
Implications of Poly(A) Tail Processing in Repeat Expansion Diseases



Abstract
1. Introduction
2. Polyadenylation in Animal Cells
2.1. Polyadenylation Process
2.2. Polyadenylation Roles
2.3. Length and Composition of Tails
2.4. Alternative Polyadenylation (APA)
3. Methods for Determining the Length of Poly(A) Tails and Analyzing APA Sites
3.1. Transcript-Specific Analyses of Poly(A) Tails
3.2. Transcriptome-Wide Methods for Measurement of Poly(A) Tails
3.3. APA-Identification Techniques and Databases
4. Repeat Expansion Diseases
4.1. APA and Poly(A) Tail Lengths in Neurons and Muscles
4.2. Characteristics and Perturbations in Poly(A) Tail Processing in Repeat Expansion Diseases
4.2.1. Oculopharyngeal Muscular Dystrophy
4.2.2. Fragile X-Associated Tremor/Ataxia Syndrome
4.2.3. Amyotrophic Lateral Sclerosis
4.2.4. Myotonic Dystrophy Type 1
4.2.5. Huntington’s Disease
4.3. Potential Perturbations in Poly(A) Tail Processing in Other PolyQ Diseases
5. Conclusions and Perspectives
5.1. Methodological Aspects
5.2. Implications for Pathogenesis of Repeat Expansion Diseases
5.3. Implications into Therapy of Repeat Expansion Diseases
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| APA | alternative polyadenylation |
| DM | myotonic dystrophy |
| HD | Huntington’s disease |
| OPMD | oculopharyngeal muscular dystrophy |
| PABPN1 | poly(A) binding protein nuclear 1 |
| PAS | polyadenylation signal |
| PAT | poly(A) test |
| polyQ diseases | polyglutamine diseases |
References
- Casañal, A.; Kumar, A.; Hill, C.H.; Easter, A.D.; Emsley, P.; Degliesposti, G.; Gordiyenko, Y.; Santhanam, B.; Wolf, J.; Wiederhold, K.; et al. Architecture of eukaryotic mRNA 3′-end processing machinery. Science 2017, 358, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Passmore, L.A.; Coller, J. Roles of mRNA poly(A) tails in regulation of eukaryotic gene expression. Nat. Rev. Mol. Cell Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Nourse, J.; Spada, S.; Danckwardt, S. Emerging roles of RNA 3′-end cleavage and polyadenylation in pathogenesis, diagnosis and therapy of human disorders. Biomolecules 2020, 10, 915. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M. Polyadenylation and nuclear export of mRNAs. J. Biol. Chem. 2019, 294, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and Consequences of Alternative Polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef]
- Sadek, J.; Omer, A.; Hall, D.; Ashour, K.; Gallouzi, I.E. Alternative polyadenylation and the stress response. Wiley Interdiscip. Rev. RNA 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Ren, F.; Zhang, N.; Zhang, L.; Miller, E.; Pu, J.J. Alternative Polyadenylation: A new frontier in post transcriptional regulation. Biomark. Res. 2020, 8, 67. [Google Scholar] [CrossRef]
- Danckwardt, S.; Hentze, M.W.; Kulozik, A.E. 3′ end mRNA processing: Molecular mechanisms and implications for health and disease. EMBO J. 2008, 27, 482–498. [Google Scholar] [CrossRef]
- Proudfoot, N.J. Ending the message: Poly(A) signals then and now. Genes Dev. 2011, 25, 1770–1782. [Google Scholar] [CrossRef]
- Derti, A.; Garrett-Engele, P.; MacIsaac, K.D.; Stevens, R.C.; Sriram, S.; Chen, R.; Rohl, C.A.; Johnson, J.M.; Babak, T. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012, 22, 1173–1183. [Google Scholar] [CrossRef]
- Gruber, A.J.; Zavolan, M. Alternative cleavage and polyadenylation in health and disease. Nat. Rev. Genet. 2019, 20, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Hyman, L.; Moore, C. Formation of mRNA 3′ Ends in Eukaryotes: Mechanism, Regulation, and Interrelationships with Other Steps in mRNA Synthesis. Microbiol. Mol. Biol. Rev. 1999, 63, 405–445. [Google Scholar] [CrossRef]
- Eckmann, C.R.; Rammelt, C.; Wahle, E. Control of poly(A) tail length. Wiley Interdiscip. Rev. RNA 2011, 2, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Natalizio, B.J.; Wente, S.R. Postage for the messenger: Designating routes for nuclear mRNA export. Trends Cell Biol. 2013, 23, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Bresson, S.M.; Conrad, N.K. The Human Nuclear Poly(A)-Binding Protein Promotes RNA Hyperadenylation and Decay. PLoS Genet. 2013, 9, 1003893. [Google Scholar] [CrossRef]
- Eisen, T.J.; Eichhorn, S.W.; Subtelny, A.O.; Lin, K.S.; McGeary, S.E.; Gupta, S.; Bartel, D.P. The Dynamics of Cytoplasmic mRNA Metabolism. Mol. Cell 2020, 77, 786–799. [Google Scholar] [CrossRef]
