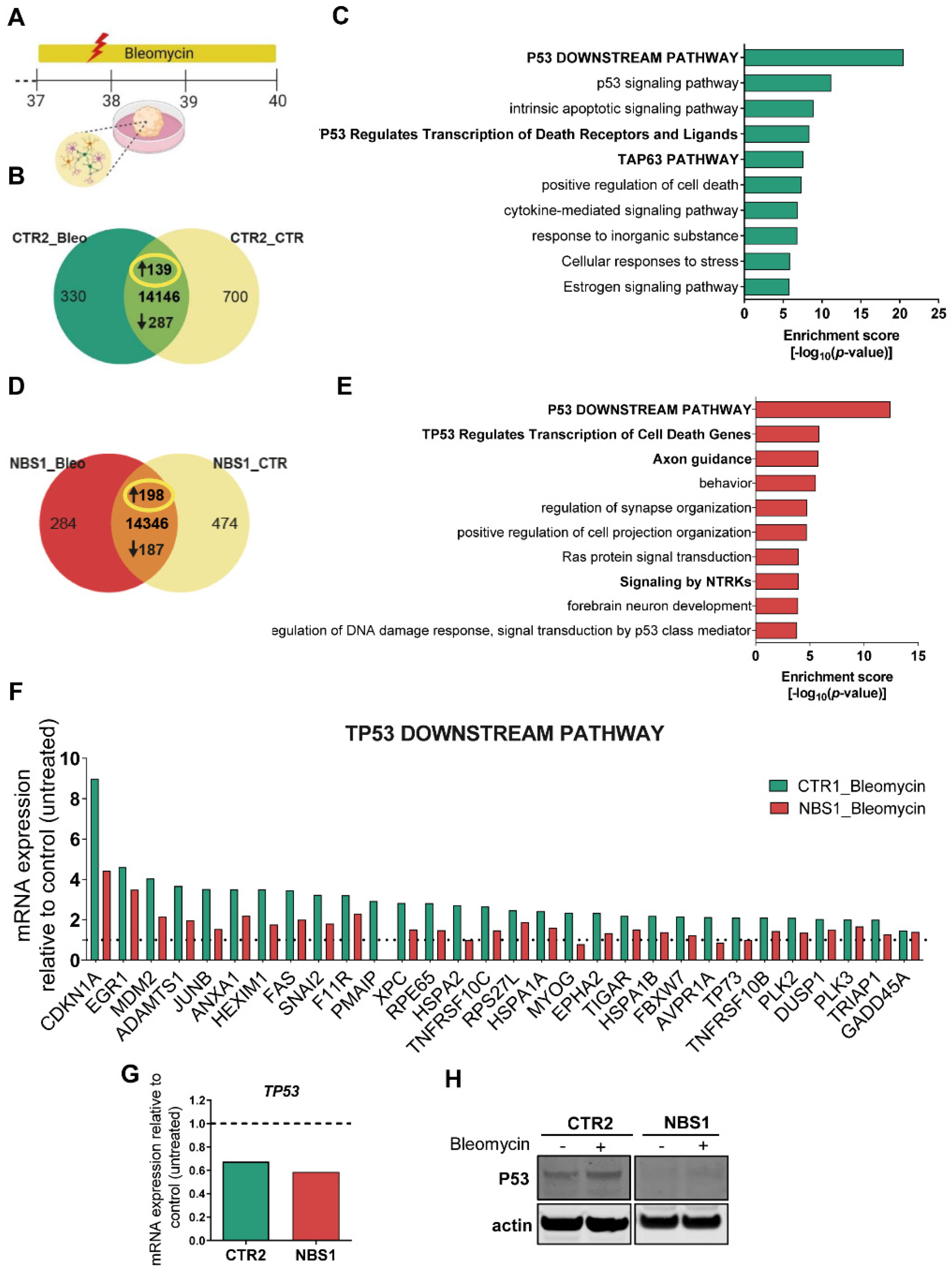
3.8. Bleomycin-Induced Cytotoxicity Highlights the Aberrant NBS Phenotype
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barzilai, A.; Biton, S.; Shiloh, Y. The role of the DNA damage response in neuronal development, organization and maintenance. DNA Repair 2008, 7, 1010–1027. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska, K.H.; Gregorek, H.; Dembowska-Bagińska, B.; Kalina, M.A.; Digweed, M. Nijmegen breakage syndrome (NBS). Orphanet J. Rare Dis. 2012, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Varon, R.; Demuth, I.; Chrzanowska, K.H. Nijmegen Breakage Syndrome; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Digweed, M.; Sperling, K. Nijmegen breakage syndrome: Clinical manifestation of defective response to DNA double-strand breaks. DNA Repair 2004, 3, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska, K.H.; Stumm, M.; Bekiesiska-Figatowska, M.; Varon, R.; Biaecka, M.; Gregorek, H.; Michakiewicz, J.; Krajewska-Walasek, M.; Jówiak, S.; Reis, A. Atypical clinical picture of the Nijmegen breakage syndrome associated with developmental abnormalities of the brain. J. Med. Genet. 2001, 38, E3. [Google Scholar] [CrossRef]
- Seemanova, E.; Jarolim, P.; Seeman, P.; Varon, R.; Digweed, M.; Swift, M.; Sperling, K. Cancer Risk of Heterozygotes with the NBN Founder Mutation. JNCI J. Natl. Cancer Inst. 2007, 99, 1875–1880. [Google Scholar] [CrossRef]
- Varon, R.; Seemanova, E.; Chrzanowska, K.; Hnateyko, O.; Piekutowska-Abramczuk, D.; Krajewska-Walasek, M.; Sykut-Cegielska, J.; Sperling, K.; Reis, A. Clinical ascertainment of Nijmegen breakage syndrome (NBS) and prevalence of the major mutation, 657del5, in three Slav populations. Eur. J. Hum. Genet. 2000, 8, 900–902. [Google Scholar] [CrossRef]
- Maser, R.S.; Zinkel, R.; Petrini, J.H.J. An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat. Genet. 2001, 27, 417–421. [Google Scholar] [CrossRef]
- Stracker, T.H.; Petrini, J.H.J. The MRE11 complex: Starting from the ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef]
- Jongmans, W.; Vuillaume, M.; Chrzanowska, K.; Smeets, D.; Sperling, K.; Hall, J. Nijmegen breakage syndrome cells fail to induce the p53-mediated DNA damage response following exposure to ionizing radiation. Mol. Cell. Biol. 1997, 17, 5016–5022. [Google Scholar] [CrossRef][Green Version]
