
Altered RBP1 Gene Expression Impacts Epithelial Cell Retinoic Acid, Proliferation, and Microenvironment

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Human Tissue Samples
2.3. Preparation of All-Trans Retinol Bound to Human Retinol-Binding Protein 4 (holoRBP4)
2.4. Decitabine (5-Aza-2′-deoxycytidine) Treatment
2.5. Treatment of Cells in Culture to Mimic Tumor Microenvironmental Factors
2.6. RARα Agonist AM580 in Culture Condition
2.7. Retinoids Extraction and Quantification
2.8. Total RNA Isolation and Q-PCR
2.9. Overexpression RBP1 in MCF-7 and MDA-MB-231 Cell Lines
2.10. Knock-down RBP1 in MCF-10-2A Cell Line by shRNA
2.11. Immunofluorescence Staining and Quantification for Ki-67
2.12. Collagen Staining and Quantification by Picrosirius Red in Cell Line Model
2.13. Statistical Analysis
3. Results
3.1. RBP1 and atRA Are Reduced in Mammary Tumor Tissue and Tumorigenic Epithelial Cell Lines
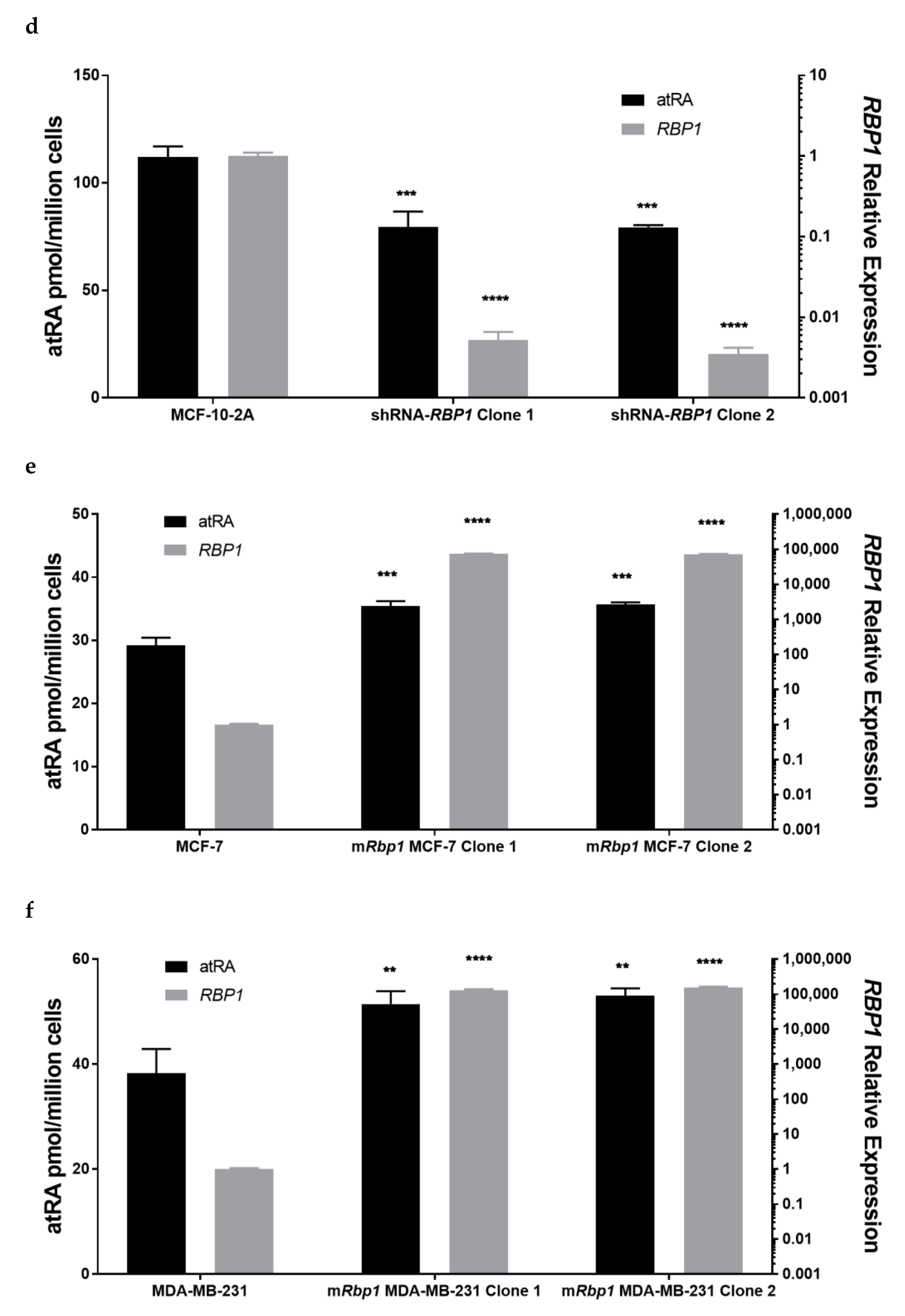
3.2. Altered RBP1 Expression Has a Direct Relationship with Altered RA Biosynthesis
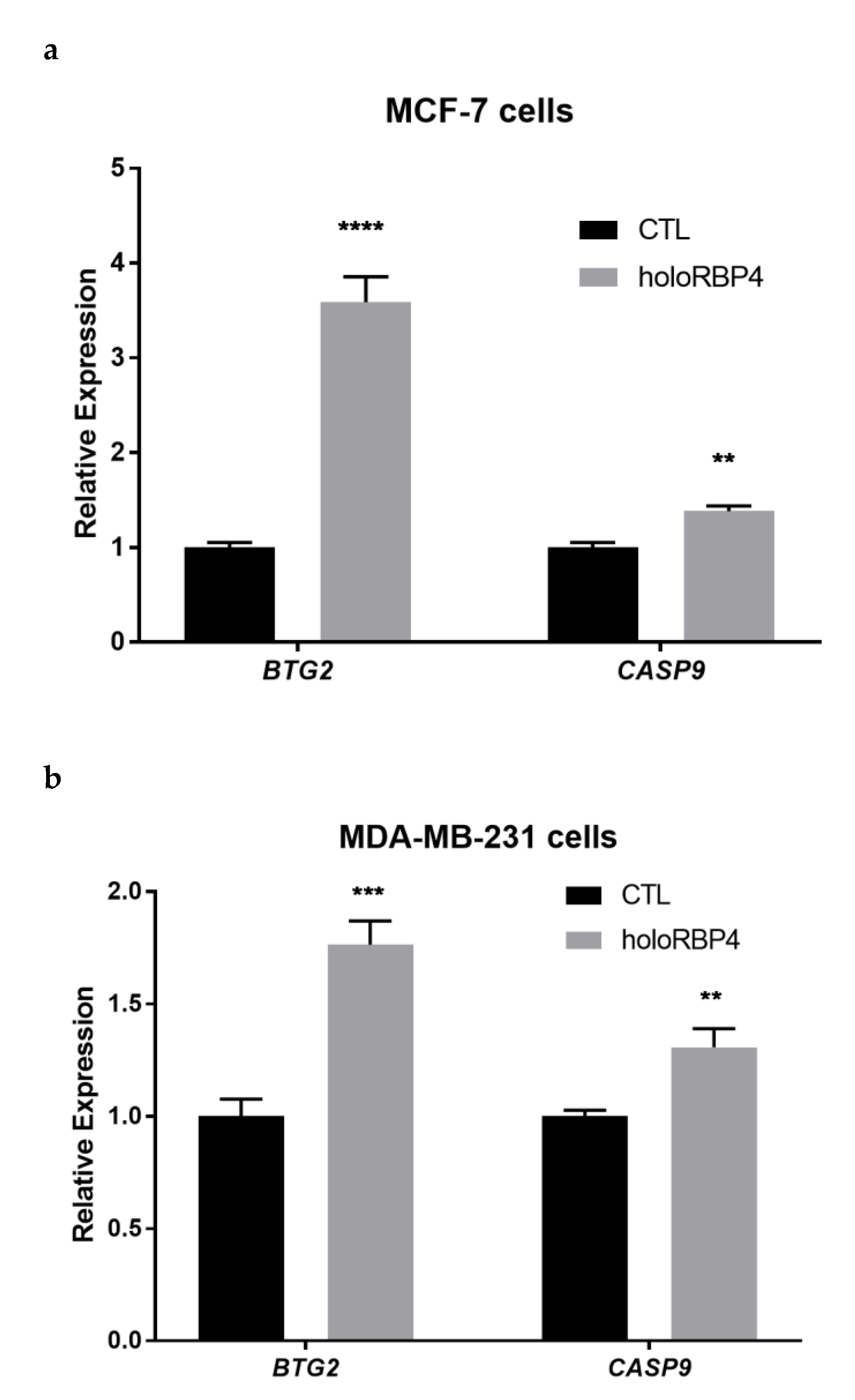
3.3. atRA Produced from Physiological Substrate RBP4-Retinol Can Activate RA Direct Targets in Epithelial Cells
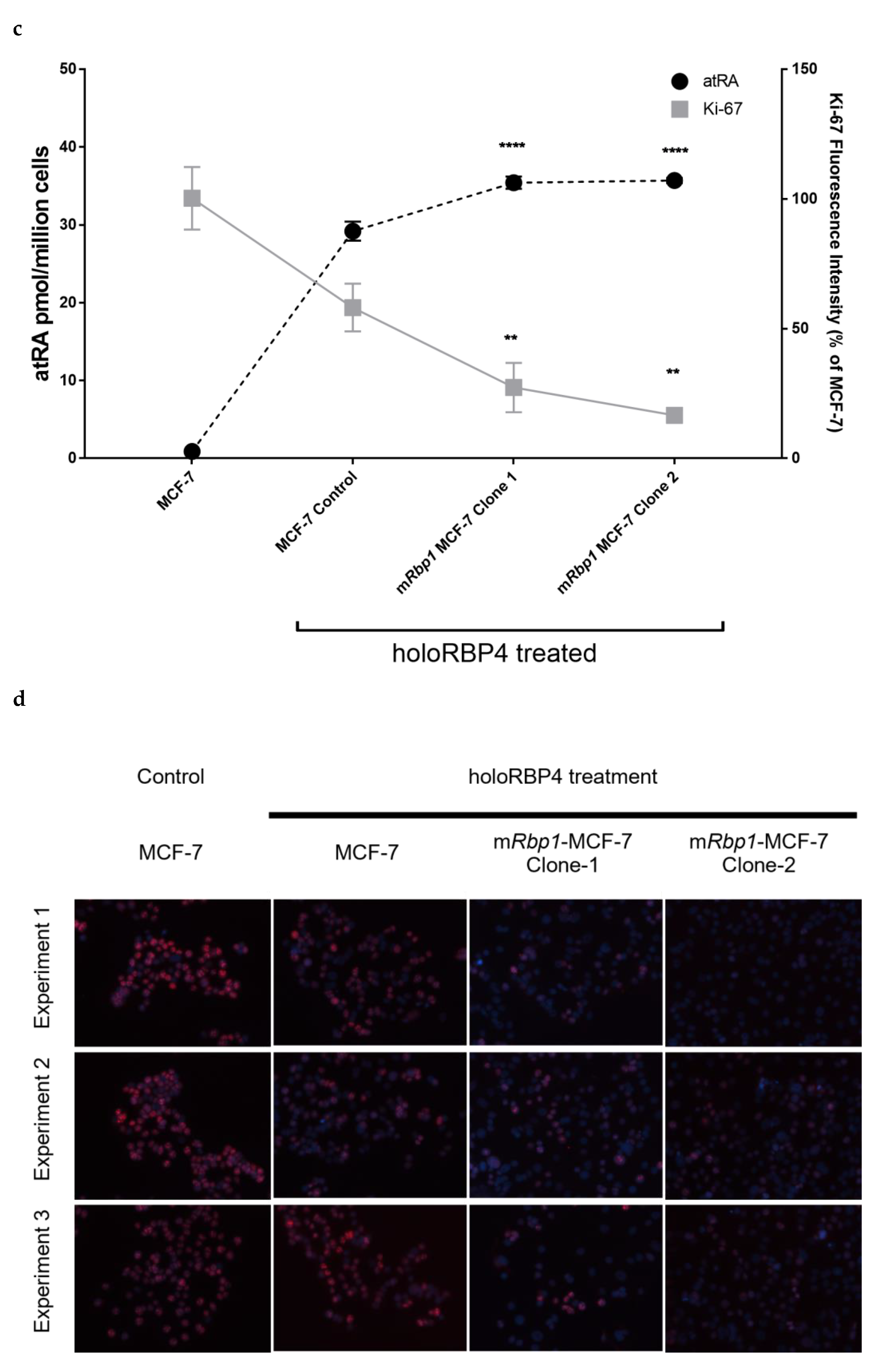
3.4. Overexpression of RBP1 and atRA Inhibits Cell Proliferation in MCF-7 Cells
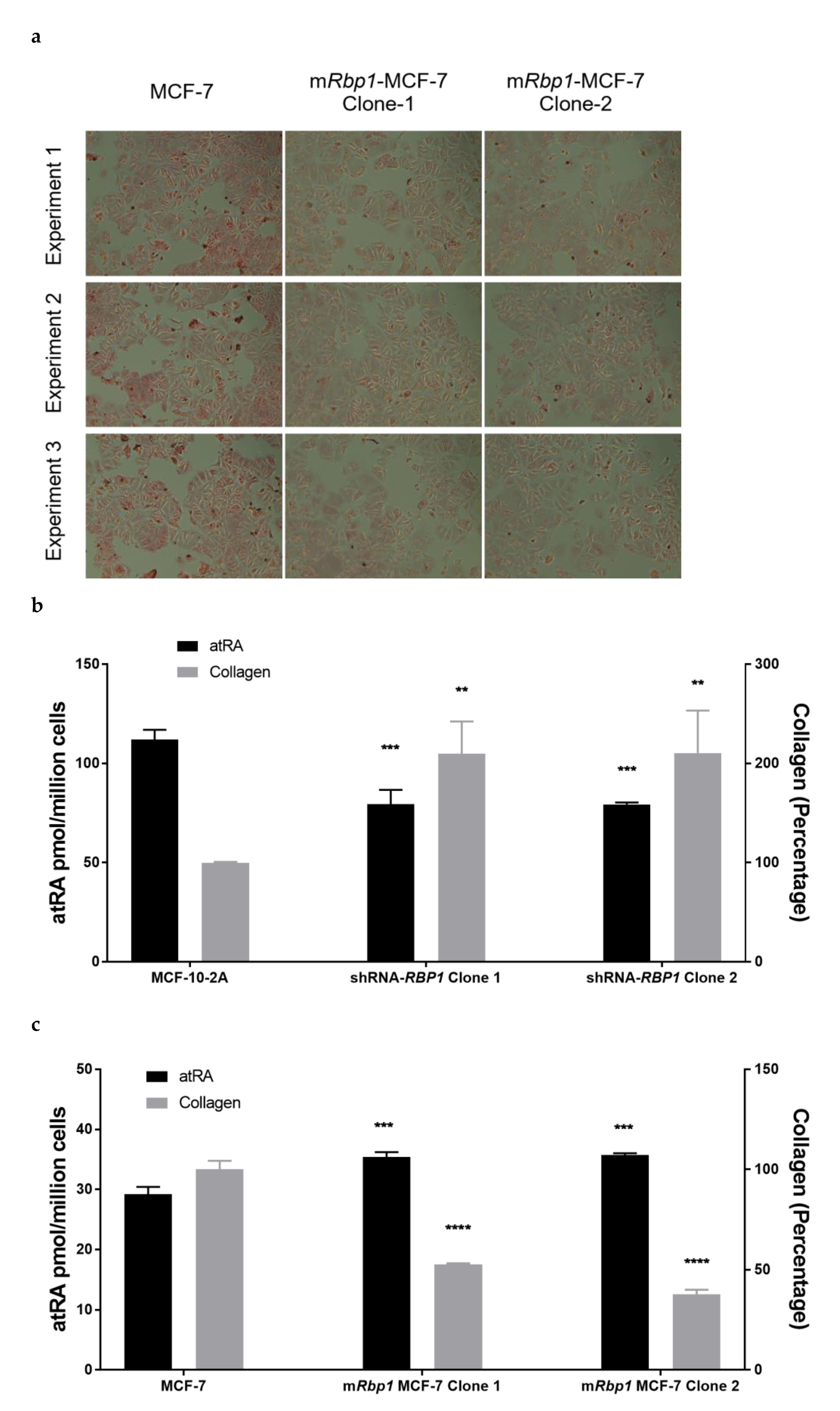
3.5. RBP1 Expression and atRA Inhibited Collagen Deposition in Cell Culture
3.6. Tumor Microenvironmental Factors in Culture Condition Alter RBP1 Expression and atRA Homeostasis in Cell Lines
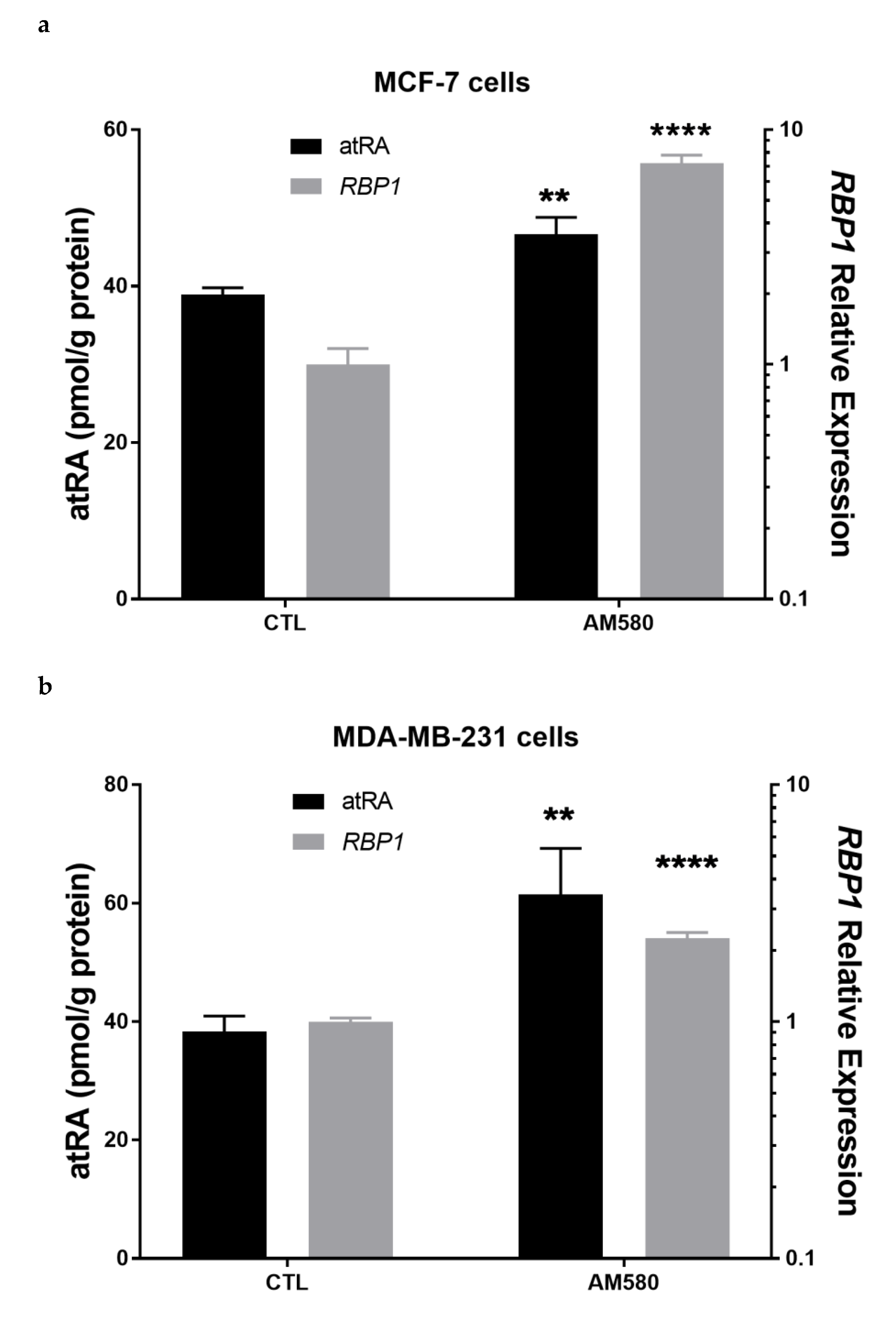
3.7. Therapeutic Treatment Increases RBP1 Expression and atRA Production
3.8. Epithelial atRA Production Capacity Impacts Neighboring Fibroblast Cell atRA Supply
4. Discussion
4.1. RBP1 and atRA Homeostasis
4.2. RBP1 and atRA Impact the Microenvironment
4.3. Microenvironmental Factors Impact RBP1 Expression
4.4. Therapeutic Potential for Restoring RBP1 Expression and Endogenous atRA Production
4.5. Limitations and Future Opportunities
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Napoli, J.L. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim. Biophys. Acta 2012, 1821, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Napoli, J.L. Post-natal all-trans-retinoic acid biosynthesis. Methods Enzymol. 2020, 637, 27–54. [Google Scholar] [CrossRef]
- Fields, A.L.; Soprano, D.R.; Soprano, K.J. Retinoids in biological control and cancer. J. Cell. Biochem. 2007, 102, 886–898. [Google Scholar] [CrossRef]