- Weill, L.; Belloc, E.; Bava, F.-A.; Méndez, R. Translational control by changes in poly(A) tail length: Recycling mRNAs. Nat. Struct. Mol. Biol. 2012, 19, 577–585. [Google Scholar] [CrossRef]
- Xiang, K.; Bartel, D.P. The molecular basis of coupling between poly(A)-tail length and translational efficiency. eLife 2021, 10, 66493. [Google Scholar] [CrossRef]
- Nicholson, A.L.; Pasquinelli, A.E. Tales of Detailed Poly(A) Tails. Trends Cell Biol. 2019, 29, 191–200. [Google Scholar] [CrossRef]
- Dominski, Z.; Yang, X.-C.; Kaygun, H.; Dadlez, M.; Marzluff, W.F. A 3′ Exonuclease that Specifically Interacts with the 3′ End of Histone mRNA. Mol. Cell 2003, 12, 295–305. [Google Scholar] [CrossRef]
- Lorenzi, L.; Chiu, H.S.; Avila Cobos, F.; Gross, S.; Volders, P.J.; Cannoodt, R.; Nuytens, J.; Vanderheyden, K.; Anckaert, J.; Lefever, S.; et al. The RNA Atlas expands the catalog of human non-coding RNAs. Nat. Biotechnol. 2021, 39, 1453–1465. [Google Scholar] [CrossRef]
- Chen, L.L. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef]
- Lima, S.A.; Chipman, L.B.; Nicholson, A.L.; Chen, Y.H.; Yee, B.A.; Yeo, G.W.; Coller, J.; Pasquinelli, A.E. Short poly(A) tails are a conserved feature of highly expressed genes. Nat. Struct. Mol. Biol. 2017, 24, 1057–1063. [Google Scholar] [CrossRef]
- Subtelny, A.O.; Eichhorn, S.W.; Chen, G.R.; Sive, H.; Bartel, D.P. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature 2014, 508, 66–71. [Google Scholar] [CrossRef]
- Gu, H.; Gupta, J.D.; Schoenberg, D.R. The poly(A)-limiting element is a conserved cis-acting sequence that regulates poly(A) tail length on nuclear pre-mRNAs. Proc. Natl. Acad. Sci. USA 1999, 96, 8943–8948. [Google Scholar] [CrossRef] [PubMed]
- Jalkanen, A.L.; Coleman, S.J.; Wilusz, J. Determinants and implications of mRNA poly(A) tail size—Does this protein make my tail look big? Semin. Cell Dev. Biol. 2014, 34, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, A.; Meijer, H.A.; de Moor, C.H. Specificity factors in cytoplasmic polyadenylation. Wiley Interdiscip. Rev. RNA 2013, 4, 437. [Google Scholar] [CrossRef] [PubMed]
- Ivshina, M.; Lasko, P.; Richter, J.D. Cytoplasmic polyadenylation element binding proteins in development, health, and disease. Annu. Rev. Cell Dev. Biol. 2014, 30, 393–415. [Google Scholar] [CrossRef]
- Mendez, R.; Richter, J.D. Translational control by CPEB: A means to the end. Nat. Rev. Mol. Cell Biol. 2001, 2, 521–529. [Google Scholar] [CrossRef]
- Udagawa, T.; Swanger, S.A.; Takeuchi, K.; Kim, J.H.; Nalavadi, V.; Shin, J.; Lorenz, L.J.; Zukin, R.S.; Bassell, G.J.; Richter, J.D. Bidirectional Control of mRNA Translation and Synaptic Plasticity by the Cytoplasmic Polyadenylation Complex. Mol. Cell 2012, 47, 253–266. [Google Scholar] [CrossRef]
- Lim, J.; Kim, D.; Lee, Y.S.; Ha, M.; Lee, M.; Yeo, J.; Chang, H.; Song, J.; Ahn, K.; Kim, V.N. Mixed tailing by TENT4A and TENT4B shields mRNA from rapid deadenylation. Science 2018, 361, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Ha, M.; Chang, H.; Kwon, S.C.; Simanshu, D.K.; Patel, D.J.; Kim, V.N. Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 2014, 159, 1365–1376. [Google Scholar] [CrossRef]
- Morgan, M.; Much, C.; DiGiacomo, M.; Azzi, C.; Ivanova, I.; Vitsios, D.M.; Pistolic, J.; Collier, P.; Moreira, P.N.; Benes, V.; et al. MRNA 3′ uridylation and poly(A) tail length sculpt the mammalian maternal transcriptome. Nature 2017, 548, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Sher-Chen, E.L.; Green, C.B. Circadian control of mRNA polyadenylation dynamics regulates rhythmic protein expression. Genes Dev. 2012, 26, 2724–2736. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nie, H.; Lu, F. Dynamic RNA 3′ Uridylation and Guanylation during Mitosis. iScience 2020, 23. [Google Scholar] [CrossRef]
- Park, J.E.; Yi, H.; Kim, Y.; Chang, H.; Kim, V.N. Regulation of Poly(A) Tail and Translation during the Somatic Cell Cycle. Mol. Cell 2016, 62, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2016, 18, 18–30. [Google Scholar] [CrossRef]
- Sommerkamp, P.; Cabezas-Wallscheid, N.; Trumpp, A. Alternative Polyadenylation in Stem Cell Self-Renewal and Differentiation. Trends Mol. Med. 2021, 27, 660–672. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef]
