
Repeated Social Defeat Enhances CaCl2-Induced Abdominal Aortic Aneurysm Expansion by Inhibiting the Early Fibrotic Response via the MAPK-MKP-1 Pathway

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Repeated Social Defeat
2.2. Behavior Analysis
2.3. Mouse Aneurysm Model
2.4. Hemodynamic Analysis
2.5. Serum Concentration of Corticosterone
2.6. Aneurysm Measurement and Histological Analysis
2.7. Immunohistochemical Analysis
2.8. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
2.9. Ex Vivo MMP Activity
2.10. Primary Culture of VSMCs in the Thoracic Aorta
2.11. Western Blot Analysis
2.12. BrdU Assay
2.13. Statistical Analysis
3. Results
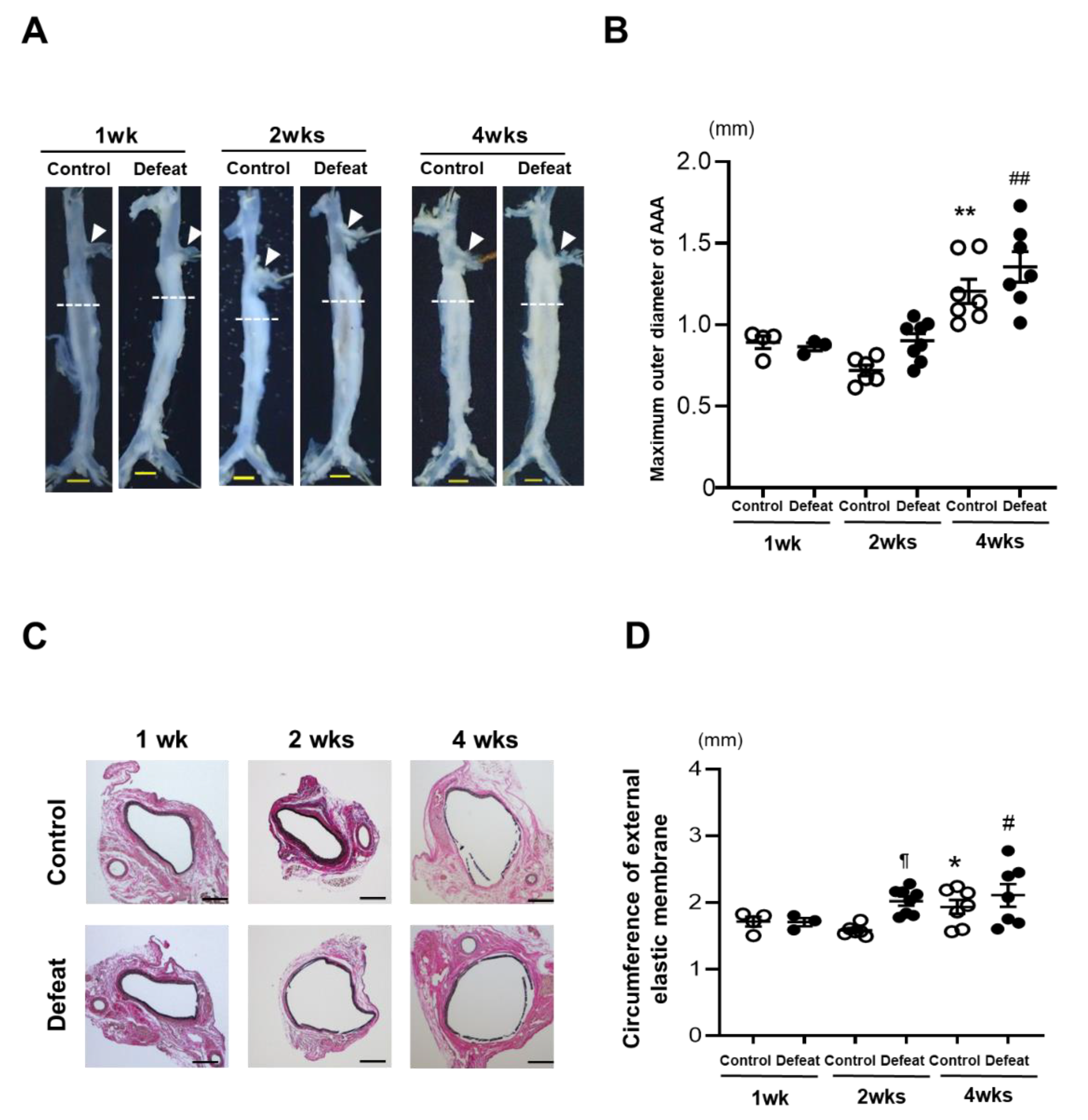
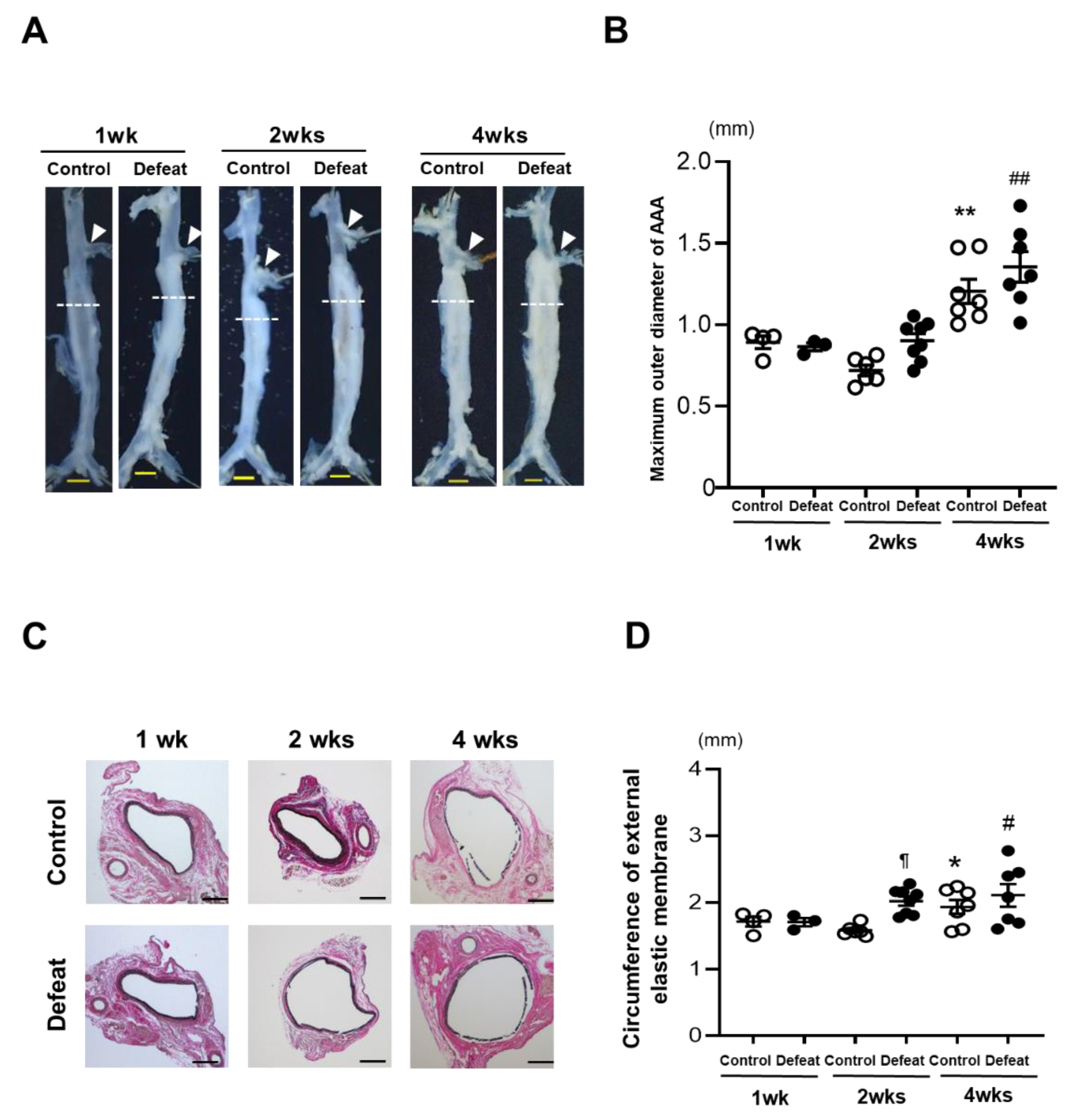
3.1. Development of AAA Is Promoted in Defeated Mice
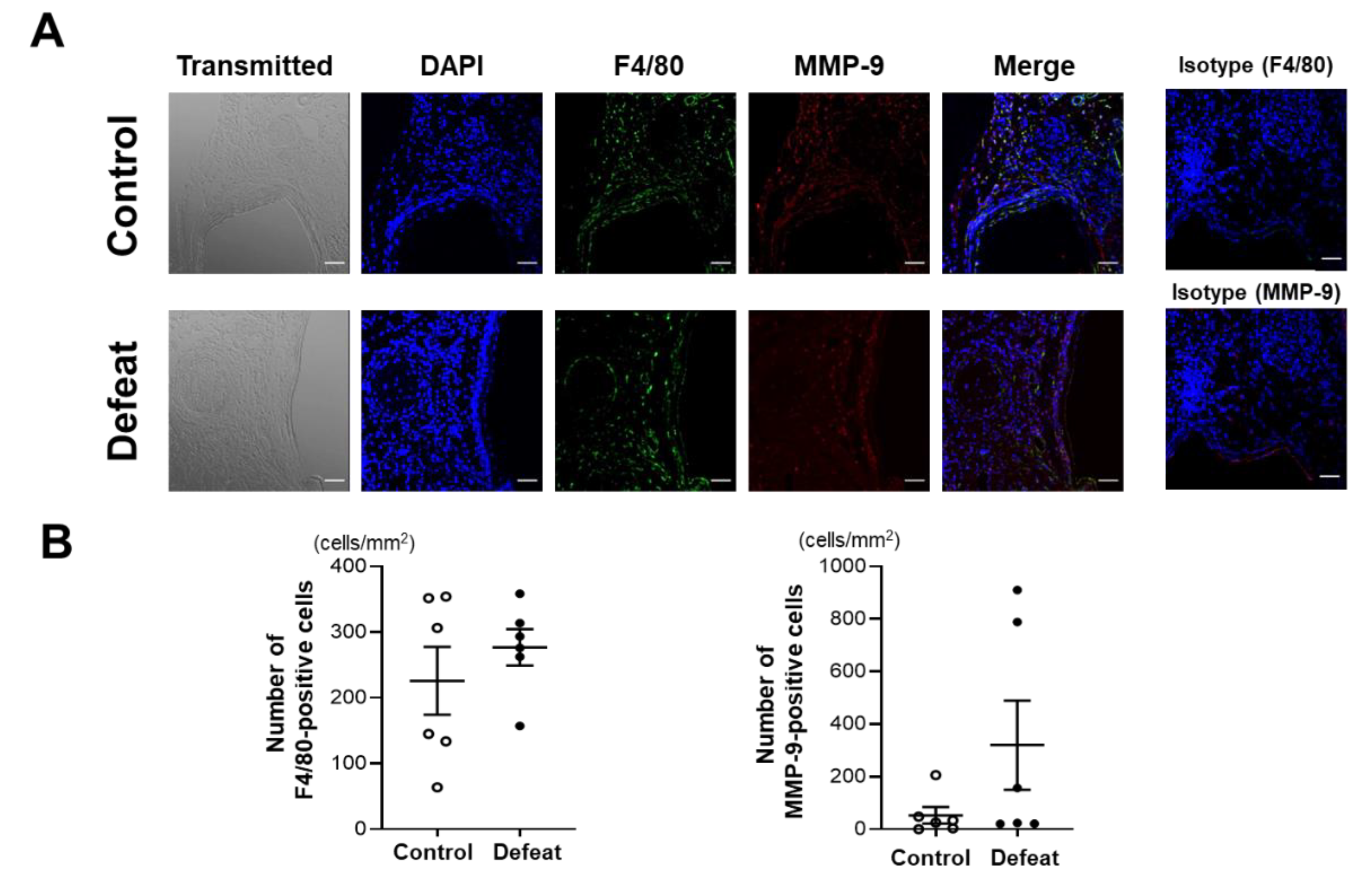
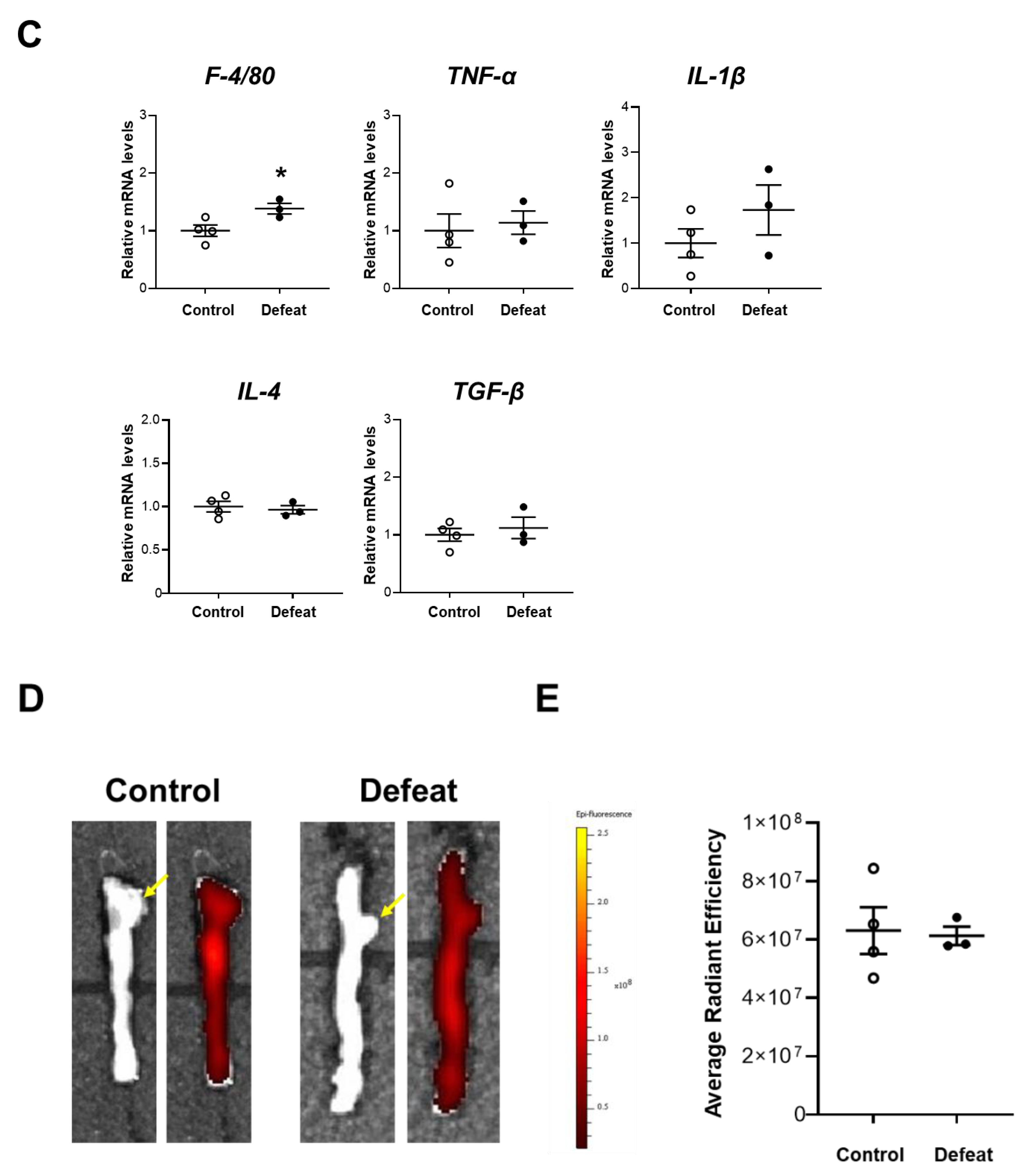
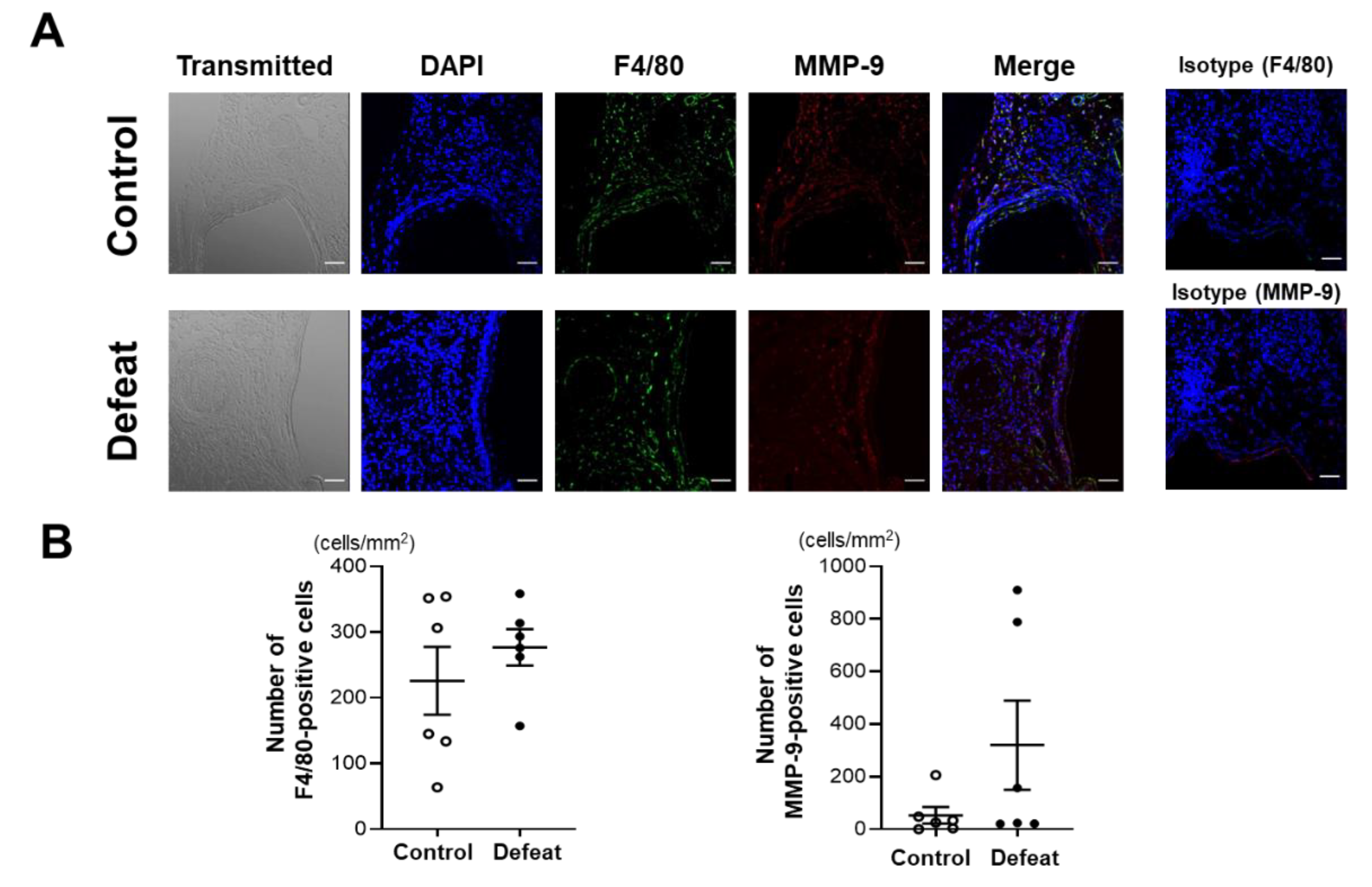
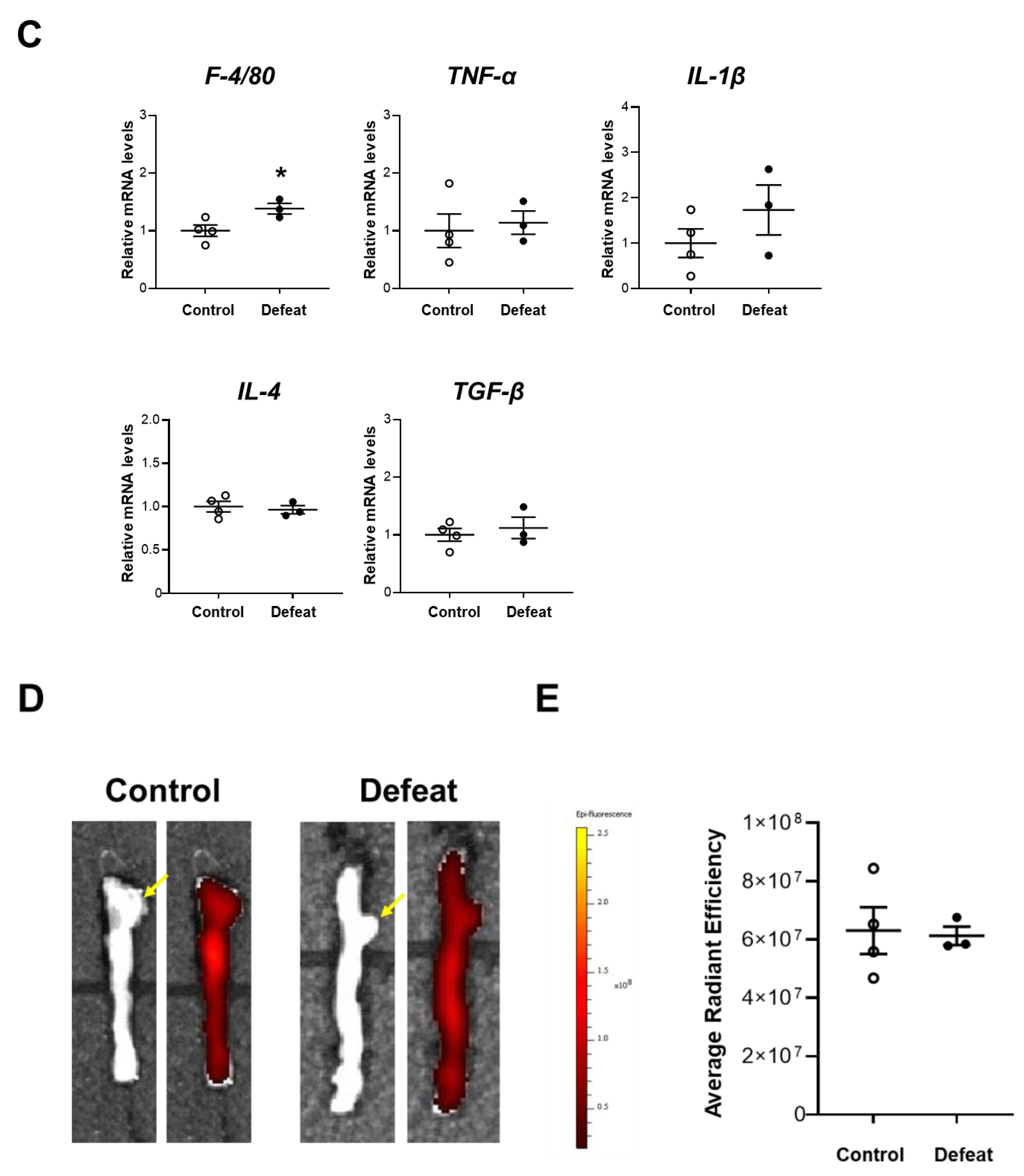
3.2. Repeated Social Defeat Does Not Enhance the Acute Inflammatory Response after CaCl2 Application
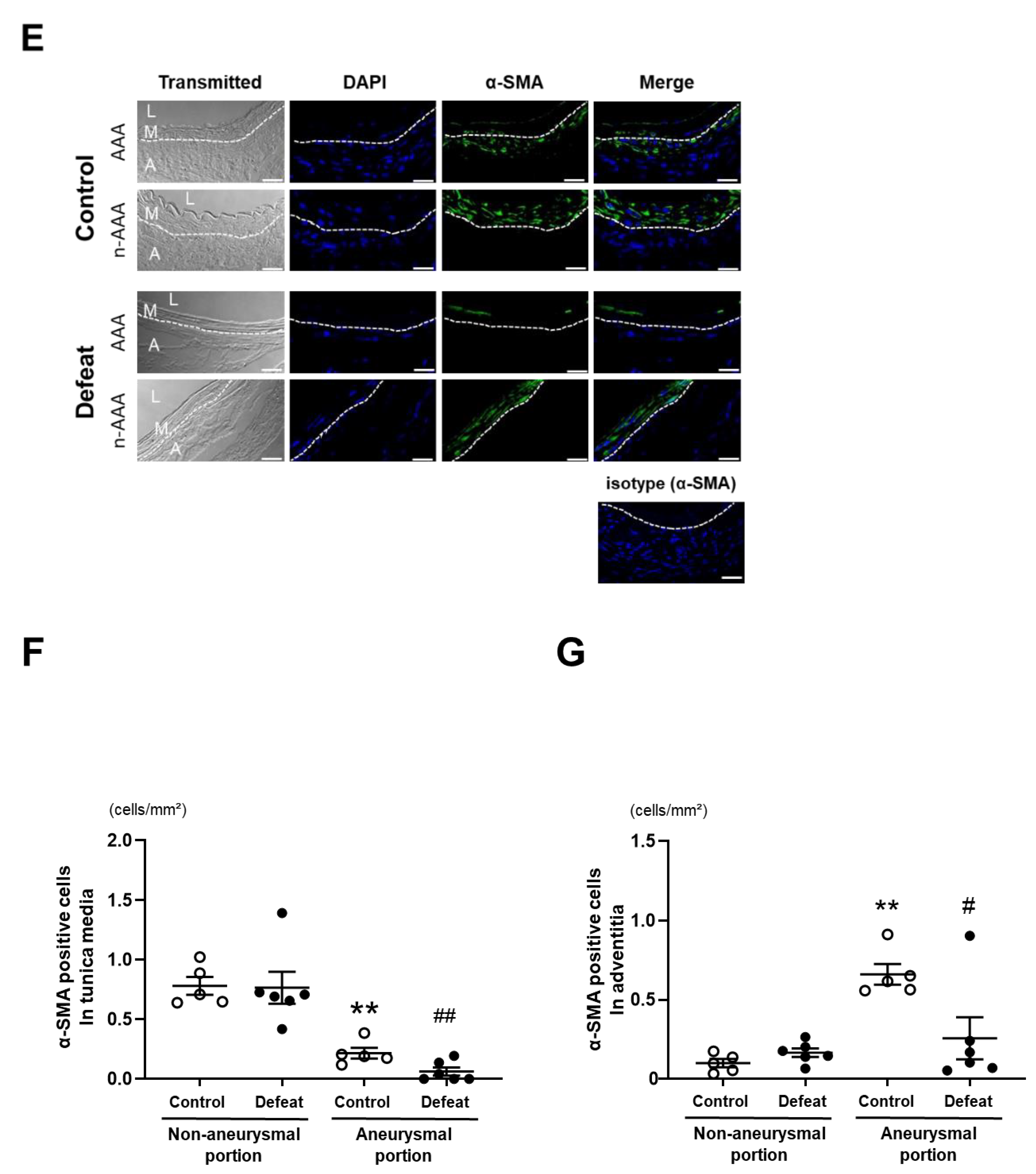
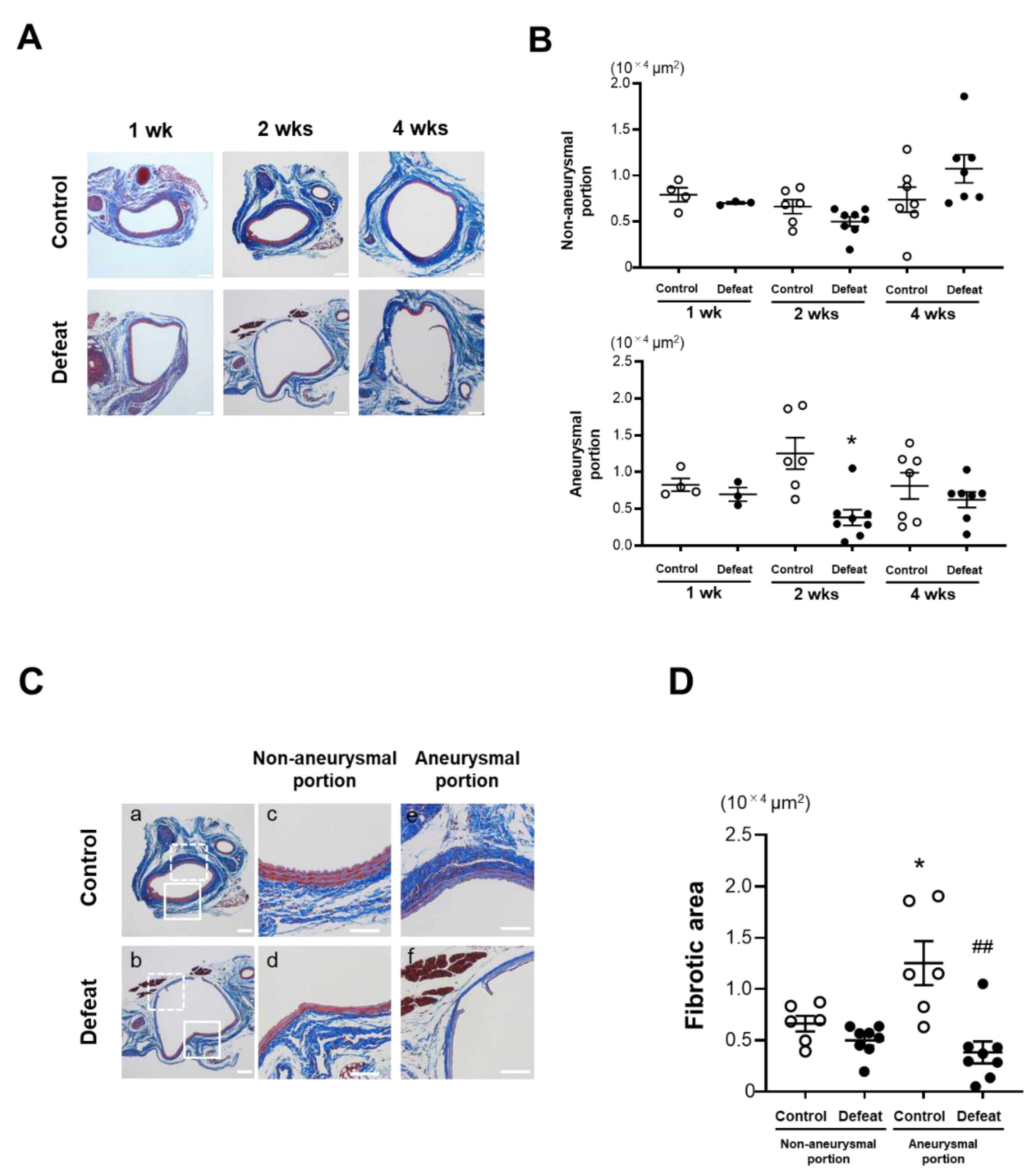
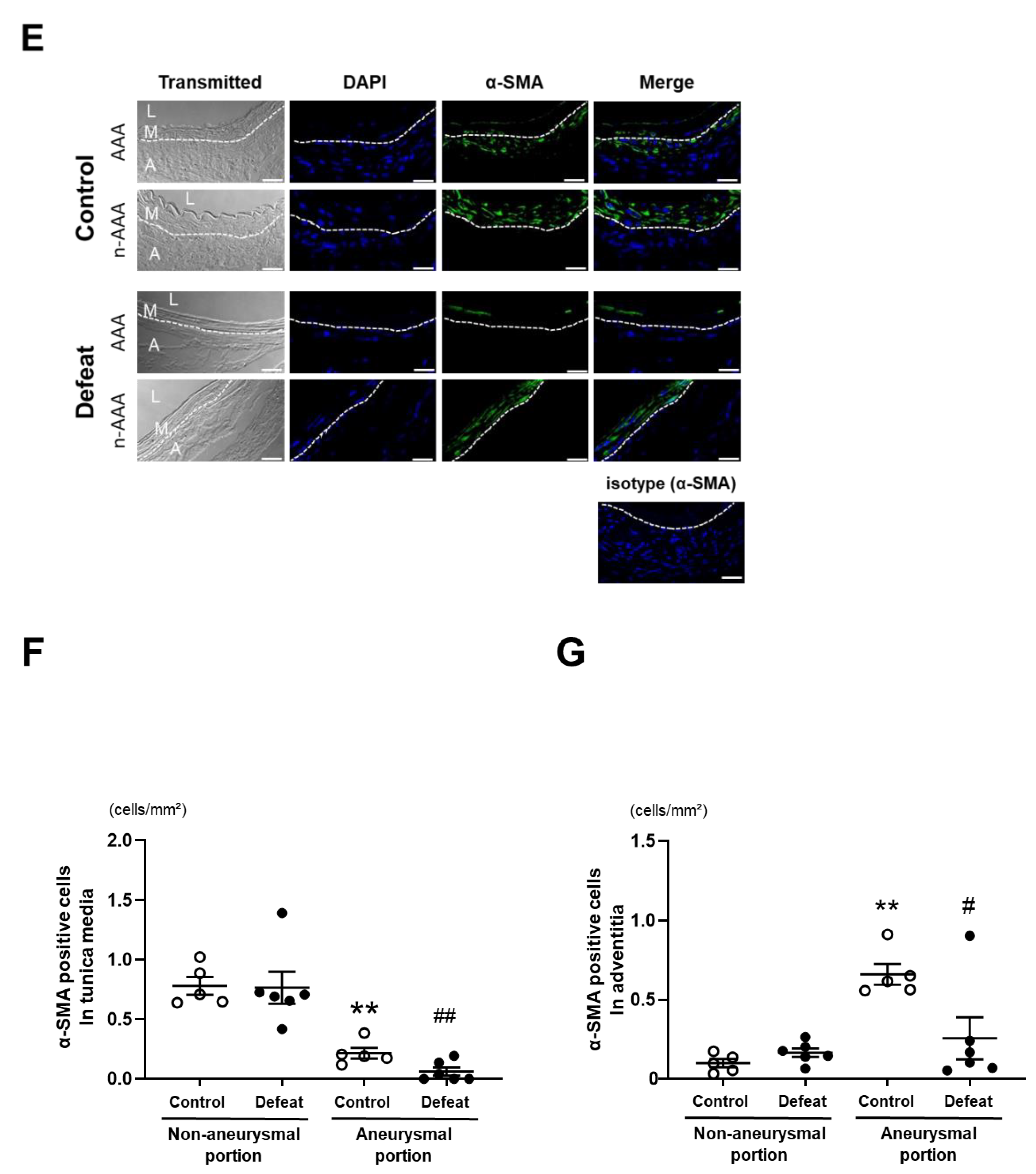
3.3. Repeated Social Defeat Inhibits Perivascular Fibrotic Healing after the Acute Inflammatory Response
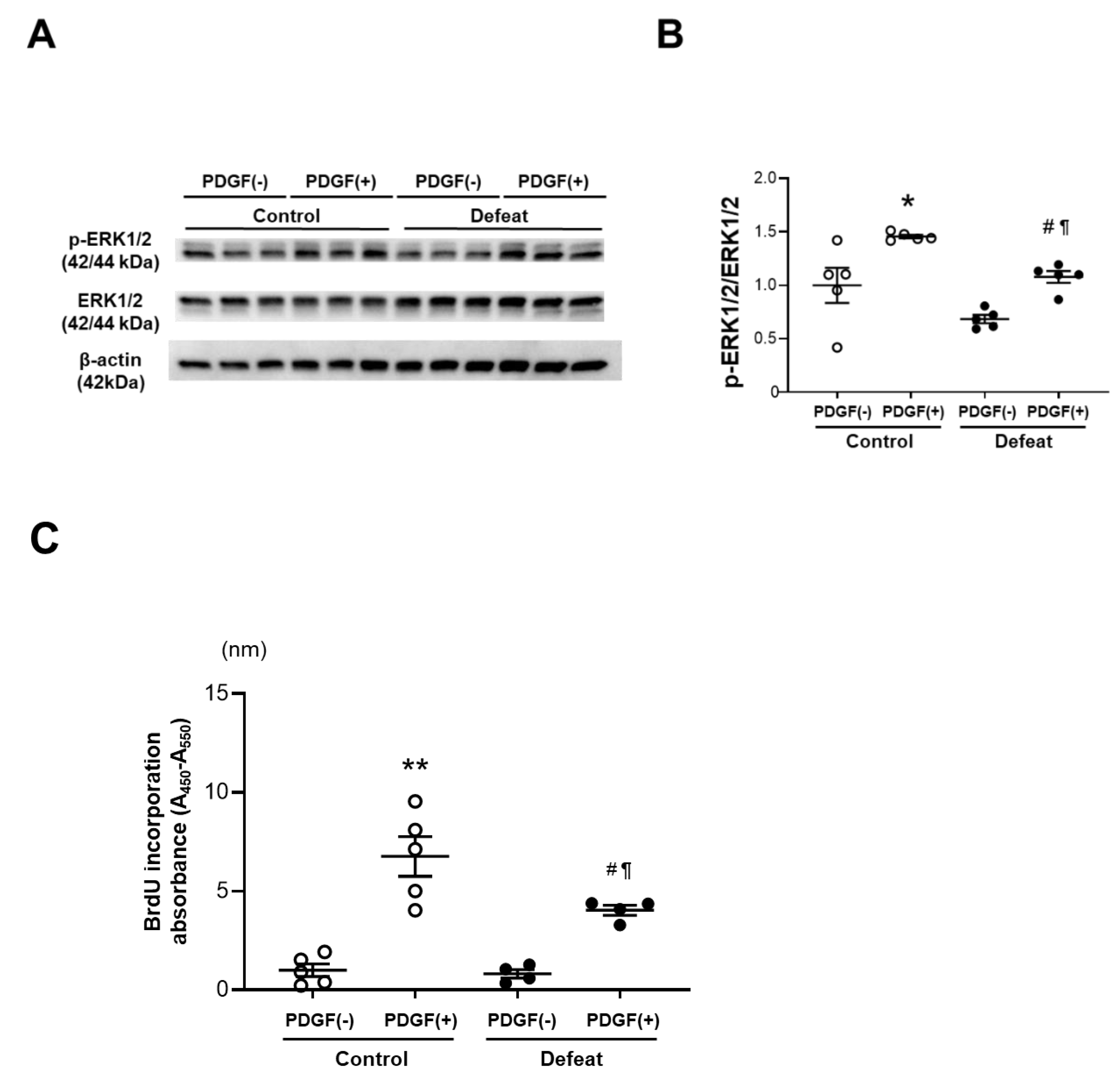
3.4. PDGF-Induced ERK Phosphorylation and BrdU Incorporation in VSMCs Are Inhibited in Defeated Mice
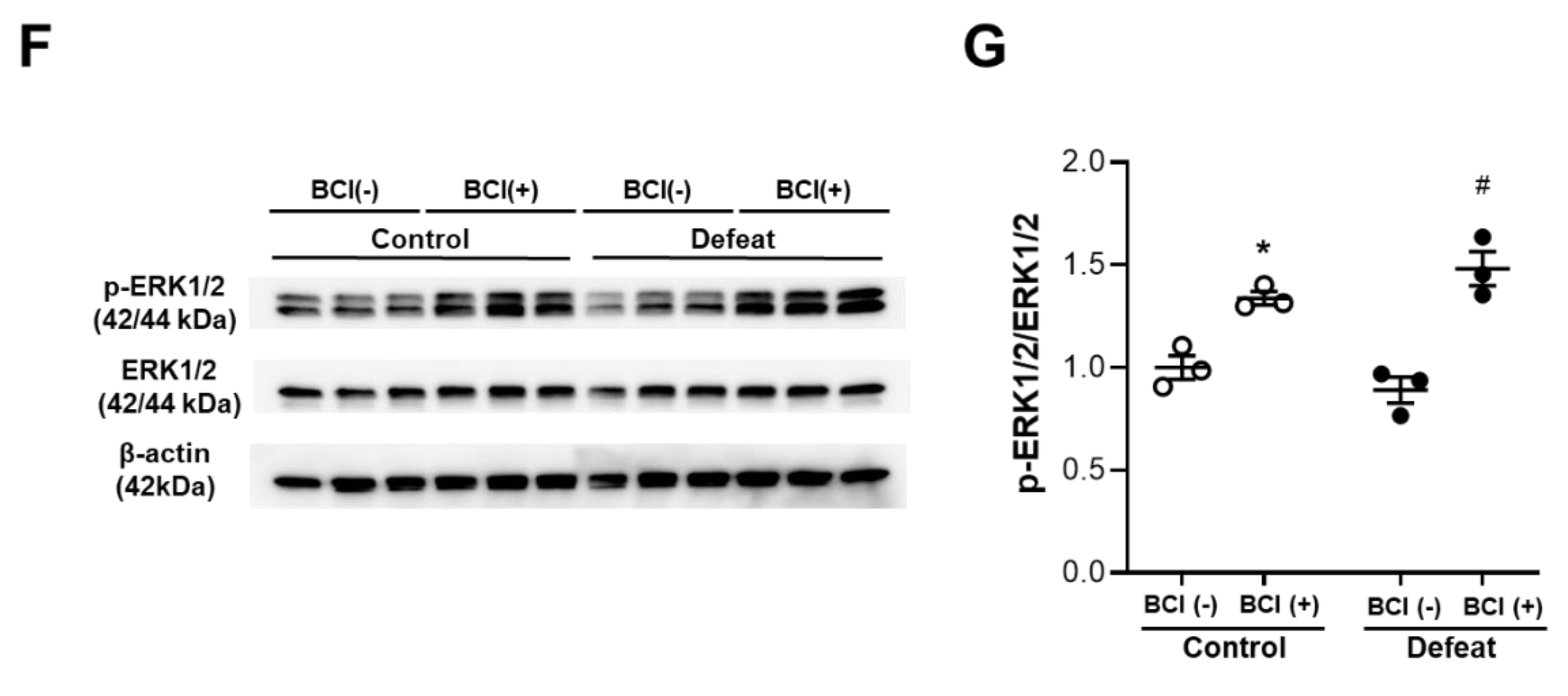
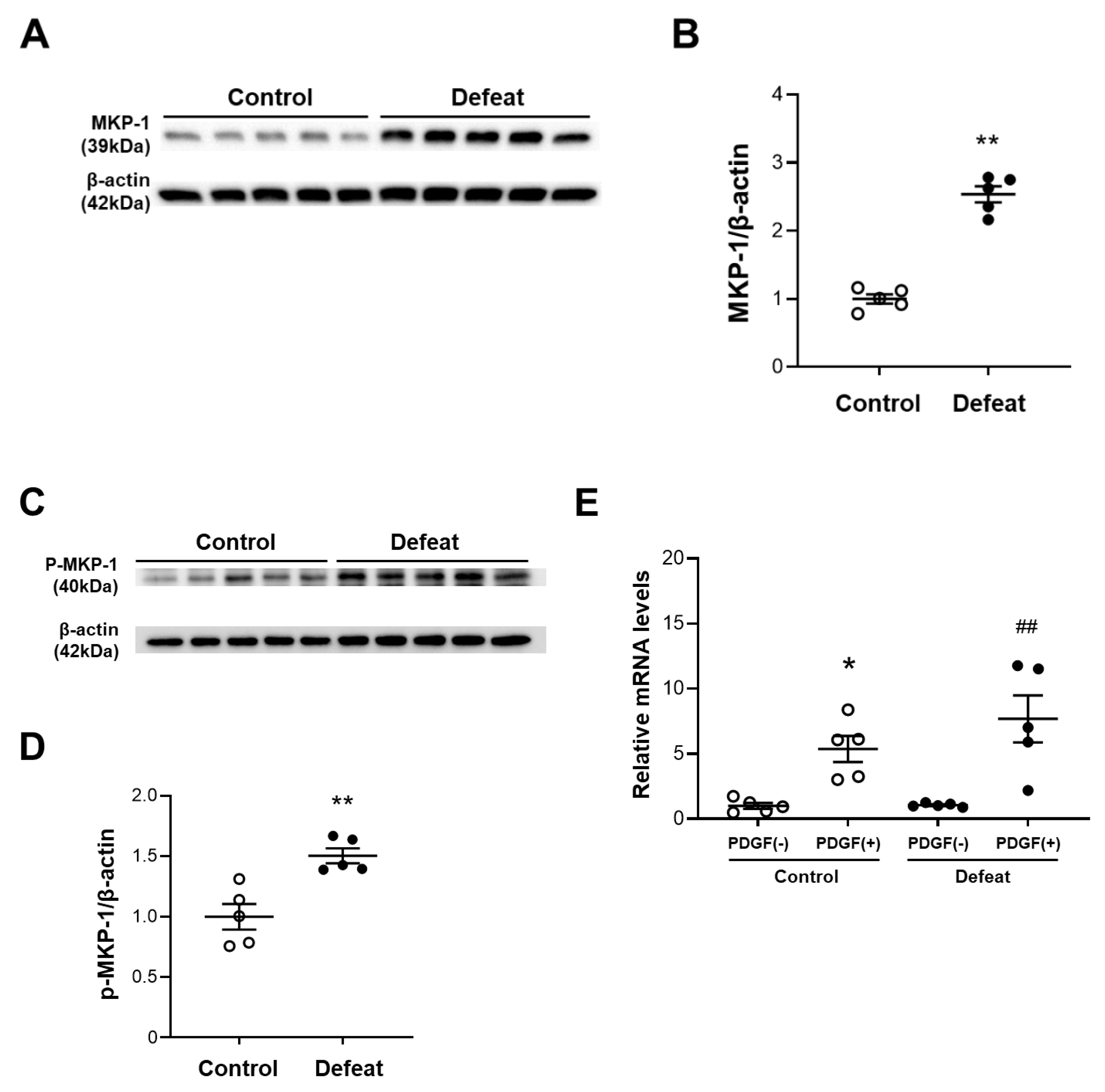
3.5. PDGF-Induced MKP-1 Expression in VSMCs Is Augmented in Defeated Mice
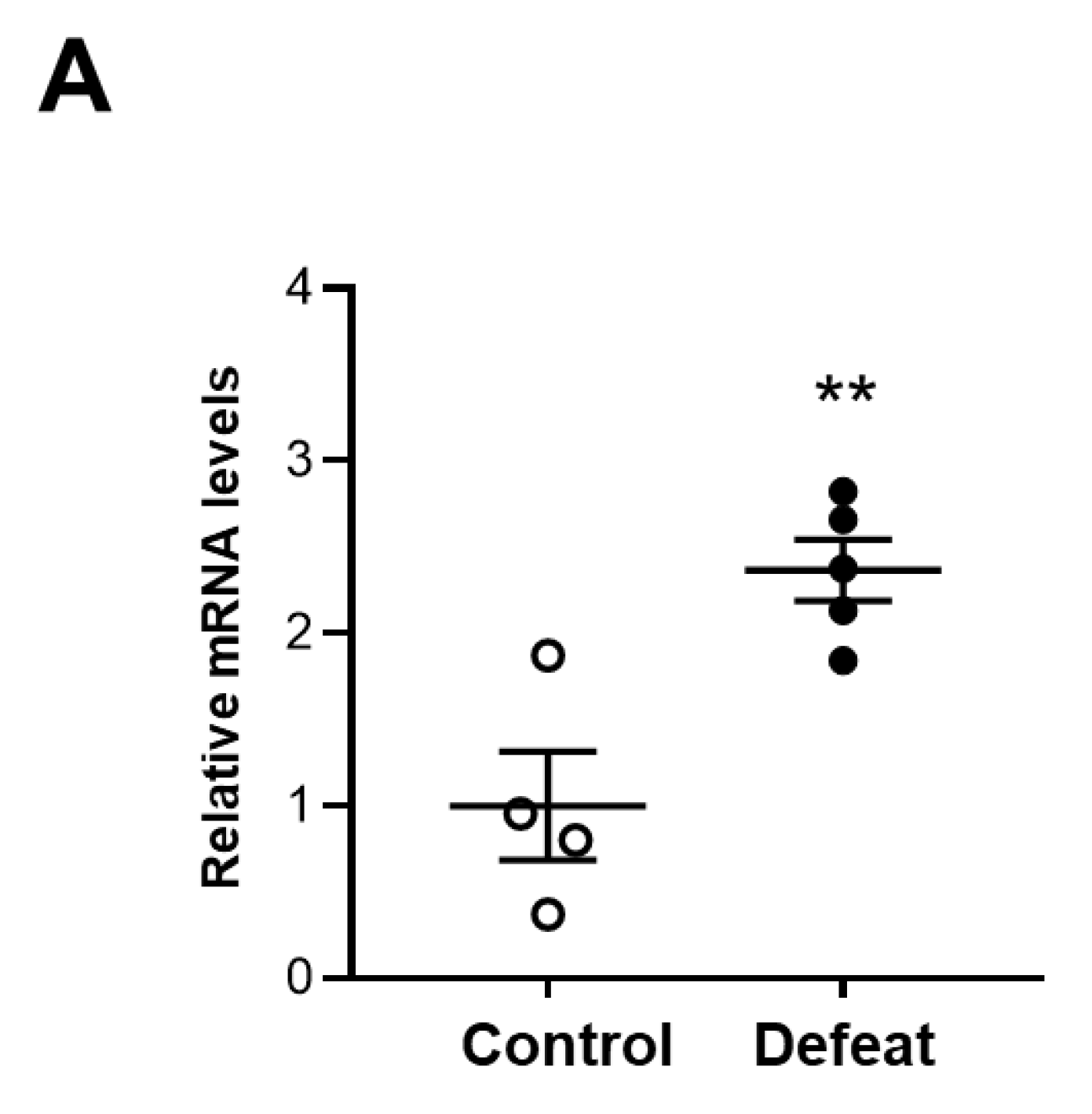
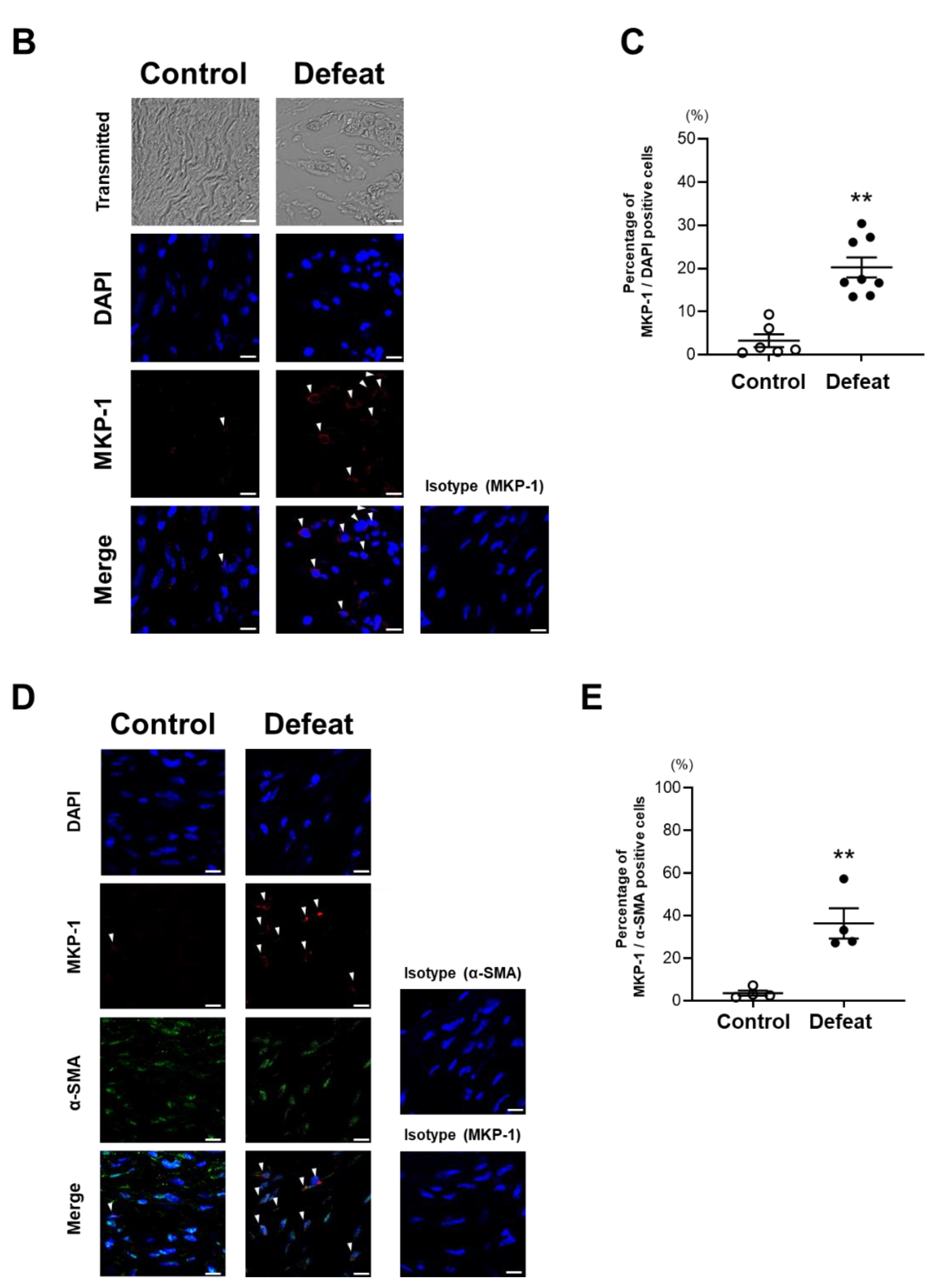
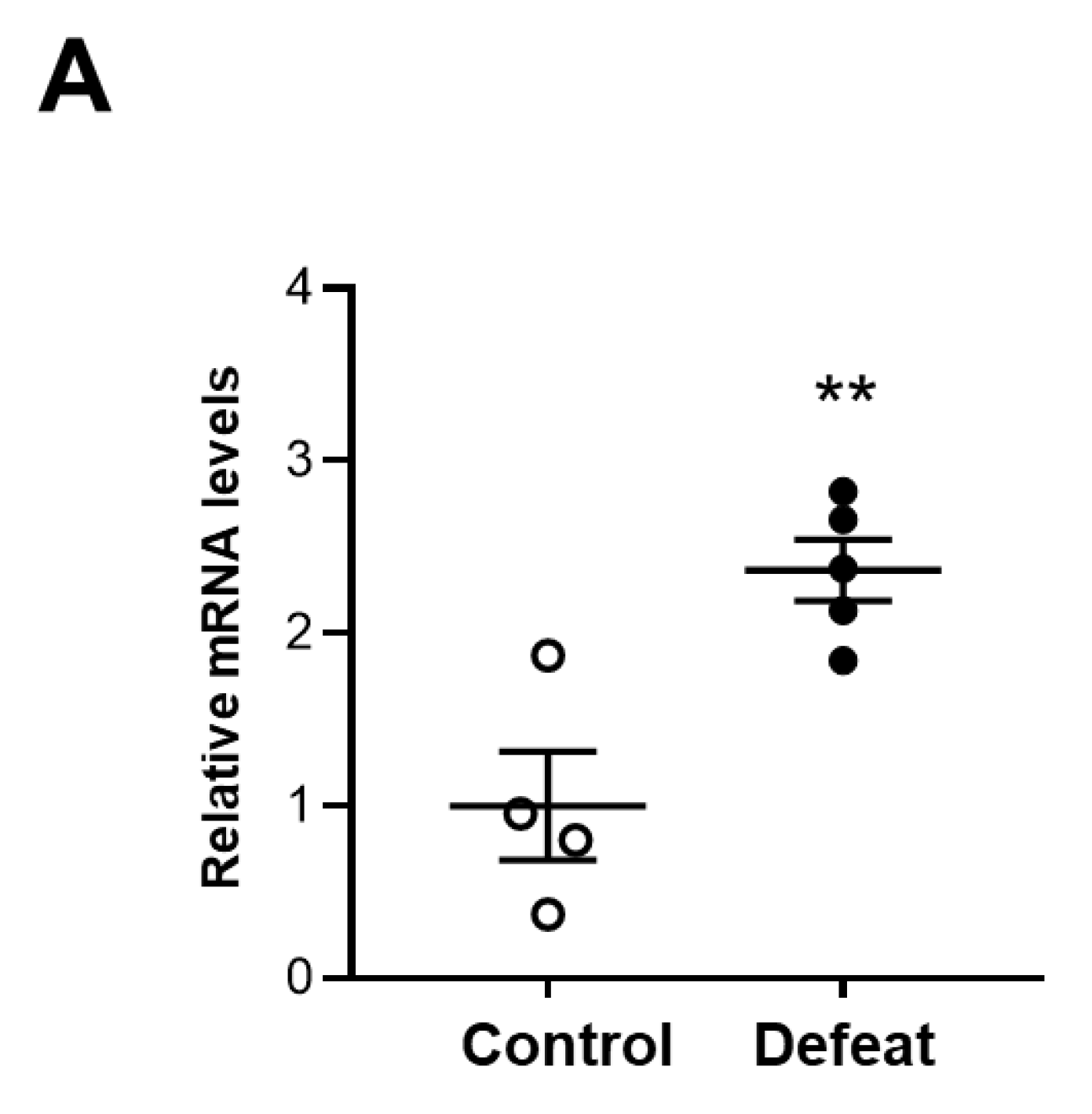
3.6. MKP-1 Expression in AAA Is Exaggerated in Defeated Mice
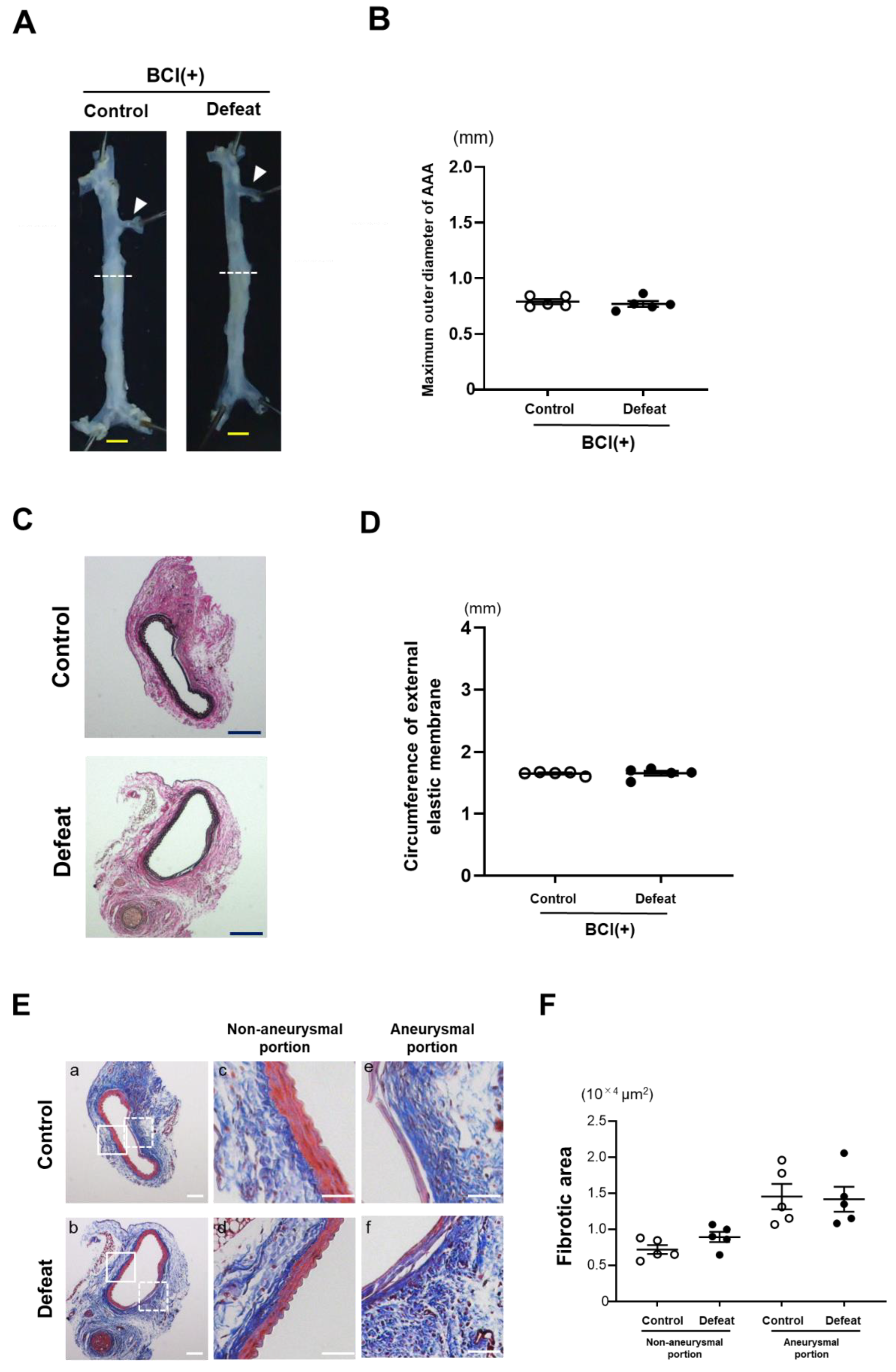
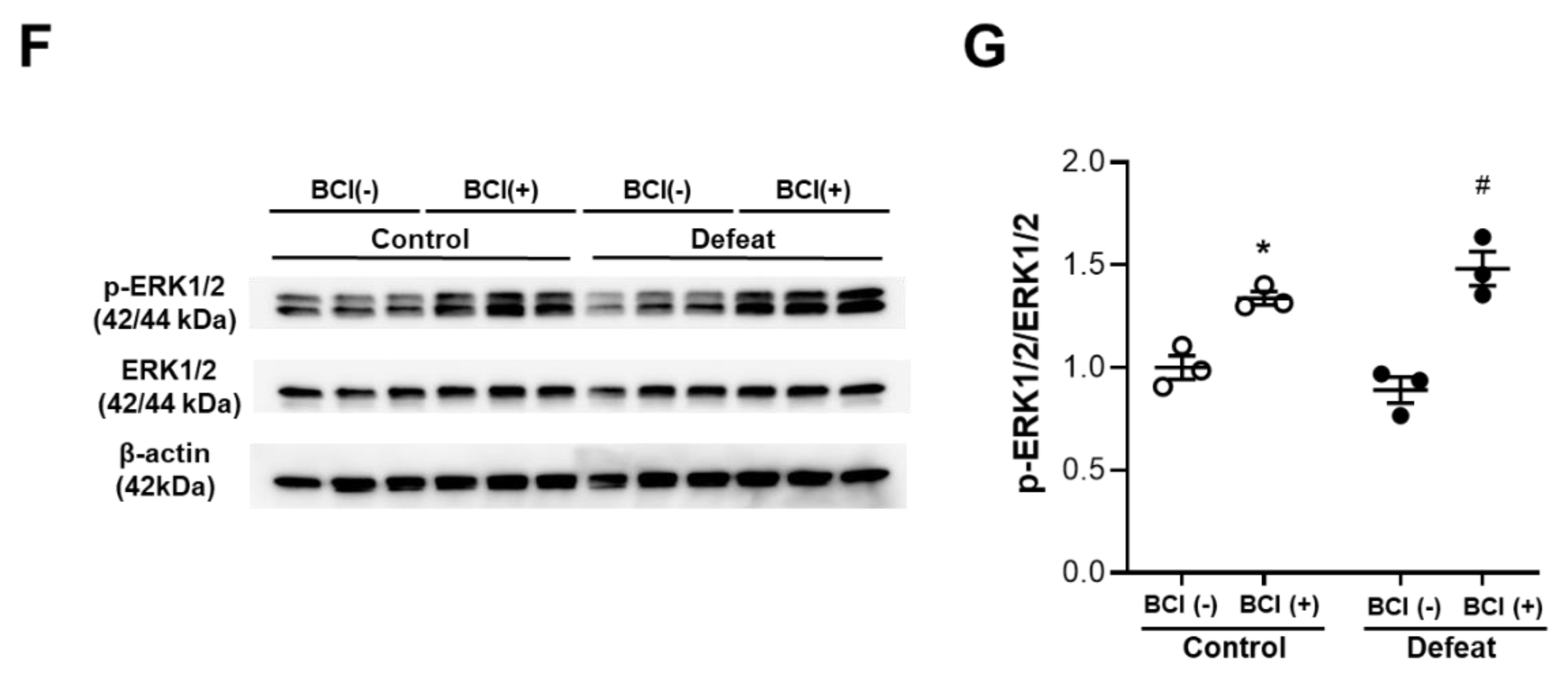
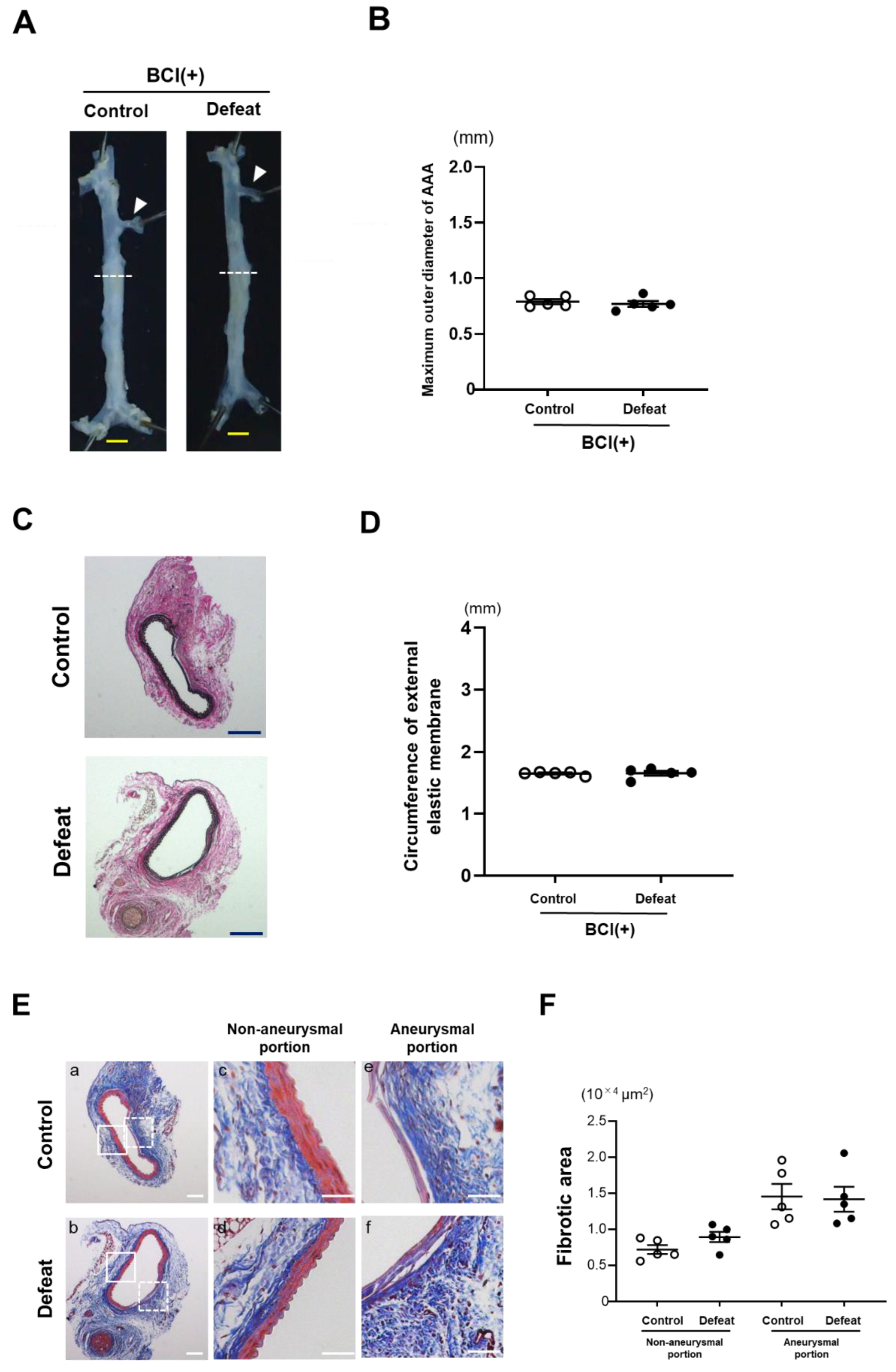