- Germain, P.; Chambon, P.; Eichele, G.; Evans, R.M.; Lazar, M.A.; Leid, M.; De Lera, A.R.; Lotan, R.; Mangelsdorf, D.J.; Gronemeyer, H. International Union of Pharmacology. LX. Retinoic acid receptors. Pharmacol. Rev. 2006, 58, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 2007, 129, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Napoli, J.L. All-trans-retinoic acid stimulates translation and induces spine formation in hippocampal neurons through a membrane-associated RARalpha. FASEB J. 2008, 22, 236–245. [Google Scholar] [CrossRef]
- Iskakova, M.; Karbyshev, M.; Piskunov, A.; Rochette-Egly, C. Nuclear and extranuclear effects of vitamin A. Can. J. Physiol. Pharmacol. 2015, 93, 1065–1075. [Google Scholar] [CrossRef]
- Germain, P.; Chambon, P.; Eichele, G.; Evans, R.M.; Lazar, M.A.; Leid, M.; De Lera, A.R.; Lotan, R.; Mangelsdorf, D.J.; Gronemeyer, H. International Union of Pharmacology. LXIII. Retinoid X receptors. Pharmacol. Rev. 2006, 58, 760–772. [Google Scholar] [CrossRef]
- Kane, M.A.; Folias, A.E.; Pingitore, A.; Perri, M.; Obrochta, K.M.; Krois, C.R.; Cione, E.; Ryu, J.Y.; Napoli, J.L. Identification of 9-cis-retinoic acid as a pancreas-specific autacoid that attenuates glucose-stimulated insulin secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 21884–21889. [Google Scholar] [CrossRef]
- Kane, M.A.; Folias, A.E.; Pingitore, A.; Perri, M.; Krois, C.R.; Ryu, J.Y.; Cione, E.; Napoli, J.L. CrbpI modulates glucose homeostasis and pancreas 9-cis-retinoic acid concentrations. Mol. Cell. Biol. 2011, 31, 3277–3285. [Google Scholar] [CrossRef]
- Kane, M.A. Analysis, occurrence, and function of 9-cis-retinoic acid. Biochim. Biophys. Acta 2012, 1821, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.A.; Folias, A.E.; Wang, C.; Napoli, J.L. Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Anal. Chem. 2008, 80, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.A.; Napoli, J.L. Quantification of endogenous retinoids. In Retinoids: Methods and Protocols; Methods in Molecular Biology Series; Humana Press: Totowa, NJ, USA, 2010; Volume 652, pp. 1–54. [Google Scholar] [CrossRef]
- Altucci, L.; Leibowitz, M.D.; Ogilvie, K.M.; de Lera, A.R.; Gronemeyer, H. RAR and RXR modulation in cancer and metabolic disease. Nat. Rev. Drug Discov. 2007, 6, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Napoli, J.L. Functions of Intracellular Retinoid Binding-Proteins. Subcell. Biochem. 2016, 81, 21–76. [Google Scholar] [CrossRef]
- Napoli, J.L. Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacol. Ther. 2017, 173, 19–33. [Google Scholar] [CrossRef]
- Kuppumbatti, Y.S.; Bleiweiss, I.J.; Mandeli, J.P.; Waxman, S.; Mira, Y.L.R. Cellular retinol-binding protein expression and breast cancer. J. Natl. Cancer Inst. 2000, 92, 475–480. [Google Scholar] [CrossRef]