- Lutz, C.S.; Moreira, A. Alternative mRNA polyadenylation in eukaryotes: An effective regulator of gene expression. Wiley Interdiscip. Rev. RNA 2011, 2, 22–31. [Google Scholar] [CrossRef]
- Agarwal, V.; Lopez-Darwin, S.; Kelley, D.R.; Shendure, J. The landscape of alternative polyadenylation in single cells of the developing mouse embryo. Nat. Commun. 2021, 12, 5101. [Google Scholar] [CrossRef]
- Berkovits, B.D.; Mayr, C. Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Jenal, M.; Elkon, R.; Loayza-Puch, F.; Van Haaften, G.; Kühn, U.; Menzies, F.M.; Vrielink, J.A.F.O.; Bos, A.J.; Drost, J.; Rooijers, K.; et al. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 2012, 149, 538–553. [Google Scholar] [CrossRef]
- Batra, R.; Manchanda, M.; Swanson, M.S. Global insights into alternative polyadenylation regulation. RNA Biol. 2015, 12, 597–602. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, G.; Makeyev, E.V. RNA-binding protein HuR autoregulates its expression by promoting alternative polyadenylation site usage. Nucleic Acids Res. 2012, 40, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Batra, R.; Charizanis, K.; Manchanda, M.; Mohan, A.; Li, M.; Finn, D.J.; Goodwin, M.; Zhang, C.; Sobczak, K.; Thornton, C.A.; et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell 2014, 56, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Sippel, A.E.; Stavrianopoulos, J.G.; Schutz, G.; Feigelson, P. Translational properties of rabbit globin mRNA after specific removal of poly(A) with ribonuclease H. Proc. Natl. Acad. Sci. USA 1974, 71, 4635–4639. [Google Scholar] [CrossRef]
- Sallés, F.J.; Richards, W.G.; Strickland, S. Assaying the polyadenylation state of mRNAs. Methods 1999, 17, 38–45. [Google Scholar] [CrossRef]
- Jänicke, A.; Vancuylenberg, J.; Boag, P.R.; Traven, A.; Beilharz, T.H. ePAT: A simple method to tag adenylated RNA to measure poly(A)-tail length and other 3′ RACE applications. RNA 2012, 18, 1289–1295. [Google Scholar] [CrossRef]
- Murray, E.L.; Schoenberg, D.R. Chapter 24 Assays for Determining Poly(A) Tail Length and the Polarity of mRNA Decay in Mammalian Cells. Methods Enzymol. 2008, 448, 483–504. [Google Scholar]
- Kusov, Y.Y.; Shatirishvili, G.; Dzagurov, G.; Gauss-Müller, V. A new G-tailing method for the determination of the poly(A) tail length applied to hepatitis A virus RNA. Nucleic Acids Res. 2001, 29, e57. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RN A sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef]
- Chang, H.; Lim, J.; Ha, M.; Kim, V.N. TAIL-seq: Genome-wide determination of poly(A) tail length and 3′ end modifications. Mol. Cell 2014, 53, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Lee, M.; Son, A.; Chang, H.; Kim, V.N. MTAIL-seq reveals dynamic poly(A) tail regulation in oocyte-to-embryo development. Genes Dev. 2016, 30, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Legnini, I.; Alles, J.; Karaiskos, N.; Ayoub, S.; Rajewsky, N. FLAM-seq: Full-length mRNA sequencing reveals principles of poly(A) tail length control. Nat. Methods 2019, 16, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Tushev, G.; Glock, C.; Heumüller, M.; Biever, A.; Jovanovic, M.; Schuman, E.M. Alternative 3′ UTRs Modify the Localization, Regulatory Potential, Stability, and Plasticity of mRNAs in Neuronal Compartments. Neuron 2018, 98, 495–511.e6. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Long, Y.; Ji, G.; Li, Q.Q.; Wu, X. APAtrap: Identification and quantification of alternative polyadenylation sites from RNA-seq data. Bioinformatics 2018, 34, 1841–1849. [Google Scholar] [CrossRef]
- Xia, Z.; Donehower, L.A.; Cooper, T.A.; Neilson, J.R.; Wheeler, D.A.; Wagner, E.J.; Li, W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat. Commun. 2014, 5, 5274. [Google Scholar] [CrossRef]
- Ye, C.; Zhou, Q.; Wu, X.; Yu, C.; Ji, G.; Saban, D.R.; Li, Q.Q. ScDAPA: Detection and visualization of dynamic alternative polyadenylation from single cell RNA-seq data. Bioinformatics 2020, 36, 1262–1264. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Qiu, Q.; Zhou, Q.; Ding, J.; Lu, Y.; Liu, P. Alternative polyadenylation: Methods, mechanism, function, and role in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 51. [Google Scholar] [CrossRef]