3.7. Treatment with MKP-1 Inhibitor Attenuated Exaggerated AAA Development in Defeated Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walker, E.R.; McGee, R.E.; Druss, B.G. Mortality in mental disorders and global disease burden implications: A systematic review and meta-analysis. JAMA Psychiatry 2015, 72, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.L.; Toukhsati, S.R.; Johansson, P.; Jaarsma, T. Depression and cardiovascular disease: A clinical review. Eur. Heart J. 2014, 35, 1365–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtman, J.H.; Froelicher, E.S.; Blumenthal, J.A.; Carney, R.M.; Doering, L.V.; Frasure-Smith, N.; Freedland, K.E.; Jaffe, A.S.; Leifheit-Limson, E.C.; Sheps, D.S.; et al. American Heart Association Statistics Committee of the Council on Epidemiology and Prevention and the Council on Cardiovascular and Stroke Nursing. Depression as a risk factor for poor prognosis among patients with acute coronary syndrome: Systematic review and recommendations: A scientific statement from the American Heart Association. Circulation 2014, 129, 1350–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daskalopoulou, M.; George, J.; Walters, K.; Osborn, D.P.; Batty, G.D.; Stogiannis, D.; Rapsomaniki, E.; Pujades-Rodriguez, M.; Denaxas, S.; Udumyan, R.; et al. Depression as a risk factor for the initial presentation of twelve cardiac, cerebrovascular, and peripheral arterial diseases: Data linkage study of 1.9 million women and men. PLoS ONE 2016, 11, e0153838. [Google Scholar] [CrossRef]
- Harshfield, E.L.; Pennells, L.; Schwartz, J.E.; Willeit, P.; Kaptoge, S.; Bell, S.; Shaffer, J.A.; Bolton, T.; Spackman, S.; Wassertheil-Smoller, S.; et al. Emerging Risk Factors Collaboration. Association between depressive symptoms and incident cardiovascular diseases. JAMA 2020, 324, 2396–2405. [Google Scholar] [CrossRef]
- Goldstein, B.I.; Carnethon, M.R.; Matthews, K.A.; McIntyre, R.S.; Miller, G.E.; Raghuveer, G.; Stoney, C.M.; Wasiak, H.; McCrindle, B.W.; American Heart Association Atherosclerosis; et al. Major depressive disorder and bipolar disorder predispose youth to accelerated atherosclerosis and early cardiovascular disease: A scientific statement from the American Heart Association. Circulation 2015, 132, 965–986. [Google Scholar] [CrossRef]