- Esteller, M.; Guo, M.; Moreno, V.; Peinado, M.A.; Capella, G.; Galm, O.; Baylin, S.B.; Herman, J.G. Hypermethylation-associated Inactivation of the Cellular Retinol-Binding-Protein 1 Gene in Human Cancer. Cancer Res. 2002, 62, 5902–5905. [Google Scholar]
- Jeronimo, C.; Henrique, R.; Oliveira, J.; Lobo, F.; Pais, I.; Teixeira, M.R.; Lopes, C. Aberrant Cellular Retinol Binding Protein 1 (CRBP1) gene expression and promoter methylation in prostate cancer. J. Clin. Pathol. 2004, 57, 872–876. [Google Scholar] [CrossRef]
- Palan, P.R.; Romney, S.L. Cellular binding proteins for vitamin A in human carcinomas and in normal tissues. Cancer Res. 1980, 40, 4221–4224. [Google Scholar]
- Palan, P.R.; Duttagupta, C.; Romney, S.L. Sex difference in cellular retinol- and retinoic acid-binding proteins in human colon adenocarcinomas. Cancer Lett. 1980, 11, 97–101. [Google Scholar] [CrossRef]
- Ong, D.E.; Markert, C.; Chiu, J.F. Cellular binding proteins for vitamin A in colorectal adenocarcinoma of rat. Cancer Res. 1978, 38, 4422–4426. [Google Scholar]
- Toki, K.; Enokida, H.; Kawakami, K.; Chiyomaru, T.; Tatarano, S.; Yoshino, H.; Uchida, Y.; Kawahara, K.; Nishiyama, K.; Seki, N.; et al. CpG hypermethylation of cellular retinol-binding protein 1 contributes to cell proliferation and migration in bladder cancer. Int. J. Oncol. 2010, 37, 1379–1388. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Colvin, E.K.; Susanto, J.M.; Kench, J.G.; Ong, V.N.; Mawson, A.; Pinese, M.; Chang, D.K.; Rooman, I.; O’Toole, S.A.; Segara, D.; et al. Retinoid signaling in pancreatic cancer, injury and regeneration. PLoS ONE 2011, 6, e29075. [Google Scholar] [CrossRef] [PubMed]
- Daxecker, F.; Marth, C.; Daxenbichler, G. Retinoic binding in melanomas of the eye and in normal choroid. Cancer Lett. 1988, 41, 119–122. [Google Scholar] [CrossRef]
- Orlandi, A.; Ferlosio, A.; Ciucci, A.; Francesconi, A.; Lifschitz-Mercer, B.; Gabbiani, G.; Spagnoli, L.G.; Czernobilsky, B. Cellular retinol binding protein-1 expression in endometrial hyperplasia and carcinoma: Diagnostic and possible therapeutic implications. Mod. Pathol. 2006, 19, 797–803. [Google Scholar] [CrossRef]
- Jones, J.W.; Pierzchalski, K.; Yu, J.; Kane, M.A. Use of fast HPLC multiple reaction monitoring cubed for endogenous retinoic acid quantification in complex matrices. Anal. Chem. 2015, 87, 3222–3230. [Google Scholar] [CrossRef]