- Herrmann, C.J.; Schmidt, R.; Kanitz, A.; Artimo, P.; Gruber, A.J.; Zavolan, M. PolyASite 2.0: A consolidated atlas of polyadenylation sites from 3′ end sequencing. Nucleic Acids Res. 2020, 48, D174–D179. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Rycak, L.; Afonso-Grunz, F.; Winter, P.; Zawada, A.M.; Damrath, E.; Scheider, J.; Schmäh, J.; Koch, I.; Kahl, G.; et al. APADB: A database for alternative polyadenylation and microRNA regulation events. Database 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wei, Z.; Li, H. A change-point model for identifying 3′UTR switching by next-generation RNA sequencing. Bioinformatics 2014, 30, 2162–2170. [Google Scholar] [CrossRef] [PubMed]
- Grassi, E.; Mariella, E.; Lembo, A.; Molineris, I.; Provero, P. Roar: Detecting alternative polyadenylation with standard mRNA sequencing libraries. BMC Bioinform. 2016, 17, 423. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.C.H.; Blencowe, B.J.; Morris, Q. QAPA: A new method for the systematic analysis of alternative polyadenylation from RNA-seq data. Genome Biol. 2018, 19, 45. [Google Scholar] [CrossRef]
- Arefeen, A.; Liu, J.; Xiao, X.; Jiang, T. TAPAS: Tool for alternative polyadenylation site analysis. Bioinformatics 2018, 34, 2521–2529. [Google Scholar] [CrossRef]
- Gruber, A.J.; Schmidt, R.; Ghosh, S.; Martin, G.; Gruber, A.R.; van Nimwegen, E.; Zavolan, M. Discovery of physiological and cancer-related regulators of 3′ UTR processing with KAPAC. Genome Biol. 2018, 19, 44. [Google Scholar] [CrossRef]
- You, L.; Wu, J.; Feng, Y.; Fu, Y.; Guo, Y.; Long, L.; Zhang, H.; Luan, Y.; Tian, P.; Chen, L.; et al. APASdb: A database describing alternative poly(A) sites and selection of heterogeneous cleavage sites downstream of poly(A) signals. Nucleic Acids Res. 2015, 43, D59–D67. [Google Scholar] [CrossRef]
- Wang, R.; Nambiar, R.; Zheng, D.; Tian, B. PolyA-DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res. 2018, 46, D315–D319. [Google Scholar] [CrossRef]
- Hong, W.; Ruan, H.; Zhang, Z.; Ye, Y.; Liu, Y.; Li, S.; Jing, Y.; Zhang, H.; Diao, L.; Liang, H.; et al. APAatlas: Decoding alternative polyadenylation across human tissues. Nucleic Acids Res. 2020, 48, D34–D39. [Google Scholar] [CrossRef]
- Jin, W.; Zhu, Q.; Yang, Y.; Yang, W.; Wang, D.; Yang, J.; Niu, X.; Yu, D.; Gong, J. Animal-APAdb: A comprehensive animal alternative polyadenylation database. Nucleic Acids Res. 2020, 49, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Ishiura, H.; Tsuji, S. Advances in repeat expansion diseases and a new concept of repeat motif–phenotype correlation. Curr. Opin. Genet. Dev. 2020, 65, 176–185. [Google Scholar] [CrossRef]
- Paulson, H. Repeat Expansion Diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar] [PubMed]
- Krzyzosiak, W.J.; Sobczak, K.; Wojciechowska, M.; Fiszer, A.; Mykowska, A.; Kozlowski, P. Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res. 2012, 40, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Zhang, Y.; Xu, P.; Man, V.H.; Roland, C.; Weninger, K.; Sagui, C. Molecular conformations and dynamics of nucleotide repeats associated with neurodegenerative diseases: Double helices and CAG hairpin loops. Comput. Struct. Biotechnol. J. 2021, 19, 2819–2832. [Google Scholar] [CrossRef]
- Jae, E.K.; Drier, E.; Barbee, S.A.; Ramaswami, M.; Yin, J.C.P.; Wickens, M. GLD2 poly(A) polymerase is required for long-term memory. Proc. Natl. Acad. Sci. USA 2008, 105, 14644–14649. [Google Scholar] [CrossRef]
- Wheeler, J.M.; McMillan, P.; Strovas, T.J.; Liachko, N.F.; Amlie-Wolf, A.; Kow, R.L.; Klein, R.L.; Szot, P.; Robinson, L.; Guthrie, C.; et al. Activity of the poly(A) binding protein MSUT2 determines susceptibility to pathological tau in the mammalian brain. Sci. Transl. Med. 2019, 11, aao6545. [Google Scholar] [CrossRef]
- Morris, K.J.; Corbett, A.H. The polyadenosine RNA-binding protein ZC3H14 interacts with the THO complex and coordinately regulates the processing of neuronal transcripts. Nucleic Acids Res. 2018, 46, 6561–6575. [Google Scholar] [CrossRef]
- Lee, J.; Kim, M.; Itoh, T.Q.; Lim, C. Ataxin-2: A versatile posttranscriptional regulator and its implication in neural function. Wiley Interdiscip. Rev. RNA 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Rüb, U.; Auburger, G. Spinocerebellar ataxia 2 (SCA2). Cerebellum 2008, 7, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G.; et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef]