- Nielsen, R.E.; Banner, J.; Jensen, S.E. Cardiovascular disease in patients with severe mental illness. Nat. Rev. Cardiol. 2021, 18, 136–145. [Google Scholar] [CrossRef]
- Levine, G.N.; Cohen, B.E.; Commodore-Mensah, Y.; Fleury, J.; Huffman, J.C.; Khalid, U.; Labarthe, D.R.; Lavretsky, H.; Michos, E.D.; Spatz, E.S.; et al. Psychological health, well-being, and the mind-heart-body connection: A scientific statement from the American Heart Association. Circulation 2021, 143, e763–e783. [Google Scholar] [CrossRef]
- Kent, K.C. Clinical practice. Abdominal aortic aneurysms. N. Engl. J. Med. 2014, 371, 2101–2108. [Google Scholar] [CrossRef]
- Golledge, J. Abdominal aortic aneurysm: Update on pathogenesis and medical treatments. Nat. Rev. Cardiol. 2019, 16, 225–242. [Google Scholar] [CrossRef]
- Lindeman, J.H.; Matsumura, J.S. Pharmacologic management of aneurysms. Circ. Res. 2019, 124, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Raffort, J.; Lareyre, F.; Clément, M.; Hassen-Khodja, R.; Chinetti, G.; Mallat, Z. Diabetes and aortic aneurysm: Current state of the art. Cardiovasc. Res. 2018, 114, 1702–1713. [Google Scholar] [CrossRef] [PubMed]
- Nyrønning, L.Å.; Stenman, M.; Hultgren, R.; Albrektsen, G.; Videm, V.; Mattsson, E. Symptoms of depression and risk of abdominal aortic aneurysm: A HUNT Study. J. Am. Heart Assoc. 2019, 8, e012535. [Google Scholar] [CrossRef]
- Kim, M.H.; Yoo, J.H.; Cho, H.J.; Ko, K.J.; Jun, K.W.; Han, K.D.; Hwang, J.K. Increased depression risk in patients with abdominal aortic aneurysm: A nationwide cohort study. Ann. Surg. Treat. Res. 2021, 101, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Yamada, H.; Wakana, N.; Kikai, M.; Terada, K.; Wada, N.; Motoyama, S.; Saburi, M.; Sugimoto, T.; Kami, D.; et al. Augmented neutrophil extracellular traps formation promotes atherosclerosis development in socially defeated apoE−/− mice. Biochem. Biophys. Res. Commun. 