- Kane, M.A.; Chen, N.; Sparks, S.; Napoli, J.L. Quantification of endogenous retinoic acid in limited biological samples by LC/MS/MS. Biochem. J. 2005, 388 Pt 1, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.A.; Folias, A.E.; Wang, C.; Napoli, J.L. Ethanol elevates physiological all-trans-retinoic acid levels in select loci through altering retinoid metabolism in multiple loci: A potential mechanism of ethanol toxicity. FASEB J. 2010, 24, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Pierzchalski, K.; Taylor, R.N.; Nezhat, C.; Jones, J.W.; Napoli, J.L.; Yang, G.; Kane, M.A.; Sidell, N. Retinoic acid biosynthesis is impaired in human and murine endometriosis. Biol. Reprod. 2014, 91, 84. [Google Scholar] [CrossRef]
- Pierzchalski, K.; Yu, J.; Norman, V.; Kane, M.A. CrbpI regulates mammary retinoic acid homeostasis and the mammary microenvironment. FASEB J. 2013, 27, 1904–1916. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Bosch, A.; Leibovitch, B.; Pereira, L.; Cubedo, E.; Yu, J.; Pierzchalski, K.; Jones, J.W.; Fishel, M.; Kane, M.; et al. Blocking the PAH2 domain of Sin3A inhibits tumorigenesis and confers retinoid sensitivity in triple negative breast cancer. Oncotarget 2016, 7, 43689–43702. [Google Scholar] [CrossRef]
- Farias, E.F.; Ong, D.E.; Ghyselinck, N.B.; Nakajo, S.; Kuppumbatti, Y.S.; Mira y Lopez, R. Cellular retinol-binding protein I, a regulator of breast epithelial retinoic acid receptor activity, cell differentiation, and tumorigenicity. J. Natl. Cancer Inst. 2005, 97, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Kuppumbatti, Y.S.; Rexer, B.; Nakajo, S.; Nakaya, K.; Mira-y-Lopez, R. CRBP suppresses breast cancer cell survival and anchorage-independent growth. Oncogene 2001, 20, 7413–7419. [Google Scholar] [CrossRef] [PubMed]
- Farias, E.F.; Marzan, C.; Mira-y-Lopez, R. Cellular retinol-binding protein-I inhibits PI3K/Akt signaling through a retinoic acid receptor-dependent mechanism that regulates p85-p110 heterodimerization. Oncogene 2005, 24, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Lashuel, H.A.; Miroy, G.J.; Dikler, S.; Kelly, J.W. Recombinant human retinol-binding protein refolding, native disulfide formation, and characterization. Protein Expr. Purif. 1998, 14, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Heller, J.; Horwitz, J. Conformational changes following interaction between retinol isomers and human retinol-binding protein and between the retinol-binding protein and prealbumin. J. Biol. Chem. 1973, 248, 6308–6316. [Google Scholar] [CrossRef]
- Kastl, L.; Brown, I.; Schofield, A.C. Altered DNA methylation is associated with docetaxel resistance in human breast cancer cells. Int. J. Oncol. 2010, 36, 1235–1241. [Google Scholar] [CrossRef]
- Doldo, E.; Costanza, G.; Ferlosio, A.; Passeri, D.; Bernardini, S.; Scioli, M.G.; Mazzaglia, D.; Agostinelli, S.; Del Bufalo, D.; Czernobilsky, B.; et al. CRBP-1 expression in ovarian cancer: A potential therapeutic target. Anticancer Res. 2014, 34, 3303–3312. [Google Scholar]
