
E-Cigarette Aerosols Promote Oral S. aureus Colonization by Delaying an Immune Response and Bacterial Clearing



Abstract
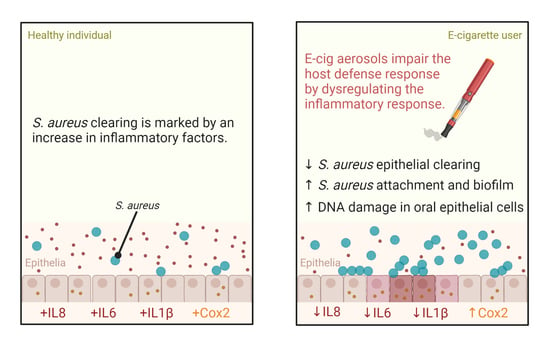
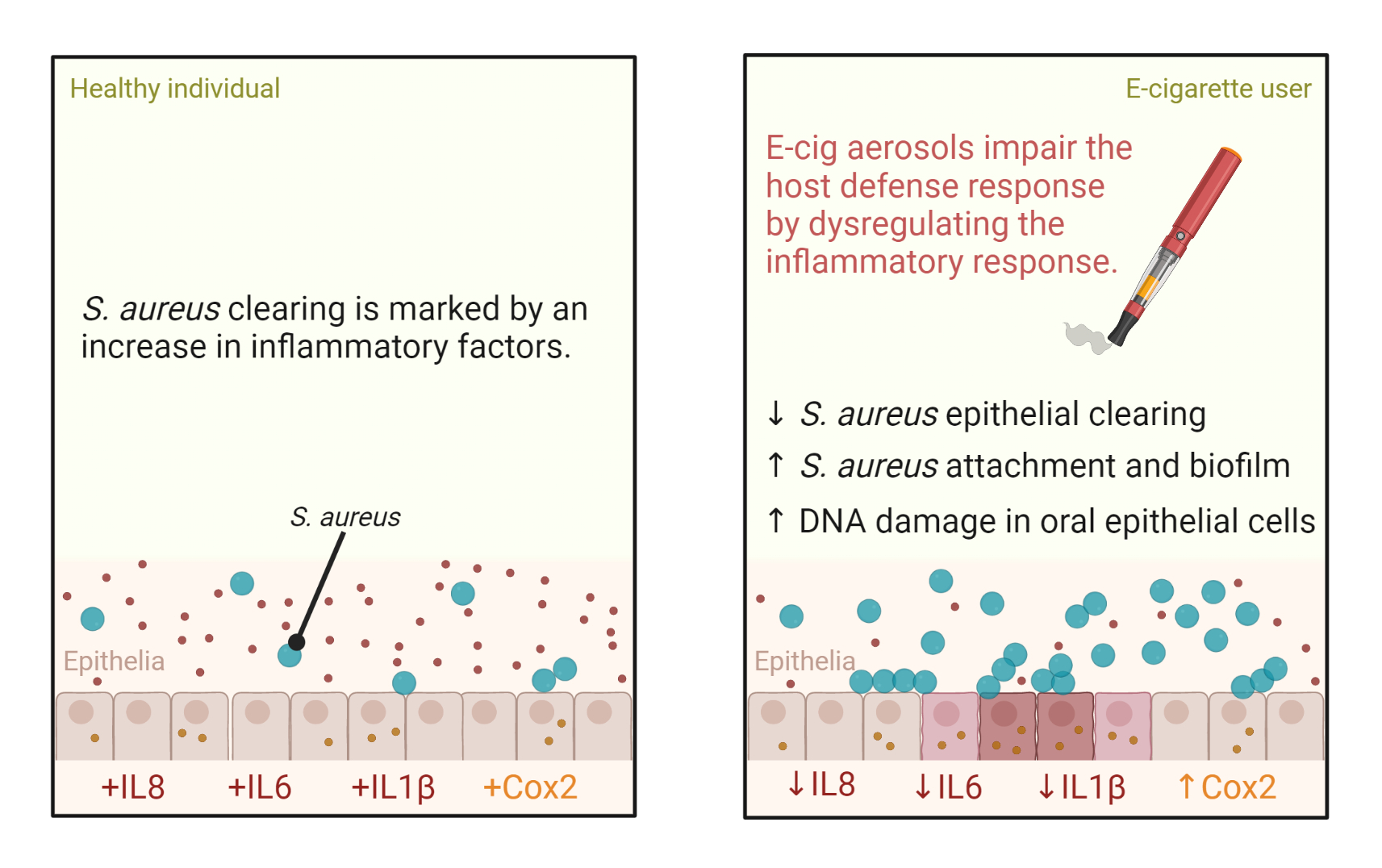
:
1. Introduction
2. Materials and Methods
2.1. Cultivation of Cell Lines
2.2. Bacterial Cultures
2.3. Electronic Cigarette Aerosol
2.4. Bacterial and Epithelial Cells Co-Culture
2.5. Epithelial and Bacterial Cell Viability
2.5.1. Cell Titer Blue
2.5.2. Click-iT EdU
2.5.3. BacTiter Glow
2.6. Attachment and Clearing Assays
2.7. Biofilm Formation
2.8. ELISA
2.9. qRT-PCR
2.10. Immunofluorescence Staining
2.11. Statistical Analyses
3. Results
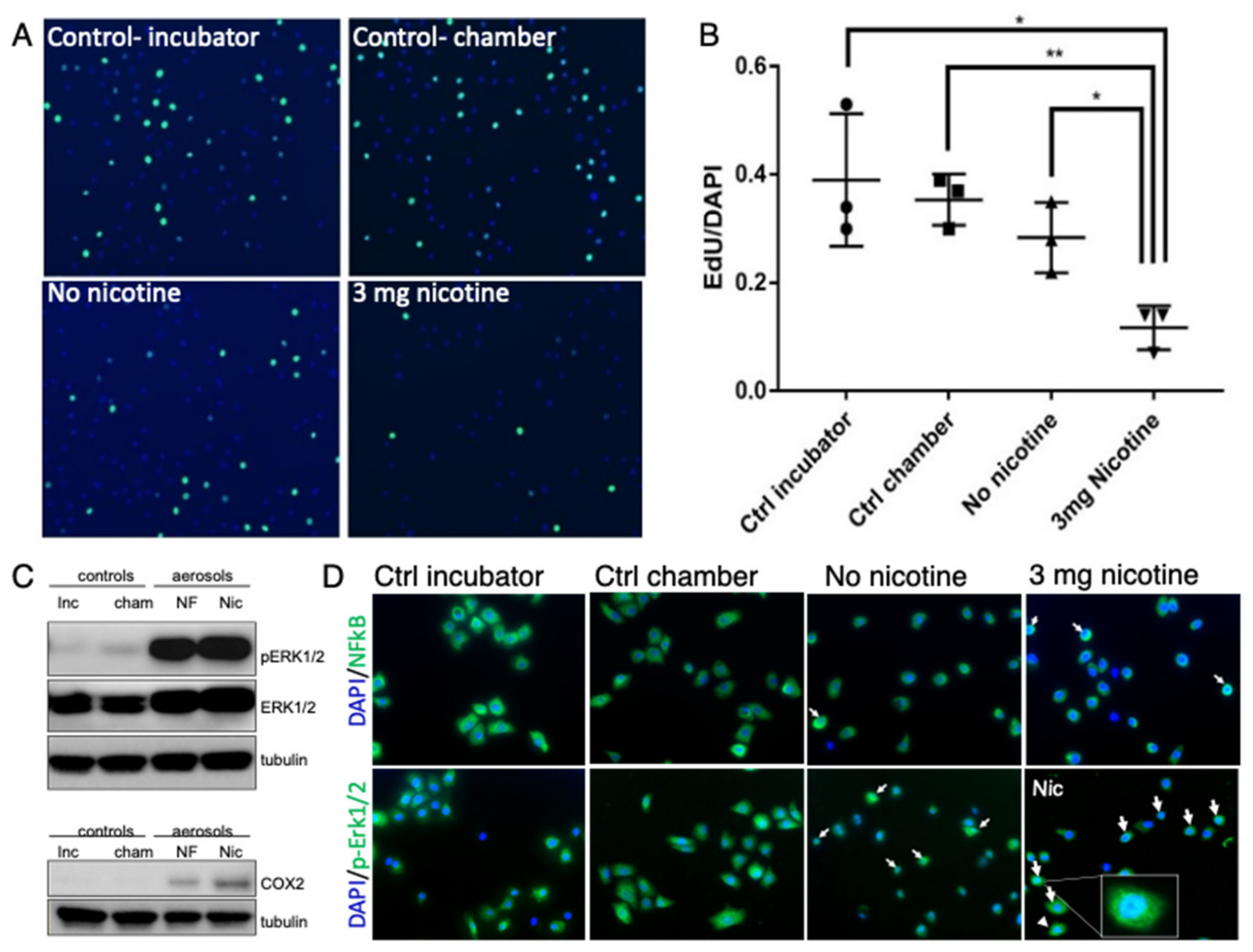
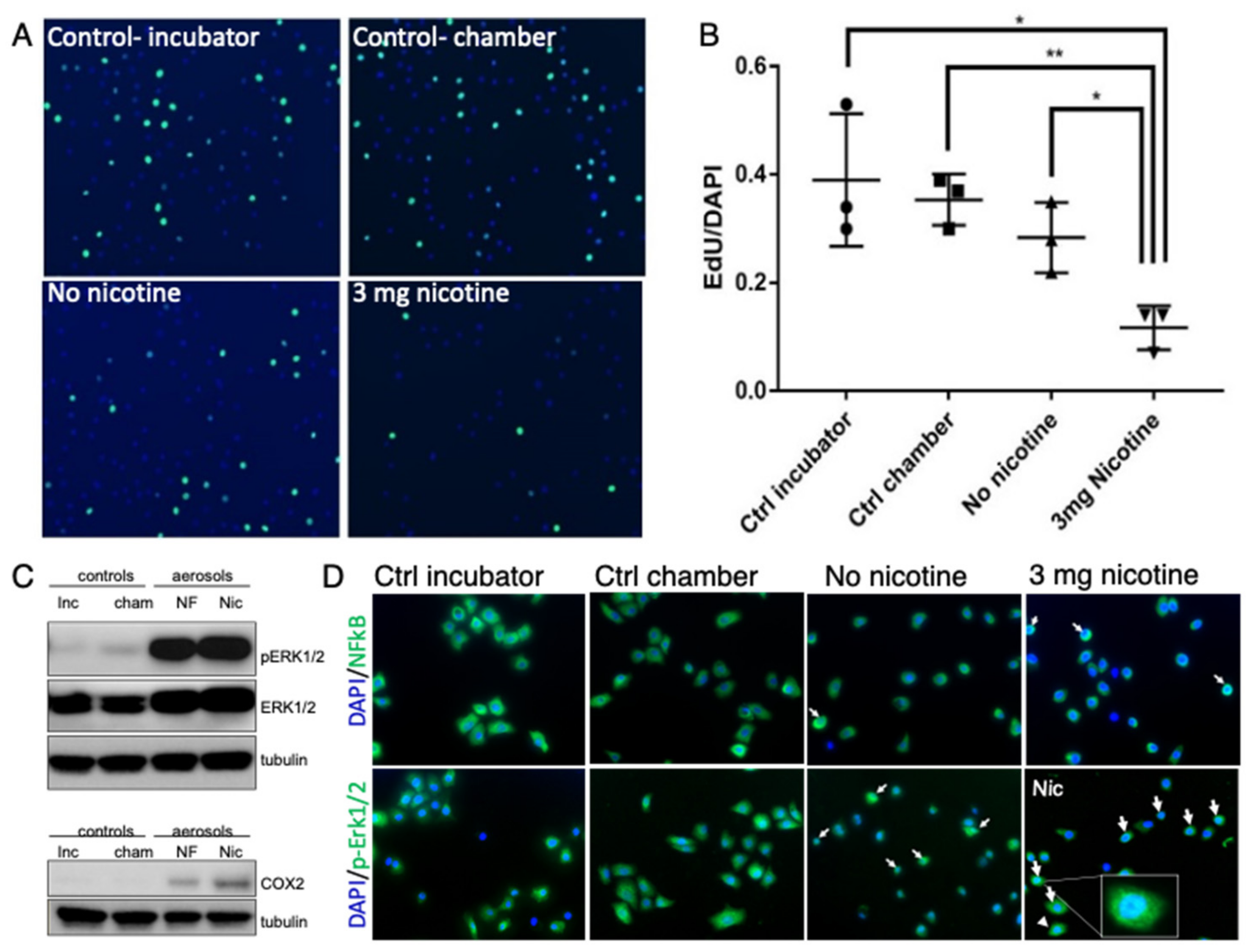
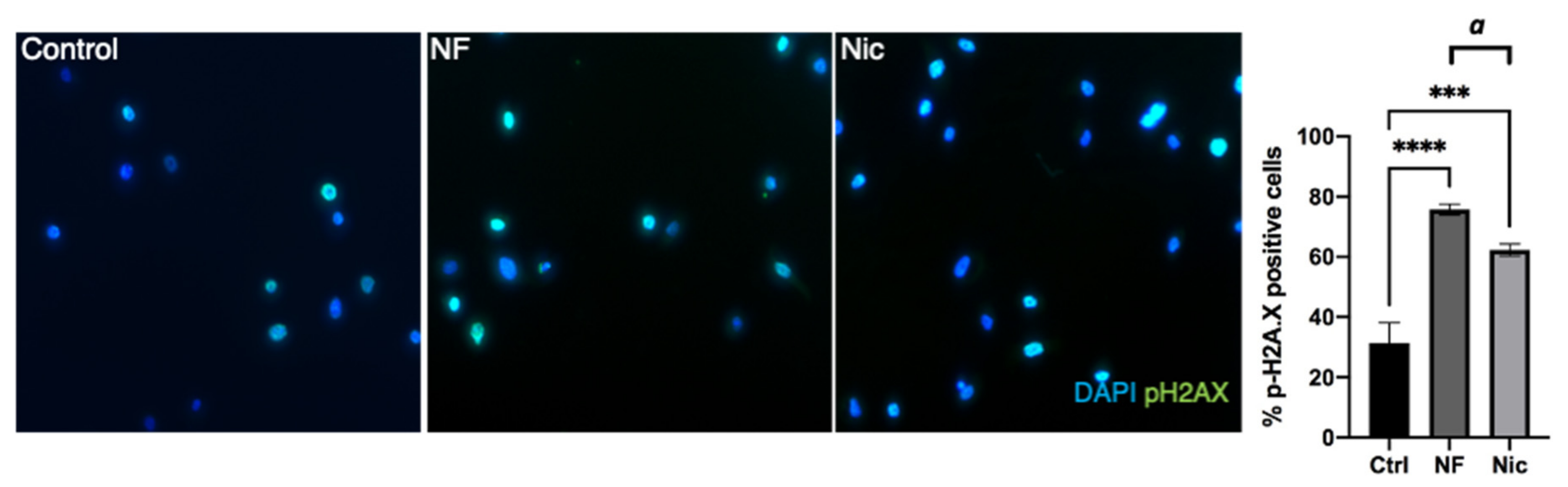
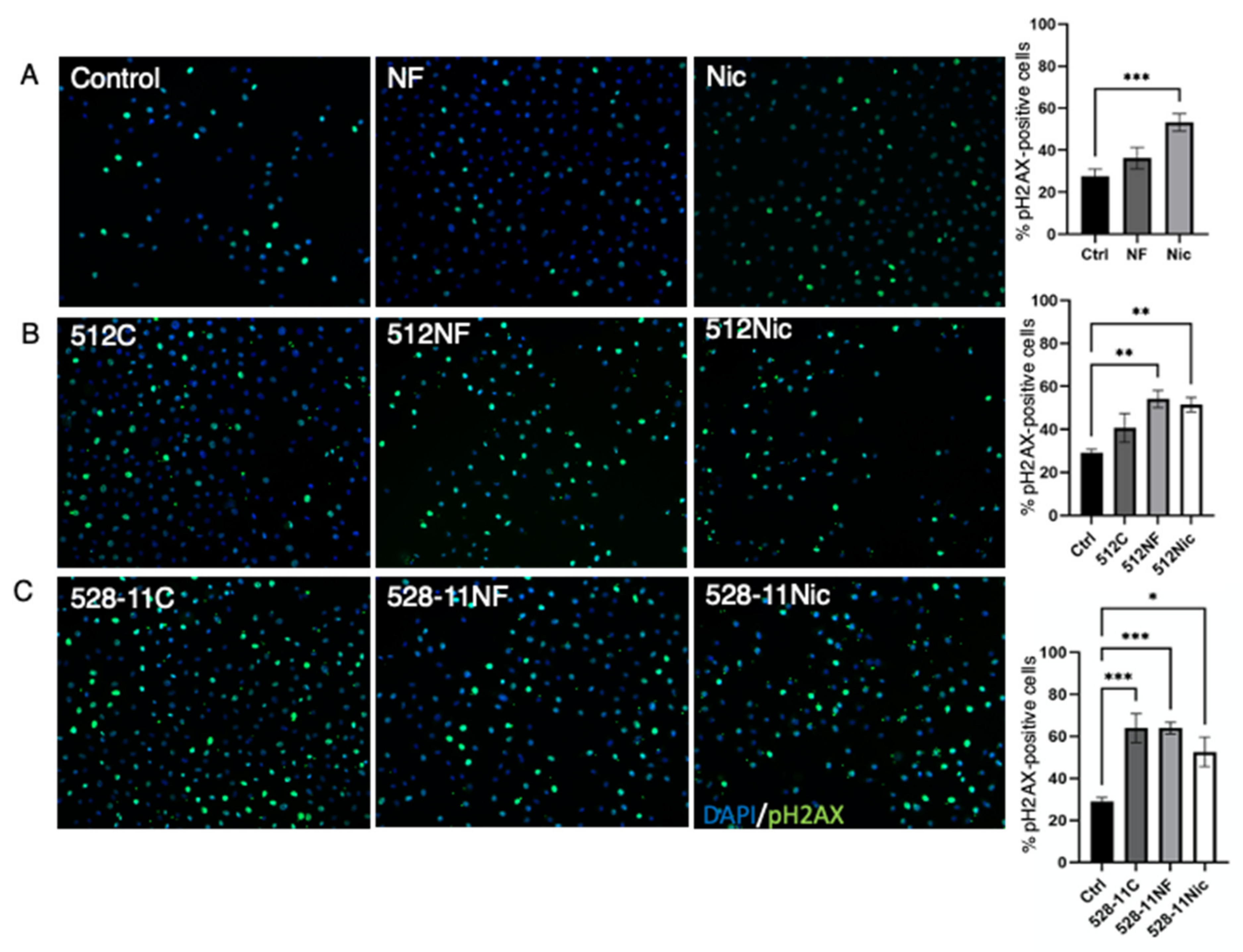
3.1. E-Cig Vape Activates Proinflammatory pERK½ and NF-KB Signaling and Induces DNA Damage
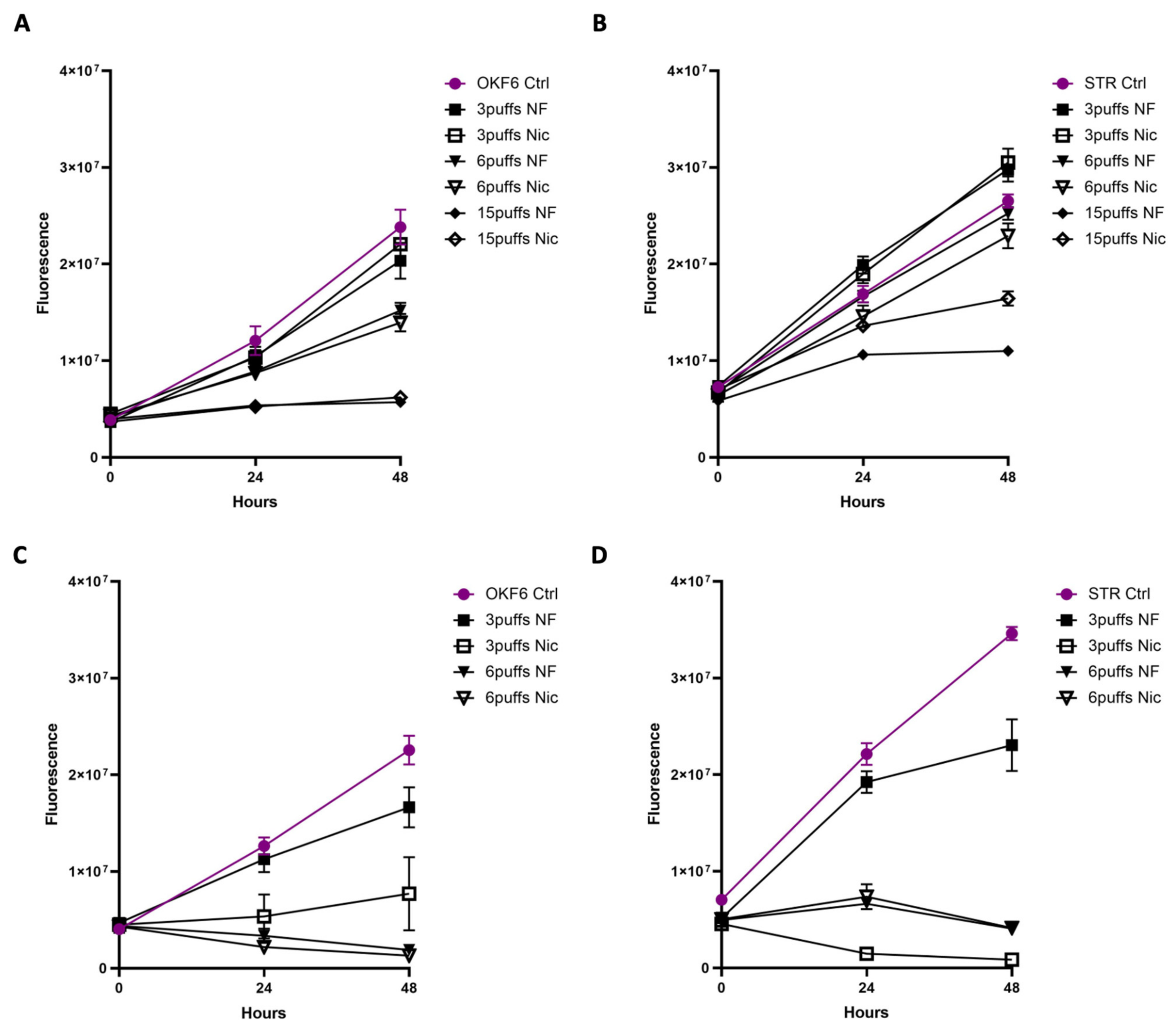
3.2. Acute Exposure to E-Cig Aerosol Decreases Epithelial Cell Viability in a Dose-Dependent Manner
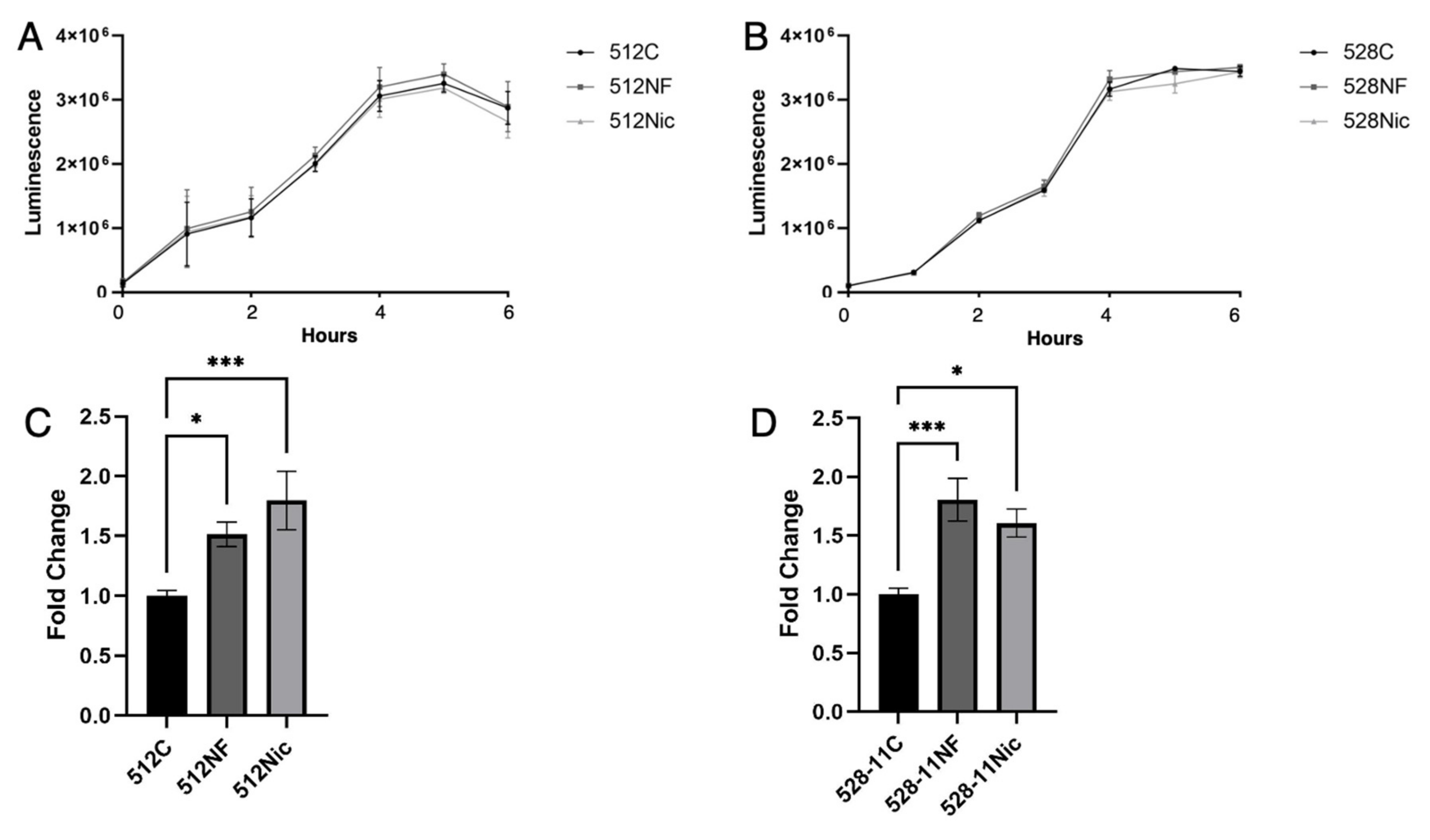
3.3. E-Cig Aerosols Do Not Alter S. aureus Growth but Promote Biofilm Formation and Attachment to Oral Cells Aiding in Oral Colonization
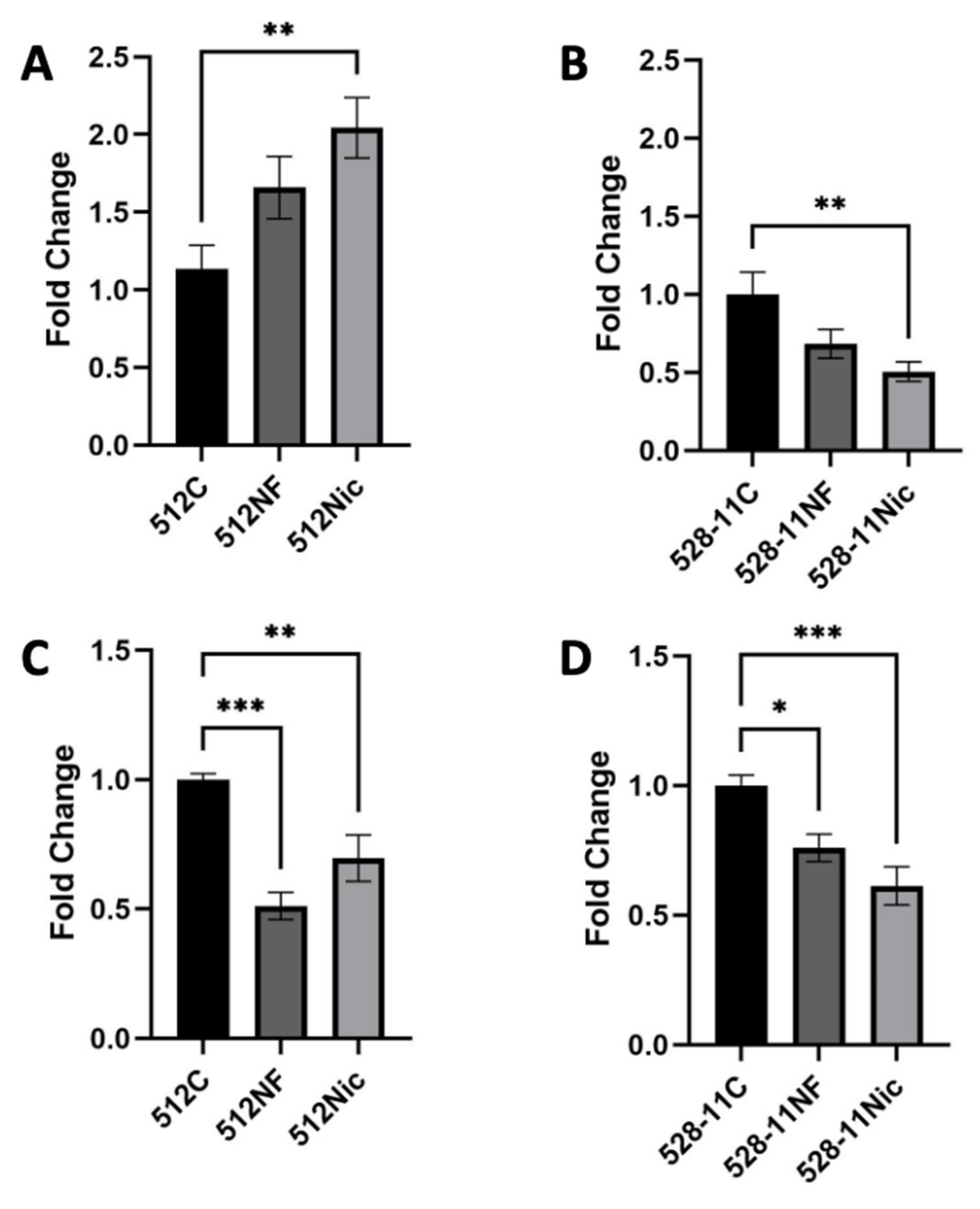
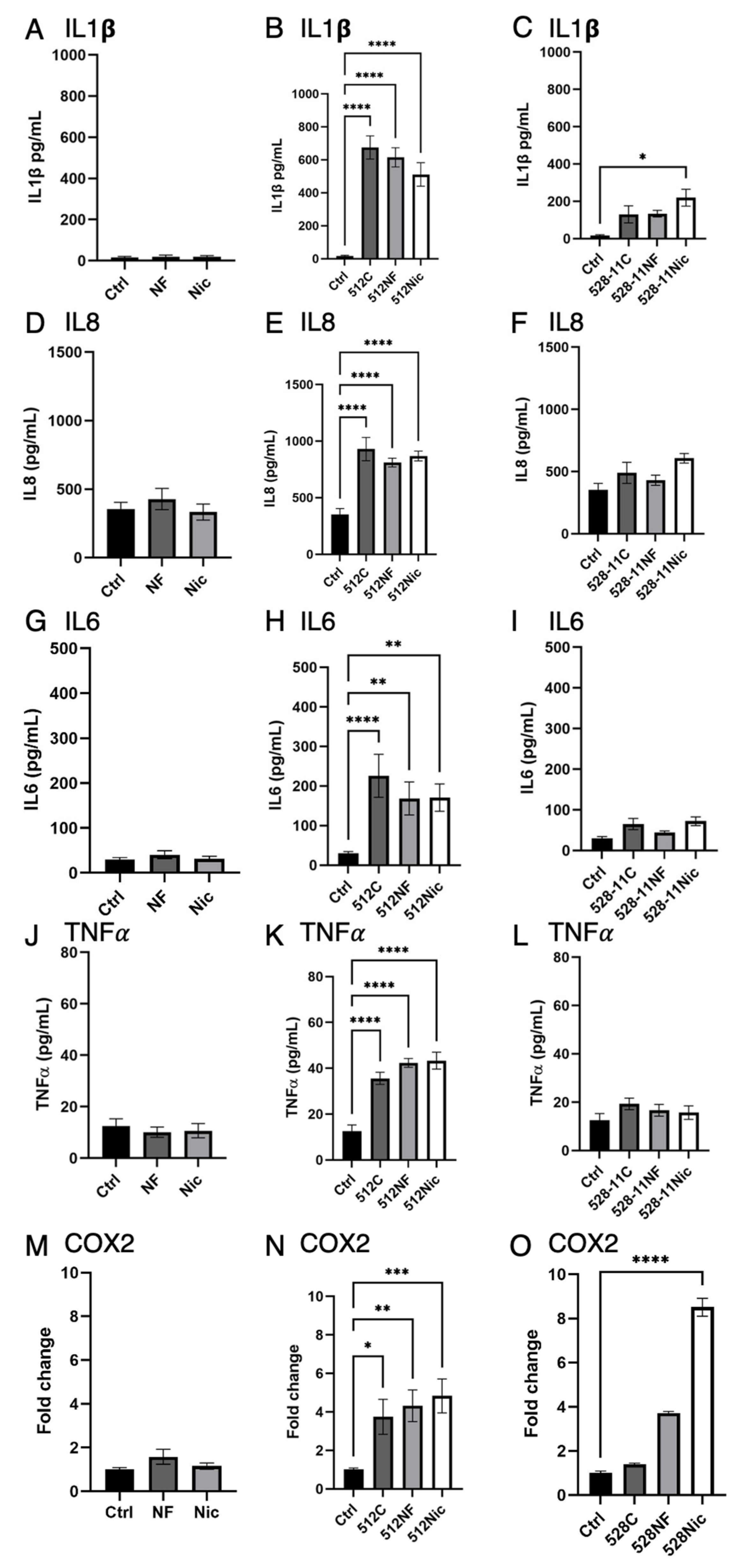
3.4. E-Cig Aerosol Decreased the Release of Proinflammatory Cytokines IL8, IL6, and IL1β
3.5. DNA Damage Is Further Enhanced in the Presence of E-Cig Aerosols and S. aureus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stanton, C.A.; Sharma, E.; Seaman, E.L.; A Kasza, K.; Edwards, K.C.; Halenar, M.J.; Taylor, K.A.; Day, H.; Anic, G.; Hull, L.C.; et al. Initiation of any tobacco and five tobacco products across 3 years among youth, young adults and adults in the USA: Findings from the PATH Study Waves 1–3 (2013–2016). Tob. Control. 2020, 29, s178–s190. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.W.; Gentzke, A.; Sharapova, S.; Cullen, K.A.; Ambrose, B.K.; Jamal, A. Tobacco Product Use Among Middle and High School Students—United States, 2011–2017. Morb. Mortal. Wkly. Rep. 2018, 67, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Grana, R.A.; Ling, P.M. Smoking Revolution A Content Analysis of Electronic Cigarette Retail Websites. Am. J. Prev. Med. 2014, 46, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.; Sandeep, S.; Rodriguez, J. The oral health impact of electronic cigarette use: A systematic review. Crit. Rev. Toxicol. 2020, 50, 97–127. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, G.M.; da Silva, H.F.; de Oliveira Campos, I.J.; de Medeiros, E.R.; Colares, D.F.; de Oliveira Guedes, T.Y.; da Cruz Lima, J.G.; Leite, R.B. Assessment of oral changes from the use of electronic cigarettes: Literature review. Res. Soc. Dev. 2021, 10, e14410212146. [Google Scholar] [CrossRef]
- Javed, F.; Kellesarian, S.V.; Sundar, I.K.; Romanos, G.E.; Rahman, I. Recent updates on electronic cigarette aerosol and inhaled nicotine effects on periodontal and pulmonary tissues. Oral Dis. 2017, 23, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Vora, M.V.; Chaffee, B.W. Tobacco-use patterns and self-reported oral health outcomes: A cross-sectional assessment of the Population Assessment of Tobacco and Health study (2013–2014). J. Am. Dent. Assoc. 2019, 176, 139–148. [Google Scholar] [CrossRef]
- Iskandar, A.R.; Zanetti, F.; Kondylis, A.; Martin, F.; Leroy, P.; Majeed, S.; Steiner, S.; Xiang, Y.; Torres, L.O.; Trivedi, K.; et al. A lower impact of an acute exposure to electronic cigarette aerosols than to cigarette smoke in human organotypic buccal and small airway cultures was demonstrated using systems toxicology assessment. Intern. Emerg. Med. 2019, 14, 863–883. [Google Scholar] [CrossRef] [Green Version]
- Lopez, A.A.; Hiler, M.M.; Soule, E.K.; Ramôa, C.P.; Karaoghlanian, N.V.; Lipato, T.; Breland, A.B.; Shihadeh, A.L.; Eissenberg, T. Effects of Electronic Cigarette Liquid Nicotine Concentration on Plasma Nicotine and Puff Topography in Tobacco Cigarette Smokers: A Preliminary Report. Nicotine Tob. Res. 2015, 18, 720–723. [Google Scholar] [CrossRef] [Green Version]