2018, 500, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Yamada, H.; Wada, N.; Motoyama, S.; Saburi, M.; Kubota, H.; Miyawaki, D.; Wakana, N.; Kami, D.; Ogata, T.; et al. Repeated social defeat exaggerates fibrin-rich clot formation by enhancing neutrophil extracellular trap formation via platelet–neutrophil interactions. Cells 2021, 10, 3344. [Google Scholar] [CrossRef]
- Golden, S.A.; Covington, H.E., III; Berton, O.; Russo, S.J. A standardized protocol for repeated social defeat stress in mice. Nat. Protoc. 2011, 6, 1183–1191. [Google Scholar] [CrossRef]
- Ménard, C.; Hodes, G.E.; Russo, S.J. Pathogenesis of depression: Insights from human and rodent studies. Neuroscience 2016, 321, 138–162. [Google Scholar] [CrossRef] [Green Version]
- Berton, O.; McClung, C.A.; Dileone, R.J.; Krishnan, V.; Renthal, W.; Russo, S.J.; Graham, D.; Tsankova, N.M.; Bolanos, C.A.; Rios, M.; et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 2006, 311, 864–868. [Google Scholar] [CrossRef] [Green Version]
- Can, A.; Dao, D.T.; Terrillion, C.E.; Piantadosi, S.C.; Bhat, S.; Gould, T.D. The tail suspension test. J. Vis. Exp. 2012, 59, e3769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sénémaud, J.; Caligiuri, G.; Etienne, H.; Delbosc, S.; Michel, J.B.; Coscas, R. Translational relevance and recent advances of animal models of abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.N.; Liao, Y.F.; Lu, Y.X.; Wang, Y.; Lu, J.H.; Zeng, Z.L.; Huang, Q.T.; Sheng, H.; Yun, J.P.; Xie, D.; et al. Pharmacological inhibition of DUSP6 suppresses gastric cancer growth and metastasis and overcomes cisplatin resistance. Cancer Lett. 2018, 412, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Sheth, R.A.; Maricevich, M.; Mahmood, U. In vivo optical molecular imaging of matrix metalloproteinase activity in abdominal aortic aneurysms correlates with treatment effects on growth rate. Atherosclerosis 2010, 212, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speer, M.Y.; Chien, Y.C.; Quan, M.; Yang, H.Y.; Vali, H.; McKee, M.D.; Giachelli, C.M. Smooth