- Tian, B.; Hu, J.; Zhang, H.; Lutz, C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005, 33, 201–212. [Google Scholar] [CrossRef]
- Lianoglou, S.; Garg, V.; Yang, J.L.; Leslie, C.S.; Mayr, C. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 2013, 27, 2380–2396. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, C.C. Tissue-specific mechanisms of alternative polyadenylation: Testis, brain, and beyond (2018 update). RNA 2019, 10, e1526. [Google Scholar] [CrossRef]
- Smibert, P.; Miura, P.; Westholm, J.O.; Shenker, S.; May, G.; Duff, M.O.; Zhang, D.; Eads, B.D.; Carlson, J.; Brown, J.B.; et al. Global Patterns of Tissue-Specific Alternative Polyadenylation in Drosophila. Cell Rep. 2012, 1, 277–289. [Google Scholar] [CrossRef]
- Miura, P.; Shenker, S.; Andreu-Agullo, C.; Westholm, J.O.; Lai, E.C. Widespread and extensive lengthening of 39 UTRs in the mammalian brain. Genome Res. 2013, 23, 812–825. [Google Scholar] [CrossRef]
- Wei, L.; Lee, S.; Majumdar, S.; Zhang, B.; Sanfilippo, P.; Joseph, B.; Miura, P.; Soller, M.; Lai, E.C. Overlapping Activities of ELAV/Hu Family RNA Binding Proteins Specify the Extended Neuronal 3′ UTR Landscape in Drosophila. Mol. Cell 2020, 80, 140–155.e6. [Google Scholar] [CrossRef]
- Soller, M.; White, K. ELAV inhibits 3′-end processing to promote neural splicing of ewg pre-mRNA. Genes Dev. 2003, 17, 2526–2538. [Google Scholar] [CrossRef] [PubMed]
- Bae, B.; Miura, P. Emerging roles for 3′ UTRs in neurons. Int. J. Mol. Sci. 2020, 21, 3413. [Google Scholar] [CrossRef]
- Hinman, M.N.; Lou, H. Diverse molecular functions of Hu proteins. Cell. Mol. Life Sci. 2008, 65, 3168–3181. [Google Scholar] [CrossRef]
- Fernandopulle, M.S.; Lippincott-Schwartz, J.; Ward, M.E. RNA transport and local translation in neurodevelopmental and neurodegenerative disease. Nat. Neurosci. 2021, 24, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Blazie, S.M.; Babb, C.; Wilky, H.; Rawls, A.; Park, J.G.; Mangone, M. Comparative RNA-Seq analysis reveals pervasive tissue-specific alternative polyadenylation in Caenorhabditis elegans intestine and muscles. BMC Biol. 2015, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Rocancourt, D.; Mansouri, A.; Buckingham, M. Divergent functions of murine Pax3 and Pax7 in limb muscle development. Genes Dev. 2004, 18, 1088–1105. [Google Scholar] [CrossRef]
- Der Vartanian, A.; Quétin, M.; Michineau, S.; Auradé, F.; Hayashi, S.; Dubois, C.; Rocancourt, D.; Drayton-Libotte, B.; Szegedi, A.; Buckingham, M.; et al. PAX3 Confers Functional Heterogeneity in Skeletal Muscle Stem Cell Responses to Environmental Stress. Cell Stem Cell 2019, 24, 958–973.e9. [Google Scholar] [CrossRef] [PubMed]
- Scaramozza, A.; Park, D.; Kollu, S.; Beerman, I.; Sun, X.; Rossi, D.J.; Lin, C.P.; Scadden, D.T.; Crist, C.; Brack, A.S. Lineage Tracing Reveals a Subset of Reserve Muscle Stem Cells Capable of Clonal Expansion under Stress. Cell Stem Cell 2019, 24, 944–957.e5. [Google Scholar] [CrossRef] [PubMed]
- Boutet, S.C.; Cheung, T.H.; Quach, N.L.; Liu, L.; Prescott, S.L.; Edalati, A.; Iori, K.; Rando, T.A. Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell 2012, 10, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Verma, M.; Watanabe, S.; Tastad, C.; Asakura, Y.; Asakura, A. MyoD regulates apoptosis of myoblasts through microRNA-mediated down-regulation of Pax3. J. Cell Biol. 2010, 191, 347–365. [Google Scholar] [CrossRef]
- Crist, C.G.; Montarras, D.; Pallafacchina, G.; Rocancourt, D.; Cumano, A.; Conway, S.J.; Buckinghama, M. Muscle stem cell behavior is modified by microRNA-27 regulation of Pax3 expression. Proc. Natl. Acad. Sci. USA 2009, 106, 13383–13387. [Google Scholar] [CrossRef]
- Tomé, F.M.S.; Chateau, D.; Helbling-Leclerc, A.; Fardeau, M. Morphological changes in muscle fibers in oculopharyngeal muscular dystrophy. Neuromuscul. Disord. 1997, 7, 63–69. [Google Scholar] [CrossRef]
- Malerba, A.; Klein, P.; Bachtarzi, H.; Jarmin, S.A.; Cordova, G.; Ferry, A.; Strings, V.; Espinoza, M.P.; Mamchaoui, K.; Blumen, S.C.; et al. PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nat. Commun. 2017, 8, 14848. [Google Scholar] [CrossRef]