- Buscemi, G.; Savio, C.; Zannini, L.; Miccichè, F.; Masnada, D.; Nakanishi, M.; Tauchi, H.; Komatsu, K.; Mizutani, S.; Khanna, K.; et al. Chk2 activation dependence on Nbs1 after DNA damage. Mol. Cell. Biol. 2001, 21, 5214–5222. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.E.; Veksler, E.; Lederman, H.; Lees-Miller, S.P. Cell Cycle Checkpoints and DNA Repair in Nijmegen Breakage Syndrome. Clin. Immunol. Immunopathol. 1997, 82, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Frappart, P.-O.; Tong, W.-M.; Demuth, I.; Radovanovic, I.; Herceg, Z.; Aguzzi, A.; Digweed, M.; Wang, Z.-Q. An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat. Med. 2005, 11, 538–544. [Google Scholar] [CrossRef]
- Zhou, Z.; Bruhn, C.; Wang, Z.-Q. Differential function of NBS1 and ATR in neurogenesis. DNA Repair 2012, 11, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.M.G.; Grigaravicius, P.; Remus, M.; Cavalheiro, G.R.; Gomes, A.L.; Martins, M.R.; Frappart, L.; Reuss, D.; McKinnon, P.J.; von Deimling, A.; et al. Nbn and Atm Cooperate in a Tissue and Developmental Stage-Specific Manner to Prevent Double Strand Breaks and Apoptosis in Developing Brain and Eye. PLoS ONE 2013, 8, e69209. [Google Scholar] [CrossRef]
- Li, R.; Sun, L.; Fang, A.; Li, P.; Wu, Q.; Wang, X. Recapitulating cortical development with organoid culture in vitro and modeling abnormal spindle-like (ASPM related primary) microcephaly disease. Protein Cell 2017, 8, 823–833. [Google Scholar] [CrossRef]
- Zhou, Z.W.; Kirtay, M.; Schneble, N.; Yakoub, G.; Ding, M.; Rüdiger, T.; Siniuk, K.; Lu, R.; Jiang, Y.N.; Li, T.L.; et al. NBS1 interacts with Notch signaling in neuronal homeostasis. Nucleic Acids Res. 2020, 48, 10924–10939. [Google Scholar] [CrossRef]
- Mlody, B.; Adjaye, J. Generation of iPSC lines from a Nijmegen Breakage Syndrome patient. Stem Cell Res. 2015, 15, 629–632. [Google Scholar] [CrossRef]
- Mlody, B.; Wruck, W.; Martins, S.; Sperling, K.; Adjaye, J. Nijmegen Breakage Syndrome fibroblasts and iPSCs: Cellular models for uncovering disease-associated signaling pathways and establishing a screening platform for anti-oxidants. Sci. Rep. 2017, 7, 7516. [Google Scholar] [CrossRef]
- Halevy, T.; Akov, S.; Bohndorf, M.; Mlody, B.; Adjaye, J.; Benvenisty, N.; Goldberg, M. Chromosomal Instability and Molecular Defects in Induced Pluripotent Stem Cells from Nijmegen Breakage Syndrome Patients. Cell Rep. 2016, 16, 2499–2511. [Google Scholar] [CrossRef]
- Bohndorf, M.; Ncube, A.; Spitzhorn, L.-S.; Enczmann, J.; Wruck, W.; Adjaye, J. Derivation and characterization of integration-free iPSC line ISRM-UM51 derived from SIX2-positive renal cells isolated from urine of an African male expressing the CYP2D6 *4/*17 variant which confers intermediate drug metabolizing activity. Stem Cell Res. 2017, 25, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.; Bohndorf, M.; Graffmann, N.; Wruck, W.; Chrzanowska, K.H.; Adjaye, J. Fibroblast-derived integration-free iPSC line ISRM-NBS1 from an 18-year-old Nijmegen Breakage Syndrome patient carrying the homozygous NBN c.657_661del5 mutation. Stem Cell Res. 2019, 34, 101372. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Adjaye, J. A Cyclic AMP Analog, 8-Br-cAMP, Enhances the Induction of Pluripotency in Human Fibroblast Cells. Stem Cell Rev. Rep. 2011, 7, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Sloan, S.A.; Andersen, J.; Pașca, A.M.; Birey, F.; Pașca, S.P. Generation and assembly of human brain region–specific three-dimensional cultures. Nat. Protoc. 2018, 13, 2062–2085. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Liaw, W.H.A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; Schwartz, M.; et al. gplots: Various R Programming Tools for Plotting Data. R Package Vers 2015, 2, 1–68. [Google Scholar]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- Graffmann, N.; Ring, S.; Kawala, M.-A.; Wruck, W.; Ncube, A.; Trompeter, H.-I.; Adjaye, J. Modeling Nonalcoholic Fatty Liver Disease with Human Pluripotent Stem Cell-Derived Immature Hepatocyte-Like Cells Reveals Activation of PLIN2 and Confirms Regulatory Functions of Peroxisome Proliferator-Activated Receptor Alpha. Stem Cells Dev. 2016, 25, 1119–1133. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Jones, A.R.; Overly, C.C.; Sunkin, S.M. The allen brain atlas: 5 years and beyond. Nat. Rev. Neurosci. 2009, 10, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D. A direct approach to false discovery rates. J. R. Stat. Soc. Ser. B Stat. Methodol. 2002, 64, 479–498. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Erichsen, L.; Seifert, H.-H.; Schulz, W.A.; Hoffmann, M.J.; Niegisch, G.; Araúzo-Bravo, M.J.; Bendhack, M.L.; Poyet, C.; Hermanns, T.; Beermann, A.; et al. Basic Hallmarks of Urothelial Cancer Unleashed in Primary Uroepithelium by Interference with the Epigenetic Master Regulator ODC1. Sci. Rep. 2020, 10, 3808. [Google Scholar] [CrossRef]
- Kumaki, Y.; Oda, M.; Okano, M. QUMA: Quantification tool for methylation analysis. Nucleic Acids Res. 2008, 36, W170–W175. [Google Scholar] [CrossRef]
- Miller, J.A.; Ding, S.-L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional landscape of the prenatal human brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef]
- Ottoboni, L.; von Wunster, B.; Martino, G. Therapeutic Plasticity of Neural Stem Cells. Front. Neurol. 2020, 11, 148. [Google Scholar] [CrossRef]
- Komatsu, K. NBS1 and multiple regulations of DNA damage response. J. Radiat. Res. 2016, 57, i11. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Pitale, P.M.; Howse, W.; Gorbatyuk, M. Neuronatin Protein in Health and Disease. J. Cell. Physiol. 2017, 232, 477–481. [Google Scholar] [CrossRef]