muscle cells deficient in osteopontin have enhanced susceptibility to calcification in vitro. Cardiovasc. Res. 2005, 66, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Comalada, M.; Lloberas, J.; Celada, A. MKP-1: A critical phosphatase in the biology of macrophages controlling the switch between proliferation and activation. Eur. J. Immunol. 2012, 42, 1938–1948. [Google Scholar] [CrossRef]
- Maegdefessel, L.; Azuma, J.; Toh, R.; Merk, D.R.; Deng, A.; Chin, J.T.; Raaz, U.; Schoelmerich, A.M.; Raiesdana, A.; Leeper, N.J.; et al. Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J. Clin. Investig. 2012, 122, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Lindeman, J.H.; Ashcroft, B.A.; Beenakker, J.W.; van Es, M.; Koekkoek, N.B.; Prins, F.A.; Tielemans, J.F.; Abdul-Hussien, H.; Bank, R.A.; Oosterkamp, T.H. Distinct defects in collagen microarchitecture underlie vessel-wall failure in advanced abdominal aneurysms and aneurysms in Marfan syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 862–865. [Google Scholar] [CrossRef] [Green Version]
- Cole-King, A.; Harding, K.G. Psychological factors and delayed healing in chronic wounds. Psychosom. Med. 2001, 63, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Gouin, J.P.; Kiecolt-Glaser, J.K. The impact of psychological stress on wound healing: Methods and mechanisms. Immunol. Allergy Clin. N. Am. 2011, 31, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Robinson, H.; Norton, S.; Jarrett, P.; Broadbent, E. The effects of psychological interventions on wound healing: A systematic review of randomized trials. Br. J. Health Psychol. 2017, 22, 805–835. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound healing: A cellular perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.T.; Han, Y.P.; Yan, C.; Shaw, M.C.; Garner, W.L. TNF-alpha suppresses alpha-smooth muscle actin expression in human dermal fibroblasts: An implication for abnormal wound healing. J. Investig. Dermatol. 