- Apponi, L.H.; Leung, S.W.; Williams, K.R.; Valentini, S.R.; Corbett, A.H.; Pavlath, G.K. Loss of nuclear poly(A)-binding protein 1 causes defects in myogenesis and mRNA biogenesis. Hum. Mol. Genet. 2010, 19, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Simonelig, M. PABPN1 shuts down alternative poly(A) sites. Cell Res. 2012, 22, 1419–1421. [Google Scholar] [CrossRef]
- Jacquemont, S.; Hagerman, R.J.; Hagerman, P.J.; Leehey, M.A. Fragile-X syndrome and fragile X-associated tremor/ataxia syndrome: Two faces of FMR1. Lancet Neurol. 2007, 6, 45–55. [Google Scholar] [CrossRef]
- Sunamura, N.; Iwashita, S.; Enomoto, K.; Kadoshima, T.; Isono, F. Loss of the fragile X mental retardation protein causes aberrant differentiation in human neural progenitor cells. Sci. Rep. 2018, 8, 11585. [Google Scholar] [CrossRef] [PubMed]
- Cabal-Herrera, A.M.; Tassanakijpanich, N.; Salcedo-Arellano, M.J.; Hagerman, R.J. Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS): Pathophysiology and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 4391. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; De Rubeis, S.; Carosi, C.; La Fata, G.; Serpa, G.; Raske, C.; Willemsen, R.; Hagerman, P.J.; Bagni, C. Differential usage of transcriptional start sites and polyadenylation sites in FMR1 premutation alleles. Nucleic Acids Res. 2011, 39, 6172–6185. [Google Scholar] [CrossRef]
- Melamed, Z.; Lopez-Erauskin, J.; Baughn, M.W.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.; et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180. [Google Scholar] [CrossRef]
- Patel, R.; Brophy, C.; Hickling, M.; Neve, J.; Furger, A. Alternative cleavage and polyadenylation of genes associated with protein turnover and mitochondrial function are deregulated in Parkinson’s, Alzheimer’s and ALS disease. BMC Med. Genom. 2019, 12, 1–14. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Niblock, M.; Smith, B.N.; Lee, Y.B.; Sardone, V.; Topp, S.; Troakes, C.; Al-Sarraj, S.; Leblond, C.S.; Dion, P.A.; Rouleau, G.A.; et al. Retention of hexanucleotide repeat-containing intron in C9orf72 mRNA: Implications for the pathogenesis of ALS/FTD. Acta Neuropathol. Commun. 2016, 4, 18. [Google Scholar] [CrossRef]
- Prudencio, M.; Belzil, V.V.; Batra, R.; Ross, C.A.; Gendron, T.F.; Pregent, L.J.; Murray, M.E.; Overstreet, K.K.; Piazza-Johnston, A.E.; Desaro, P.; et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 2015, 18, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Giannini, M.; Bayona-Feliu, A.; Sproviero, D.; Barroso, S.I.; Cereda, C.; Aguilera, A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLoS Genet. 2020, 16, e1009260. [Google Scholar] [CrossRef] [PubMed]
- Prashad, S.; Gopal, P.P. RNA-binding proteins in neurological development and disease. RNA Biol. 2021, 18, 972. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.; Birsa, N.; Milioto, C.; McLaughlin, M.; Ule, A.M.; Robaldo, D.; Eberle, A.B.; Kräuchi, R.; Bentham, M.; Brown, A.L.; et al. FUS ALS-causative mutations impair FUS autoregulation and splicing factor networks through intron retention. Nucleic Acids Res. 2020, 48, 6889–6905. [Google Scholar] [CrossRef] [PubMed]
- Koyama, A.; Sugai, A.; Kato, T.; Ishihara, T.; Shiga, A.; Toyoshima, Y.; Koyama, M.; Konno, T.; Hirokawa, S.; Yokoseki, A.; et al. Increased cytoplasmic TARDBP mRNA in affected spinal motor neurons in ALS caused by abnormal autoregulation of TDP-43. Nucleic Acids Res. 2016, 44, 5820–5836. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, S.; Weiner, B.; Arnold, W.D. Myotonic Dystrophies: Targeting Therapies for Multisystem Disease. Neurotherapeutics 2018, 15, 872–884. [Google Scholar] [CrossRef]
- Ozimski, L.L.; Sabater-Arcis, M.; Bargiela, A.; Artero, R. The hallmarks of myotonic dystrophy type 1 muscle dysfunction. Biol. Rev. 2021, 96, 716–730. [Google Scholar] [CrossRef]
- Kurkiewicz, A.; Cooper, A.; McIlwaine, E.; Cumming, S.A.; Adam, B.; Krahe, R.; Puymirat, J.; Schoser, B.; Timchenko, L.; Ashizawa, T.; et al. Towards development of a statistical framework to evaluate myotonic dystrophy type 1 mRNA biomarkers in the context of a clinical trial. PLoS ONE 2020, 15, e0231000. [Google Scholar] [CrossRef]
- Gudde, A.E.E.G.; van Kessel, I.D.G.; André, L.M.; Wieringa, B.; Wansink, D.G. Trinucleotide-repeat expanded and normal DMPK transcripts contain unusually long poly(A) tails despite differential nuclear residence. Biochim. Biophys. Acta—Gene Regul. Mech. 2017, 1860, 740–749. [Google Scholar] [CrossRef]