- Chan, M.C.; Ilott, N.E.; Schodel, J.; Sims, D.; Tumber, A.; Lippl, K.; Mole, D.R.; Pugh, C.W.; Ratcliffe, P.J.; Ponting, C.P.; et al. Tuning the Transcriptional Response to Hypoxia by Inhibiting Hypoxia-inducible Factor (HIF) Prolyl and Asparaginyl Hydroxylases. J. Biol. Chem. 2016, 291, 20661–20673. [Google Scholar] [CrossRef]
- Shahruzaman, S.H.; Yusof, F.Z.; Maniam, S.; Fakurazi, S.; Maniam, S. The cytotoxic effect of Baeckea frustescens extracts in eliminating hypoxic breast cancer cells. BMC Complement. Med. Ther. 2021, 21, 245. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Wilson, J.W.; Schofield, C.J.; Chen, R. Author Correction: Hypoxia-inducible factor (HIF) prolyl hydroxylase inhibitors induce autophagy and have a protective effect in an in-vitro ischaemia model. Sci. Rep. 2020, 10, 6041. [Google Scholar] [CrossRef] [PubMed]
- Ayrapetov, M.K.; Xu, C.; Sun, Y.; Zhu, K.; Parmar, K.; D’Andrea, A.D.; Price, B.D. Activation of Hif1alpha by the prolylhydroxylase inhibitor dimethyoxalyglycine decreases radiosensitivity. PLoS ONE 2011, 6, e26064. [Google Scholar] [CrossRef] [PubMed]
- Bush, J.T.; Chan, M.C.; Mohammed, S.; Schofield, C.J. Quantitative MS-Based Proteomics: Comparing the MCF-7 Cellular Response to Hypoxia and a 2-Oxoglutarate Analogue. ChemBioChem 2020, 21, 1647–1655. [Google Scholar] [CrossRef]
- Lee, H.R.; Leslie, F.; Azarin, S.M. A facile in vitro platform to study cancer cell dormancy under hypoxic microenvironments using CoCl2. J. Biol. Eng. 2018, 12, 12. [Google Scholar] [CrossRef]
- Fu, O.Y.; Hou, M.F.; Yang, S.F.; Huang, S.C.; Lee, W.Y. Cobalt chloride-induced hypoxia modulates the invasive potential and matrix metalloproteinases of primary and metastatic breast cancer cells. Anticancer Res. 2009, 29, 3131–3138. [Google Scholar]
- Nelson, B.S.; Kremer, D.M.; Lyssiotis, C.A. New tricks for an old drug. Nat. Chem. Biol. 2018, 14, 990–991. [Google Scholar] [CrossRef]
- Munoz-Sanchez, J.; Chanez-Cardenas, M.E. The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol. 2019, 39, 556–570. [Google Scholar] [CrossRef]
- Bosch, A.; Bertran, S.P.; Lu, Y.; Garcia, A.; Jones, A.M.; Dawson, M.I.; Farias, E.F. Reversal by RARalpha agonist Am580 of c-Myc-induced imbalance in RARalpha/RARgamma expression during MMTV-Myc tumorigenesis. Breast Cancer Res. 2012, 14, R121. [Google Scholar] [CrossRef]
- Lu, Y.; Bertran, S.; Samuels, T.A.; Mira-y-Lopez, R.; Farias, E.F. Mechanism of inhibition of MMTV-neu and MMTV-wnt1 induced mammary oncogenesis by RARalpha agonist AM580. Oncogene 2010, 29, 3665–3676. [Google Scholar] [CrossRef]