2007, 127, 2645–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duric, V.; Banasr, M.; Licznerski, P.; Schmidt, H.D.; Stockmeier, C.A.; Simen, A.A.; Newton, S.S.; Duman, R.S. A negative regulator of MAP kinase causes depressive behavior. Nat. Med. 2010, 16, 1328–1332. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Mao, L. The ERK pathway: Molecular mechanisms and treatment of depression. Mol. Neurobiol. 2019, 56, 6197–6205. [Google Scholar] [CrossRef] [PubMed]
- Uzui, H.; Lee, J.D.; Shimizu, H.; Tsutani, H.; Ueda, T. The role of protein-tyrosine phosphorylation and gelatinase production in the migration and proliferation of smooth muscle cells. Atherosclerosis 2000, 149, 51–59. [Google Scholar] [CrossRef]
- Zhan, Y.; Kim, S.; Izumi, Y.; Izumiya, Y.; Nakao, T.; Miyazaki, H.; Iwao, H. Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brondello, J.M.; Pouysségur, J.; McKenzie, F.R. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science 1999, 286, 2514–2517. [Google Scholar] [CrossRef]
- Kassel, O.; Sancono, A.; Krätzschmar, J.; Kreft, B.; Stassen, M.; Cato, A.C. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 2001, 20, 7108–7116. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.; Wang, H.; Lee, W.S.; Jain, M.K.; Lee, M.E.; Haber, E. Mitogen-activated protein kinase phosphatase-1 in rat arterial smooth muscle cell proliferation. J. Clin. Investig. 1996, 98, 1560–1567. [Google Scholar] [CrossRef] [Green Version]
- Koyama, H.; Olson, N.E.; Dastvan, F.F.; Reidy, M.A. Cell replication in the arterial wall: Activation of signaling pathway following in vivo injury. Circ. Res. 1998, 82, 713–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Liu, Y.; Wang, X.; Wu, J.; Liu, K.; Zhou, J.; Liu, L.; Zhang, C. Identification of differential microRNAs in cerebrospinal fluid and serum of patients with major depressive disorder. PLoS ONE 2015, 10, e0121975. [Google Scholar] [CrossRef] [PubMed]