- Kino, Y.; Mori, D.; Oma, Y.; Takeshita, Y.; Sasagawa, N.; Ishiura, S. Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum. Mol. Genet. 2004, 13, 495–507. [Google Scholar] [CrossRef][Green Version]
- Yuan, Y.; Compton, S.A.; Sobczak, K.; Stenberg, M.G.; Thornton, C.A.; Griffith, J.D.; Swanson, M.S. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 2007, 35, 5474–5486. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, P.; Stepniak-Konieczna, E.; Sobczak, K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res. 2014, 42, 10873–10887. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.; Sznajder, Ł.J.; Cywoniuk, P.; Thomas, J.D.; Swanson, M.S.; Sobczak, K. MBNL splicing activity depends on RNA binding site structural context. Nucleic Acids Res. 2018, 46, 9119–9133. [Google Scholar] [CrossRef]
- Brinegar, A.E.; Cooper, T.A. Roles for RNA-binding proteins in development and disease. Brain Res. 2016, 1647, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Cody, N.A.L.; Jog, S.; Biancolella, M.; Wang, T.T.; Treacy, D.J.; Luo, S.; Schroth, G.P.; Housman, D.E.; Reddy, S.; et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 2012, 150, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Itskovich, S.S.; Gurunathan, A.; Clark, J.; Burwinkel, M.; Wunderlich, M.; Berger, M.R.; Kulkarni, A.; Chetal, K.; Venkatasubramanian, M.; Salomonis, N.; et al. MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia. Nat. Commun. 2020, 11, 2369. [Google Scholar] [CrossRef]
- Mankodi, A. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 2001, 10, 2165–2170. [Google Scholar] [CrossRef]
- Jiang, H.; Mankodi, A.; Swanson, M.S.; Moxley, R.T.; Thornton, C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004, 13, 3079–3088. [Google Scholar] [CrossRef]
- Song, K.Y.; Guo, X.M.; Wang, H.Q.; Zhang, L.; Huang, S.Y.; Huo, Y.C.; Zhang, G.; Feng, J.Z.; Zhang, R.R.; Ma, Y.; et al. MBNL1 reverses the proliferation defect of skeletal muscle satellite cells in myotonic dystrophy type 1 by inhibiting autophagy via the mTOR pathway. Cell Death Dis. 2020, 11, 545. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Mccolgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Romo, L.; Ashar-Patel, A.; Pfister, E.; Aronin, N. Alterations in mRNA 3′ UTR Isoform Abundance Accompany Gene Expression Changes in Human Huntington’s Disease Brains. Cell Rep. 2017, 20, 3057–3070. [Google Scholar] [CrossRef]
- Lin, B.; Rommens, J.M.; Graham, R.K.; Kalchman, M.; Macdonald, H.; Nasir, J.; Delaney, A.; Goldberg, Y.P.; Hayden, M.R. Differential 3′ polyadenylation of the huntington disease gene results in two mRNA species with variable tissue expression. Hum. Mol. Genet. 1993, 2, 1541–1545. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; An, J.J.; Xu, B. Distinct cellular toxicity of two mutant huntingtin mRNA variants due to translation regulation. PLoS ONE 2017, 12, e0177610. [Google Scholar] [CrossRef] [PubMed]
- Sathasivam, K.; Neueder, A.; Gipson, T.A.; Landles, C.; Benjamin, A.C.; Bondulich, M.K.; Smith, D.L.; Faull, R.L.M.; Roos, R.A.C.; Howland, D.; et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, 2366–2370. [Google Scholar] [CrossRef] [PubMed]
- Neueder, A.; Landles, C.; Ghosh, R.; Howland, D.; Myers, R.H.; Faull, R.L.M.; Tabrizi, S.J.; Bates, G.P. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci. Rep. 2017, 7, 1307. [Google Scholar] [CrossRef]
- Neueder, A.; Dumas, A.A.; Benjamin, A.C.; Bates, G.P. Regulatory mechanisms of incomplete huntingtin mRNA splicing. Nat. Commun. 2018, 9, 3955. [Google Scholar] [CrossRef]
- Mason, M.A.; Gomez-Paredes, C.; Sathasivam, K.; Neueder, A.; Papadopoulou, A.S.; Bates, G.P. Silencing Srsf6 does not modulate incomplete splicing of the huntingtin gene in Huntington’s disease models. Sci. Rep. 2020, 10, 14057. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Shakkottai, V.G.; Albin, R.L. Polyglutamine Repeats in Neurodegenerative Diseases. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 1–27. [Google Scholar] [CrossRef]
- Kamieniarz-Gdula, K.; Proudfoot, N.J. Transcriptional Control by Premature Termination: A Forgotten Mechanism. Trends Genet. 2019, 35, 553–564. [Google Scholar] [CrossRef]