- Goniewicz, M.L.; Knysak, J.; Gawron, M.; Kosmider, L.; Sobczak, A.; Kurek, J.; Prokopowicz, A.; Jablonska-Czapla, M.; Rosik-Dulewska, C.; Havel, C.; et al. Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tob. Control 2013, 23, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Yalcin, E.; de la Monte, S. Tobacco nitrosamines as culprits in disease: Mechanisms reviewed. J. Physiol. Biochem. 2016, 165, 255–269. [Google Scholar] [CrossRef]
- Lee, Y.O.; Nonnemaker, J.M.; Bradfield, B.B.; Hensel, E.; Robinson, R.J. Examining Daily Electronic Cigarette Puff Topography Among Established and Nonestablished Cigarette Smokers in their Natural Environment. Nicotine Tob. Res. 2017, 20, 1283–1288. [Google Scholar] [CrossRef]
- Yu, M.A.; Kiang, A.; Wang-Rodriguez, J.; Rahimy, E.; Haas, M.; Yu, V.; Ellies, L.G.; Chen, J.; Fan, J.-B.; Brumund, K.T.; et al. Nicotine Promotes Acquisition of Stem Cell and Epithelial-to-Mesenchymal Properties in Head and Neck Squamous Cell Carcinoma. PLoS ONE 2012, 7, e51967. [Google Scholar] [CrossRef] [Green Version]
- Ganapathy, V.; Manyanga, J.; Brame, L.; McGuire, D.; Sadhasivam, B.; Floyd, E.; Rubenstein, D.; Ramachandran, I.; Wagener, T.; Queimado, L. Electronic cigarette aerosols suppress cellular antioxidant defenses and induce significant oxidative DNA damage. PLoS ONE 2017, 12, e0177780. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.W.; Karakasheva, T.A.; Hicks, P.D.; Bass, A.J.; Rustgi, A.K. The tumor microenvironment in esophageal cancer. Oncogene 2016, 35, 5337–5349. [Google Scholar] [CrossRef]
- Bustamante, G.; Ma, B.; Yakovlev, G.; Yershova, K.; Callejas, G.B.; Jensen, J.; Hatsukami, D.K.; Stepanov, I. Presence of the Carcinogen N′-Nitrosonornicotine in Saliva of E-cigarette Users. Chem. Res. Toxicol. 2018, 31, 731–738. [Google Scholar] [CrossRef]
- Xue, J.; Yang, S.; Seng, S. Mechanisms of Cancer Induction by Tobacco-Specific NNK and NNN. Cancers 2014, 6, 1138–1156. [Google Scholar] [CrossRef] [Green Version]
- Hanioka, T.; Morita, M.; Yamamoto, T.; Inagaki, K.; Wang, P.-L.; Ito, H.; Morozumi, T.; Takeshita, T.; Suzuki, N.; Shigeishi, H.; et al. Smoking and periodontal microorganisms. Jpn. Dent. Sci. Rev. 2019, 55, 88–94. [Google Scholar] [CrossRef]