- Ferrúa, C.P.; Giorgi, R.; da Rosa, L.C.; do Amaral, C.C.; Ghisleni, G.C.; Pinheiro, R.T.; Nedel, F. MicroRNAs expressed in depression and their associated pathways: A systematic review and a bioinformatics analysis. J. Chem. Neuroanat. 2019, 100, 101650. [Google Scholar] [CrossRef]
- Ortega, M.A.; Alvarez-Mon, M.A.; García-Montero, C.; Fraile-Martinez, O.; Lahera, G.; Monserrat, J.; Muñoz-Merida, L.; Mora, F.; Rodríguez-Jiménez, R.; Fernandez-Rojo, S.; et al. MicroRNAs as critical biomarkers of major depressive disorder: A comprehensive perspective. Biomedicines 2021, 9, 1659. [Google Scholar] [CrossRef]
- Hung, Y.Y.; Wu, M.K.; Tsai, M.C.; Huang, Y.L.; Kang, H.Y. Aberrant expression of intracellular let-7e, miR-146a, and miR-155 correlates with severity of depression in patients with major depressive disorder and is ameliorated after antidepressant treatment. Cells 2019, 8, 647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.P.; Fiori, L.M.; Cruceanu, C.; Lin, R.; Labonte, B.; Cates, H.M.; Heller, E.A.; Vialou, V.; Ku, S.M.; Gerald, C.; et al. MicroRNAs 146a/b-5 and 425-3p and 24-3p are markers of antidepressant response and regulate MAPK/Wnt-system genes. Nat. Commun. 2017, 8, 15497. [Google Scholar] [CrossRef] [Green Version]
- Kin, K.; Miyagawa, S.; Fukushima, S.; Shirakawa, Y.; Torikai, K.; Shimamura, K.; Daimon, T.; Kawahara, Y.; Kuratani, T.; Sawa, Y. Tissue- and plasma-specific MicroRNA signatures for atherosclerotic abdominal aortic aneurysm. J. Am. Heart Assoc. 2012, 1, e000745. [Google Scholar] [CrossRef] [Green Version]
- Holm, T.M.; Habashi, J.P.; Doyle, J.J.; Bedja, D.; Chen, Y.; van Erp, C.; Lindsay, M.E.; Kim, D.; Schoenhoff, F.; Cohn, R.D.; et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011, 332, 358–361. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; DiMusto, P.D.; Ehrlichman, L.K.; Sadiq, O.; McEvoy, B.; Futchko, J.S.; Henke, P.K.; Eliason, J.L.; Upchurch, G.R., Jr. The role of extracellular signal-related kinase during abdominal aortic aneurysm formation. J. Am. Coll. Surg. 2012, 215, 668–680. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Zhang, K.; Liu, Z.; Xu, Q.; You, B.; Li, C.; Cao, J.; Zhou, H.; Li, X.; Chen, J.; et al. VPO1 Modulates Vascular Smooth Muscle Cell Phenotypic Switch by Activating Extracellular Signal-regulated Kinase 1/2 (ERK 1/2) in Abdominal Aortic Aneurysms. J. Am. Heart Assoc. 2018, 7, e010069. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, J.T.; Tallquist, M.D. Tracking Adventitial Fibroblast Contribution to Disease: A Review of Current Methods to Identify Resident Fibroblasts. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1598–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubota, H.; Yamada, H.; Sugimoto, T.; Wada, N.; Motoyama, S.; Saburi, M.; Miyawaki, D.; Wakana, N.; Kami, D.; Ogata, T.; et al. Repeated Social Defeat Enhances CaCl2-Induced Abdominal Aortic Aneurysm Expansion by Inhibiting the Early Fibrotic Response via the MAPK-MKP-1 Pathway. Cells 2022, 11, 732. https://doi.org/10.3390/cells11040732
Kubota H, Yamada H, Sugimoto T, Wada N, Motoyama S, Saburi M, Miyawaki D, Wakana N, Kami D, Ogata T, et al. Repeated Social Defeat Enhances CaCl2-Induced Abdominal Aortic Aneurysm Expansion by Inhibiting the Early Fibrotic Response via the MAPK-MKP-1 Pathway. Cells. 2022; 11(4):732. https://doi.org/10.3390/cells11040732
Chicago/Turabian StyleKubota, Hiroshi, Hiroyuki Yamada, Takeshi Sugimoto, Naotoshi Wada, Shinichiro Motoyama, Makoto Saburi, Daisuke Miyawaki, Noriyuki Wakana, Daisuke Kami, Takehiro Ogata, and et al. 2022. "Repeated Social Defeat Enhances CaCl2-Induced Abdominal Aortic Aneurysm Expansion by Inhibiting the Early Fibrotic Response via the MAPK-MKP-1 Pathway" Cells 11, no. 4: 732. https://doi.org/10.3390/cells11040732
APA StyleKubota, H., Yamada, H., Sugimoto, T., Wada, N., Motoyama, S., Saburi, M., Miyawaki, D., Wakana, N., Kami, D., Ogata, T., Ibi, M., & Matoba, S. (2022). Repeated Social Defeat Enhances CaCl2-Induced Abdominal Aortic Aneurysm Expansion by Inhibiting the Early Fibrotic Response via the MAPK-MKP-1 Pathway. Cells, 11(4), 732. https://doi.org/10.3390/cells11040732