- Ciesiolka, A.; Jazurek, M.; Drazkowska, K.; Krzyzosiak, W.J. Structural characteristics of simple RNA repeats associated with disease and their deleterious protein interactions. Front. Cell. Neurosci. 2017, 11, 1–19. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Ridwan Amode, M.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chen, S.T.; Juan, H.F.; Huang, H.C. Lengthening of 3′UTR increases with morphological complexity in animal evolution. Bioinformatics 2012, 28, 3178–3181. [Google Scholar] [CrossRef]
- Mayr, C. What are 3′ utrs doing? Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef]
- Zhu, S.; Lian, Q.; Ye, W.; Qin, W.; Wu, Z.; Ji, G.; Wu, X. scAPAdb: A comprehensive database of alternative polyadenylation at single-cell resolution. Nucleic Acids Res. 2021, gkab795. [Google Scholar] [CrossRef]
- Gao, Y.; Li, L.; Amos, C.I.; Li, W. Analysis of alternative polyadenylation from singlecell RNA-seq using scDaPars reveals cell subpopulations invisible to gene expression. Genome Res. 2021, 31, 1856–1866. [Google Scholar] [CrossRef]
- Saxena, S.; Caroni, P. Selective Neuronal Vulnerability in Neurodegenerative Diseases: From Stressor Thresholds to Degeneration. Neuron 2011, 71, 35–48. [Google Scholar] [CrossRef]
- Ogorodnikov, A.; Levin, M.; Tattikota, S.; Tokalov, S.; Hoque, M.; Scherzinger, D.; Marini, F.; Poetsch, A.; Binder, H.; Macher-Göppinger, S.; et al. Transcriptome 3′end organization by PCF11 links alternative polyadenylation to formation and neuronal differentiation of neuroblastoma. Nat. Commun. 2018, 9, 5331. [Google Scholar] [CrossRef]
- Masamha, C.P.; Xia, Z.; Yang, J.; Albrecht, T.R.; Li, M.; Shyu, A.-B.; Li, W.; Wagner, E.J. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 2014, 510, 412–416. [Google Scholar] [CrossRef]
- Fiszer, A.; Krzyzosiak, W.J. Oligonucleotide-based strategies to combat polyglutamine diseases. Nucleic Acids Res. 2014, 42, 6787–6810. [Google Scholar] [CrossRef] [PubMed]
- Fiszer, A.; Olejniczak, M.; Galka-Marciniak, P.; Mykowska, A.; Krzyzosiak, W.J. Self-duplexing CUG repeats selectively inhibit mutant huntingtin expression. Nucleic Acids Res. 2013, 41, 10426–10437. [Google Scholar] [CrossRef]
- Ciesiolka, A.; Stroynowska-Czerwinska, A.; Joachimiak, P.; Ciolak, A.; Kozlowska, E.; Michalak, M.; Dabrowska, M.; Olejniczak, M.; Raczynska, K.D.; Zielinska, D.; et al. Artificial miRNAs targeting CAG repeat expansion in ORFs cause rapid deadenylation and translation inhibition of mutant transcripts. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
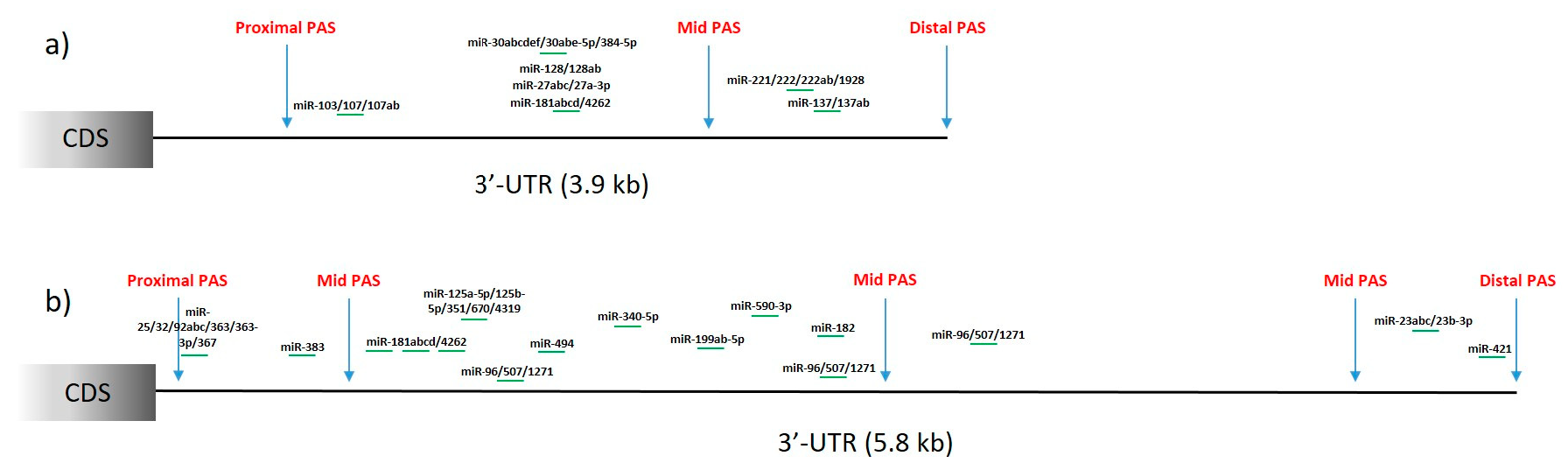{kind=link}
| Name | Year of Publication | Website | References |
|---|---|---|---|
| Computational tools for APA detection | |||
| DaPars | 2014 | https://github.com/ZhengXia/dapars | [58] |
| Change point | 2014 | http://utr.sourceforge.net | [63] |
| Roar | 2016 | https://github.com/vodkatad/roar | [64] |
| APAtrap | 2018 | https://apatrap.sourceforge.io | [57] |
| QAPA | 2018 | https://www.github.com/morrislab/qapa | [65] |
| TAPAS | 2018 | https://github.com/arefeen/TAPAS | [66] |
| KAPAC | 2018 | https://github.com/zavolanlab/PAQR_KAPAC.git | [67] |
| scDAPA | 2019 | https://scdapa.sourceforge.io | [59] |
| APA databases | |||
| PolyA-Seq Atlas | 2012 | http://genome.ucsc.edu/ | [10] |
| APADB | 2014 | http://tools.genxpro.net:9000/apadb/ | [62] |
| APASdb | 2015 | http://genome.bucm.edu.cn/utr/ | [68] |
| PolyA_DB3 | 2018 | https://exon.apps.wistar.org/PolyA_DB/v3/ | [69] |
| APAatlas | 2020 | https://hanlab.uth.edu/apa/ | [70] |
| PolyAsite 2.0 | 2020 | https://polyasite.unibas.ch | [61] |
| Animal-APAdb | 2021 | http://gong_lab.hzau.edu.cn/Animal-APAdb/ | [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joachimiak, P.; Ciesiołka, A.; Figura, G.; Fiszer, A. Implications of Poly(A) Tail Processing in Repeat Expansion Diseases. Cells 2022, 11, 677. https://doi.org/10.3390/cells11040677
Joachimiak P, Ciesiołka A, Figura G, Fiszer A. Implications of Poly(A) Tail Processing in Repeat Expansion Diseases. Cells. 2022; 11(4):677. https://doi.org/10.3390/cells11040677
Chicago/Turabian StyleJoachimiak, Paweł, Adam Ciesiołka, Grzegorz Figura, and Agnieszka Fiszer. 2022. "Implications of Poly(A) Tail Processing in Repeat Expansion Diseases" Cells 11, no. 4: 677. https://doi.org/10.3390/cells11040677
APA StyleJoachimiak, P., Ciesiołka, A., Figura, G., & Fiszer, A. (2022). Implications of Poly(A) Tail Processing in Repeat Expansion Diseases. Cells, 11(4), 677. https://doi.org/10.3390/cells11040677