- Pushalkar, S.; Paul, B.; Li, Q.; Yang, J.; Vasconcelos, R.; Makwana, S.; González, J.M.; Shah, S.; Xie, C.; Janal, M.N.; et al. Electronic Cigarette Aerosol Modulates the Oral Microbiome and Increases Risk of Infection. iScience 2020, 23, 100884. [Google Scholar] [CrossRef] [Green Version]
- Nalawade, T.M.; Sogi, S.H.P.; Bhat, K. Bactericidal activity of propylene glycol, glycerine, polyethylene glycol 400, and polyethylene glycol 1000 against selected microorganisms. J. Int. Soc. Prev. Community Dent. 2015, 5, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.H.; Lyes, M.; Sladewski, K.; Enany, S.; McEachern, E.; Mathew, D.P.; Das, S.; Moshensky, A.; Bapat, S.; Pride, D.T.; et al. Electronic cigarette inhalation alters innate immunity and airway cytokines while increasing the virulence of colonizing bacteria. Klin. Wochenschr. 2016, 94, 667–679. [Google Scholar] [CrossRef]
- Crotty Alexander, L.E.; Drummond, C.A.; Hepokoski, M.; Mathew, D.; Moshensky, A.; Willeford, A.; Das, S.; Singh, P.; Yong, Z.; Lee, J.H.; et al. Chronic inhalation of e-cigarette vapor containing nicotine disrupts airway barrier function and induces systemic inflammation and multiorgan fibrosis in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, 834–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Striz, I.; Brabcova, E.; Kolesar, L.; Sekerkova, A. Cytokine networking of innate immunity cells: A potential target of therapy. Clin. Sci. 2014, 126, 593–612. [Google Scholar] [CrossRef]
- Korniluk, A.; Koper-Lenkiewicz, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Ir. J. Med Sci. 2016, 186, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Fritschi, B.Z.; Albert-Kiszely, A.; Persson, G.R. Staphylococcus aureus and Other Bacteria in Untreated Periodontitis. J. Dent. Res. 2008, 87, 589–593. [Google Scholar] [CrossRef]
- Uribe-García, A.; Paniagua-Contreras, G.L.; Monroy-Pérez, E.; Bustos-Martínez, J.; Hamdan-Partida, A.; Garzón, J.; Alanís, J.; Quezada, R.; Vaca-Paniagua, F.; Vaca, S. Frequency and expression of genes involved in adhesion and biofilm formation in Staphylococcus aureus strains isolated from periodontal lesions. J. Microbiol. Immunol. Infect. 2019, 54, 267–275. [Google Scholar] [CrossRef]