- Sobol, M.; Raykova, D.; Cavelier, L.; Khalfallah, A.; Schuster, J.; Dahl, N. Methods of Reprogramming to Induced Pluripotent Stem Cell Associated with Chromosomal Integrity and Delineation of a Chromosome 5q Candidate Region for Growth Advantage. Stem Cells Dev. 2015, 24, 2032–2040. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P.; Gebertová, K.; Paděrová, K.; Sperling, K.; Seemanová, E. Nijmegen breakage syndrome in 13% of age-matched Czech children with primary microcephaly. Pediatric Neurol. 2004, 30, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, E.; Wason, A.; Ramani, A.; Gooi, L.M.; Keller, P.; Pozniakovsky, A.; Poser, I.; Noack, F.; Telugu, N.S.; Calegari, F.; et al. CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J. 2016, 35, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, S.-L.; Yang, M.; Herrlinger, S.; Shao, Q.; Collar, J.L.; Fierro, E.; Shi, Y.; Liu, A.; Lu, H.; et al. Modeling microcephaly with cerebral organoids reveals a WDR62-CEP170-KIF2A pathway promoting cilium disassembly in neural progenitors. Nat. Commun. 2019, 10, 2612. [Google Scholar] [CrossRef] [PubMed]
- Esk, C.; Lindenhofer, D.; Haendeler, S.; Wester, R.A.; Pflug, F.; Schroeder, B.; Bagley, J.A.; Elling, U.; Zuber, J.; von Haeseler, A.; et al. A human tissue screen identifies a regulator of ER secretion as a brain-size determinant. Science 2020, 370, 935–941. [Google Scholar] [CrossRef]
- Dabin, J.; Fortuny, A.; Polo, S.E. Epigenome Maintenance in Response to DNA Damage. Mol. Cell 2016, 62, 712–727. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Cerosaletti, K.; Wright, J.; Concannon, P. Active Role for Nibrin in the Kinetics of Atm Activation. Mol. Cell. Biol. 2006, 26, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, C.; Zhou, Z.W.; Ai, H.; Wang, Z.Q. The essential function of the MRN complex in the resolution of endogenous replication intermediates. Cell Rep. 2014, 6, 182–195. [Google Scholar] [CrossRef][Green Version]
- Chen, Y.; Sun, J.; Ju, Z.; Wang, Z.Q.; Li, T. Nbs1-mediated DNA damage repair pathway regulates haematopoietic stem cell development and embryonic haematopoiesis. Cell Prolif. 2021, 54, e12972. [Google Scholar] [CrossRef]
- Kojima, K.; Konopleva, M.; McQueen, T.; O’Brien, S.; Plunkett, W.; Andreeff, M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood 2006, 108, 993–1000. [Google Scholar] [CrossRef]
- Fiévet, A.; Bellanger, D.; Zahed, L.; Burglen, L.; Derrien, A.; Dubois d’Enghien, C.; Lespinasse, J.; Parfait, B.; Pedespan, J.; Rieunier, G.; et al. DNA repair functional analyses of NBN hypomorphic variants associated with NBN-related infertility. Hum. Mutat. 2020, 41, 608–618. [Google Scholar] [CrossRef]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef]