- du Manoir, S.; Guillaud, P.; Camus, E.; Seigneurin, D.; Brugal, G. Ki-67 labeling in postmitotic cells defines different Ki-67 pathways within the 2c compartment. Cytometry 1991, 12, 455–463. [Google Scholar] [CrossRef] [PubMed]
- van Dierendonck, J.H.; Keijzer, R.; van de Velde, C.J.; Cornelisse, C.J. Nuclear distribution of the Ki-67 antigen during the cell cycle: Comparison with growth fraction in human breast cancer cells. Cancer Res. 1989, 49, 2999–3006. [Google Scholar] [PubMed]
- Donato, L.J.; Noy, N. Suppression of mammary carcinoma growth by retinoic acid: Proapoptotic genes are targets for retinoic acid receptor and cellular retinoic acid-binding protein II signaling. Cancer Res. 2005, 65, 8193–8199. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.J.; Suh, J.H.; Noy, N. Suppression of mammary carcinoma cell growth by retinoic acid: The cell cycle control gene Btg2 is a direct target for retinoic acid receptor signaling. Cancer Res. 2007, 67, 609–615. [Google Scholar] [CrossRef]
- He, Y.; Huang, Y.; Zhou, L.; Lu, L.M.; Zhu, Y.C.; Yao, T. All-trans retinoic acid inhibited angiotensin II-induced increase in cell growth and collagen secretion of neonatal cardiac fibroblasts. Acta Pharmacol. Sin. 2006, 27, 423–429. [Google Scholar] [CrossRef]
- Henning, T.; Kraus, M.; Brischwein, M.; Otto, A.M.; Wolf, B. Relevance of tumor microenvironment for progression, therapy and drug development. Anticancer Drugs 2004, 15, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Arapshian, A.; Bertran, S.; Kuppumbatti, Y.S.; Nakajo, S.; Mira-y-Lopez, R. Epigenetic CRBP downregulation appears to be an evolutionarily conserved (human and mouse) and oncogene-specific phenomenon in breast cancer. Mol. Cancer 2004, 3, 13. [Google Scholar] [CrossRef]
- Patra, S.K.; Bettuzzi, S. Epigenetic DNA-(cytosine-5-carbon) modifications: 5-aza-2′-deoxycytidine and DNA-demethylation. Biochemistry (Moscow) 2009, 74, 613–619. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Pestell, R.G.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Stromal-epithelial metabolic coupling in cancer: Integrating autophagy and metabolism in the tumor microenvironment. Int. J. Biochem. Cell Biol. 2011, 43, 1045–1051. [Google Scholar] [CrossRef]
- Elenbaas, B.; Weinberg, R.A. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp. Cell Res. 2001, 264, 169–184. [Google Scholar] [CrossRef]