- Passariello, C.; Lucchese, A.; Virga, A.; Pera, F.; Gigola, P. Isolation of Staphylococcus aureus and Progression of Periodontal Lesions in Aggressive Periodontitis. Eur. J. Inflamm. 2012, 10, 501–513. [Google Scholar] [CrossRef]
- Dickson, M.A.; Hahn, W.C.; Ino, Y.; Ronfard, V.; Wu, J.Y.; Weinberg, R.A.; Louis, D.N.; Li, F.P.; Rheinwald, J.G. Human Keratinocytes That Express hTERT and Also Bypass a p16 INK4a-Enforced Mechanism That Limits Life Span Become Immortal yet Retain Normal Growth and Differentiation Characteristics. Mol. Cell. Biol. 2000, 20, 1436–1447. [Google Scholar] [CrossRef] [Green Version]
- Harada, H.; Nakagawa, H.; Oyama, K.; Takaoka, M.; Andl, C.D.; Jacobmeier, B.; Von Werder, A.; Enders, G.H.; Opitz, O.G.; Rustgi, A.K. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol. Cancer Res. 2003, 1, 729–738. [Google Scholar]
- Cole, A.L.; Sundar, M.; Lopez, A.; Forsman, A.; Yooseph, S.; Cole, A.M. Identification of Nasal Gammaproteobacteria with Potent Activity against Staphylococcus aureus: Novel Insights into the “Noncarrier” State. Msphere 2021, 6, e01015-20. [Google Scholar] [CrossRef]
- Passariello, C.; Puttini, M.; Iebba, V.; Pera, P.; Gigola, P. Influence of oral conditions on colonization by highly toxigenic Staphylococcus aureus strains. Oral Dis. 2012, 18, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Letourneau, J.; Levesque, C.; Berthiaume, F.; Jacques, M.; Mourez, M. In Vitro Assay of Bacterial Adhesion onto Mammalian Epithelial Cells. J. Vis. Exp. 2011, e2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merritt, J.H.; Kadouri, D.E.; O’Toole, G.A. Growing and Analyzing Static Biofilms. Curr. Protoc. Microbiol. 2005, 22, 1B.1.1–1B.1.17. [Google Scholar] [CrossRef] [Green Version]