- Su, Y.; Ming, G.L.; Song, H. DNA damage and repair regulate neuronal gene expression. Cell Res. 2015, 25, 993–994. [Google Scholar] [CrossRef]
- Niklison-Chirou, M.V.; Agostini, M.; Amelio, I.; Melino, G. Regulation of Adult Neurogenesis in Mammalian Brain. Int. J. Mol. Sci. 2020, 21, 4869. [Google Scholar] [CrossRef]
- Lin, H.-H.; Bell, E.; Uwanogho, D.; Perfect, L.W.; Noristani, H.; Bates, T.J.D.; Snetkov, V.; Price, J.; Sun, Y.-M. Neuronatin promotes neural lineage in ESCs via Ca2+ signaling. Stem Cells (Dayt. Ohio) 2010, 28, 1950–1960. [Google Scholar] [CrossRef] [PubMed]
- Shinde, V.; Pitale, P.M.; Howse, W.; Gorbatyuk, O.; Gorbatyuk, M. Neuronatin is a stress-responsive protein of rod photoreceptors. Neuroscience 2016, 328, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.M. Neuronatin gene: Imprinted and misfolded. Studies in Lafora disease, diabetes and cancer may implicate NNAT-aggregates as a common downstream participant in neuronal loss. Genomics 2014, 103, 183–188. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, S.; Erichsen, L.; Datsi, A.; Wruck, W.; Goering, W.; Chatzantonaki, E.; de Amorim, V.C.M.; Rossi, A.; Chrzanowska, K.H.; Adjaye, J. Impaired p53-Mediated DNA Damage Response Contributes to Microcephaly in Nijmegen Breakage Syndrome Patient-Derived Cerebral Organoids. Cells 2022, 11, 802. https://doi.org/10.3390/cells11050802
Martins S, Erichsen L, Datsi A, Wruck W, Goering W, Chatzantonaki E, de Amorim VCM, Rossi A, Chrzanowska KH, Adjaye J. Impaired p53-Mediated DNA Damage Response Contributes to Microcephaly in Nijmegen Breakage Syndrome Patient-Derived Cerebral Organoids. Cells. 2022; 11(5):802. https://doi.org/10.3390/cells11050802
Chicago/Turabian StyleMartins, Soraia, Lars Erichsen, Angeliki Datsi, Wasco Wruck, Wolfgang Goering, Eleftheria Chatzantonaki, Vanessa Cristina Meira de Amorim, Andrea Rossi, Krystyna H. Chrzanowska, and James Adjaye. 2022. "Impaired p53-Mediated DNA Damage Response Contributes to Microcephaly in Nijmegen Breakage Syndrome Patient-Derived Cerebral Organoids" Cells 11, no. 5: 802. https://doi.org/10.3390/cells11050802
APA StyleMartins, S., Erichsen, L., Datsi, A., Wruck, W., Goering, W., Chatzantonaki, E., de Amorim, V. C. M., Rossi, A., Chrzanowska, K. H., & Adjaye, J. (2022). Impaired p53-Mediated DNA Damage Response Contributes to Microcephaly in Nijmegen Breakage Syndrome Patient-Derived Cerebral Organoids. Cells, 11(5), 802. https://doi.org/10.3390/cells11050802