- Yang, N.; Parker, L.; Yu, J.; Jones, J.W.; Liu, T.; Papanicolaou, K.N.; Talbot, C.C., Jr.; Margulies, K.B.; O’Rourke, B.; Kane, M.; et al. Cardiac retinoic acid levels decline in heart failure. JCI Insight 2021, 6, e137593. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Flomenberg, N.; Tsirigos, A.; Howell, A.; et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: Evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010, 9, 2412–2422. [Google Scholar] [CrossRef] [PubMed]
- Trimmer, C.; Sotgia, F.; Whitaker-Menezes, D.; Balliet, R.M.; Eaton, G.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Iozzo, R.V.; Pestell, R.G.; et al. Caveolin-1 and mitochondrial SOD2 (MnSOD) function as tumor suppressors in the stromal microenvironment: A new genetically tractable model for human cancer associated fibroblasts. Cancer Biol. Ther. 2011, 11, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kane, M.A.; Napoli, J.L. Multiple retinol and retinal dehydrogenases catalyze all-trans-retinoic acid biosynthesis in astrocytes. J. Biol. Chem. 2011, 286, 6542–6553. [Google Scholar] [CrossRef]
- Siegenthaler, J.A.; Ashique, A.M.; Zarbalis, K.; Patterson, K.P.; Hecht, J.H.; Kane, M.A.; Folias, A.E.; Choe, Y.; May, S.R.; Kume, T.; et al. Retinoic acid from the meninges regulates cortical neuron generation. Cell 2009, 139, 597–609. [Google Scholar] [CrossRef]
- Fu, M.; Sato, Y.; Lyons-Warren, A.; Zhang, B.; Kane, M.A.; Napoli, J.L.; Heuckeroth, R.O. Vitamin A facilitates enteric nervous system precursor migration by reducing Pten accumulation. Development 2010, 137, 631–640. [Google Scholar] [CrossRef]
- Sidell, N.; Feng, Y.; Hao, L.; Wu, J.; Yu, J.; Kane, M.A.; Napoli, J.L.; Taylor, R.N. Retinoic acid is a cofactor for translational regulation of vascular endothelial growth factor in human endometrial stromal cells. Mol. Endocrinol. 2010, 24, 148–160. [Google Scholar] [CrossRef]
- Villablanca, E.J.; Wang, S.; de Calisto, J.; Gomes, D.C.; Kane, M.A.; Napoli, J.L.; Blaner, W.S.; Kagechika, H.; Blomhoff, R.; Rosemblatt, M.; et al. MyD88 and retinoic acid signaling pathways interact to modulate gastrointestinal activities of dendritic cells. Gastroenterology 2011, 141, 176–185. [Google Scholar] [CrossRef]
- Rajakumar, A.; Kane, M.A.; Yu, J.; Jones, J.W.; Qu, H.; Badell, M.; Taylor, R.N.; Sidell, N. Alternatively Activated Macrophages Are the Primary Retinoic Acid-Producing Cells in Human Decidua. Reprod. Sci. 2020, 27, 334–341. [Google Scholar] [CrossRef]
- El Andaloussi, S.; Mager, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, F.A.; Lecuyer, E. Small Luggage for a Long Journey: Transfer of Vesicle-Enclosed Small RNA in Interspecies Communication. Front. Microbiol. 2017, 8, 377. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, H.M.; Masoumi, N.; Witwer, K.W.; Pegtel, D.M. Extracellular Vesicles Exploit Viral Entry Routes for Cargo Delivery. Microbiol. Mol. Biol. Rev. 2016, 80, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef] [PubMed]