- Sundar, I.K.; Javed, F.; Romanos, G.E.; Rahman, I. E-cigarettes and flavorings induce inflammatory and pro-senescence responses in oral epithelial cells and periodontal fibroblasts. Oncotarget 2016, 7, 77196–77204. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Arcos, I.; Geraghty, P.; Baumlin, N.; Campos, M.; Dabo, A.J.; Jundi, B.; Cummins, N.; Eden, E.; Grosche, A.; Salathe, M.; et al. Chronic electronic cigarette exposure in mice induces features of COPD in a nicotine-dependent manner. Thorax 2016, 71, 1119–1129. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000, 10, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Collins, P.L.; Purman, C.; Porter, S.I.; Nganga, V.; Saini, A.; Hayer, K.E.; Gurewitz, G.L.; Sleckman, B.P.; Bednarski, J.J.; Bassing, C.H.; et al. DNA double-strand breaks induce H2Ax phosphorylation domains in a contact-dependent manner. Nat. Commun. 2020, 11, 3158. [Google Scholar] [CrossRef]
- Whittaker, C.J.; Klier, C.M.; Kolenbrander, P.E. MECHANISMS OF ADHESION BY ORAL BACTERIA. Annu. Rev. Microbiol. 1996, 50, 513–552. [Google Scholar] [CrossRef]
- Cole, A.M.; Muthukrishnan, G.; Chong, C.; Beavis, A.; Eade, C.R.; Wood, M.P.; Deichen, M.G. Host innate inflammatory factors and staphylococcal protein A influence the duration of human Staphylococcus aureus nasal carriage. Mucosal Immunol. 2016, 9, 1537–1548. [Google Scholar] [CrossRef]
- Goradel, N.H.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell. Physiol. 2018, 234, 5683–5699. [Google Scholar] [CrossRef]
- Misra, S.; Sharma, K. COX-2 signaling and cancer: New players in old arena. Curr. Drug Targets 2014, 15, 347–359. [Google Scholar] [CrossRef]
- Nasry, W.H.S.; Rodriguez-Lecompte, J.C.; Martin, C.K. Role of COX-2/PGE2 Mediated Inflammation in Oral Squamous Cell Carcinoma. Cancers 2018, 10, 348. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-W.; Park, S.-H.; Weng, M.-W.; Wang, H.-T.; Huang, W.; Lepor, H.; Wu, X.-R.; Chen, L.C.; Tang, M.-S. E-cigarette smoke damages DNA and reduces repair activity in mouse lung, heart, and bladder as well as in human lung and bladder cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1560–E1569. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.L.; Schmidt-Owens, M.; Beavis, A.C.; Chong, C.F.; Tarwater, P.M.; Schaus, J.; Deichen, M.G.; Cole, A.M. Cessation from Smoking Improves Innate Host Defense and Clearance of Experimentally Inoculated Nasal Staphylococcus aureus. Infect. Immun. 2018, 86, e00912-17. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, E.G.; Cavallo, I.; Bordignon, V.; Prignano, G.; Sperduti, I.; Gurtner, A.; Trento, E.; Toma, L.; Pimpinelli, F.; Capitanio, B.; et al. Inflammatory cytokines and biofilm production sustain Staphylococcus aureus outgrowth and persistence: A pivotal interplay in the pathogenesis of Atopic Dermatitis. Sci. Rep. 2018, 8, 9573. [Google Scholar] [CrossRef]
- Bien, J.; Sokolova, O.; Bozko, P. Characterization of Virulence Factors of Staphylococcus aureus: Novel Function of Known Virulence Factors That Are Implicated in Activation of Airway Epithelial Proinflammatory Response. J. Pathog. 2011, 2011, 601905. [Google Scholar] [CrossRef] [Green Version]
- Hooper, S.J.; Crean, S.J.; Lewis, M.A.O.; Spratt, D.A.; Wade, W.G.; Wilson, M.J. Viable Bacteria Present within Oral Squamous Cell Carcinoma Tissue. J. Clin. Microbiol. 2006, 44, 1719–1725. [Google Scholar] [CrossRef] [Green Version]
- Hooper, S.J.; Wilson, M.J.; Crean, S.J. Exploring the link between microorganisms and oral cancer: A systematic review of the literature. Head Neck 2009, 31, 1228–1239. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, S.; Li, B.; Jiang, Y.; Zhou, X.; Chen, J.; Li, M.; Ren, B.; Peng, X.; Zhou, X.; et al. Staphylococcus aureus induces COX-2-dependent proliferation and malignant transformation in oral keratinocytes. J. Oral Microbiol. 2019, 11, 1643205. [Google Scholar] [CrossRef] [Green Version]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.-E.; Malki, M.I.; Alhmoud, J.F. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef]
- Banáth, J.P.; Klokov, D.; MacPhail, S.H.; Banuelos, C.A.; Olive, P.L. Residual γH2AX foci as an indication of lethal DNA lesions. BMC Cancer 2010, 10, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar] [CrossRef]
- Leung, E.Y.; McMahon, J.D.; McLellan, D.R.; Syyed, N.; E McCarthy, C.; Nixon, C.; Orange, C.; Brock, C.; Hunter, K.D.; Adams, P.D. DNA damage marker phosphorylated histone H2AX is a potential predictive marker for progression of epithelial dysplasia of the oral cavity. Histopathology 2017, 71, 522–528. [Google Scholar] [CrossRef]
- Neville, B.W.; Day, T.A. Oral Cancer and Precancerous Lesions. CA Cancer J. Clin. 2002, 52, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Mirghani, H.; Amen, F.; Moreau, F.; Guily, J.L.S. Do high-risk human papillomaviruses cause oral cavity squamous cell carcinoma? Oral Oncol. 2015, 51, 229–236. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | 5′ to 3′ Forward | 5′ to 3′ Reverse |
|---|---|---|
| IL1β | GGA GAT TCG TAG CTG GAT GC | GAG CTC GCC AGT GAA ATG AT |
| COX2 | TGA GCA TCT ACG GTT TGC TG | TGC TTG TCT GGA ACA ACT GC |
| IL8 | CCT GAT TTC TGC AGC TCT GTG | CCA GAC AGA GCT CTC TTC CAT |
| TNFα | ACA AGC CTG TAG CCC ATG TT | AAA GTA GAC CTG CCC AGA CT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cátala-Valentín, A.R.; Almeda, J.; Bernard, J.N.; Cole, A.M.; Cole, A.L.; Moore, S.D.; Andl, C.D. E-Cigarette Aerosols Promote Oral S. aureus Colonization by Delaying an Immune Response and Bacterial Clearing. Cells 2022, 11, 773. https://doi.org/10.3390/cells11050773
Cátala-Valentín AR, Almeda J, Bernard JN, Cole AM, Cole AL, Moore SD, Andl CD. E-Cigarette Aerosols Promote Oral S. aureus Colonization by Delaying an Immune Response and Bacterial Clearing. Cells. 2022; 11(5):773. https://doi.org/10.3390/cells11050773
Chicago/Turabian StyleCátala-Valentín, Alma R., Jasmine Almeda, Joshua N. Bernard, Alexander M. Cole, Amy L. Cole, Sean D. Moore, and Claudia D. Andl. 2022. "E-Cigarette Aerosols Promote Oral S. aureus Colonization by Delaying an Immune Response and Bacterial Clearing" Cells 11, no. 5: 773. https://doi.org/10.3390/cells11050773
APA StyleCátala-Valentín, A. R., Almeda, J., Bernard, J. N., Cole, A. M., Cole, A. L., Moore, S. D., & Andl, C. D. (2022). E-Cigarette Aerosols Promote Oral S. aureus Colonization by Delaying an Immune Response and Bacterial Clearing. Cells, 11(5), 773. https://doi.org/10.3390/cells11050773