- Parmar, H.; Cunha, G.R. Epithelial-stromal interactions in the mouse and human mammary gland in vivo. Endocr. Relat. Cancer 2004, 11, 437–458. [Google Scholar] [CrossRef]
- Boyd, N.F.; Martin, L.J.; Stone, J.; Greenberg, C.; Minkin, S.; Yaffe, M.J. Mammographic densities as a marker of human breast cancer risk and their use in chemoprevention. Curr. Oncol. Rep. 2001, 3, 314–321. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef]
- Aiello, E.J.; Buist, D.S.; White, E.; Porter, P.L. Association between mammographic breast density and breast cancer tumor characteristics. Cancer Epidemiol. Biomark. Prev. 2005, 14, 662–668. [Google Scholar] [CrossRef]
- Abbas, R.; Larisch, S. Killing by Degradation: Regulation of Apoptosis by the Ubiquitin-Proteasome-System. Cells 2021, 10, 3465. [Google Scholar] [CrossRef]
- Das, P.M.; Singal, R. DNA methylation and cancer. J. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef]
- Farias, E.F.; Petrie, K.; Leibovitch, B.; Murtagh, J.; Chornet, M.B.; Schenk, T.; Zelent, A.; Waxman, S. Interference with Sin3 function induces epigenetic reprogramming and differentiation in breast cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11811–11816. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yu, J.; Kane, M.A. Quantitation of the Noncovalent Cellular Retinol-Binding Protein, Type 1 Complex Through Native Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 29–37. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Perri, M.; Jones, J.W.; Pierzchalski, K.; Ceaicovscaia, N.; Cione, E.; Kane, M.A. Altered RBP1 Gene Expression Impacts Epithelial Cell Retinoic Acid, Proliferation, and Microenvironment. Cells 2022, 11, 792. https://doi.org/10.3390/cells11050792
Yu J, Perri M, Jones JW, Pierzchalski K, Ceaicovscaia N, Cione E, Kane MA. Altered RBP1 Gene Expression Impacts Epithelial Cell Retinoic Acid, Proliferation, and Microenvironment. Cells. 2022; 11(5):792. https://doi.org/10.3390/cells11050792
Chicago/Turabian StyleYu, Jianshi, Mariarita Perri, Jace W. Jones, Keely Pierzchalski, Natalia Ceaicovscaia, Erika Cione, and Maureen A. Kane. 2022. "Altered RBP1 Gene Expression Impacts Epithelial Cell Retinoic Acid, Proliferation, and Microenvironment" Cells 11, no. 5: 792. https://doi.org/10.3390/cells11050792
APA StyleYu, J., Perri, M., Jones, J. W., Pierzchalski, K., Ceaicovscaia, N., Cione, E., & Kane, M. A. (2022). Altered RBP1 Gene Expression Impacts Epithelial Cell Retinoic Acid, Proliferation, and Microenvironment. Cells, 11(5), 792. https://doi.org/10.3390/cells11050792