
Analysis of the Glucose-Dependent Transcriptome in Murine Hypothalamic Cells


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Transfection
2.3. Total RNA-seq
2.4. Quantitative RT-PCR (qRT-PCR)
2.5. Glucose Stimulation
2.6. Reporter Gene Assay
2.7. Whole-Cell ELISA
2.8. Cholesterol Detection
2.9. cAMP Accumulation
2.10. Quantification and Statistical Analysis
3. Results
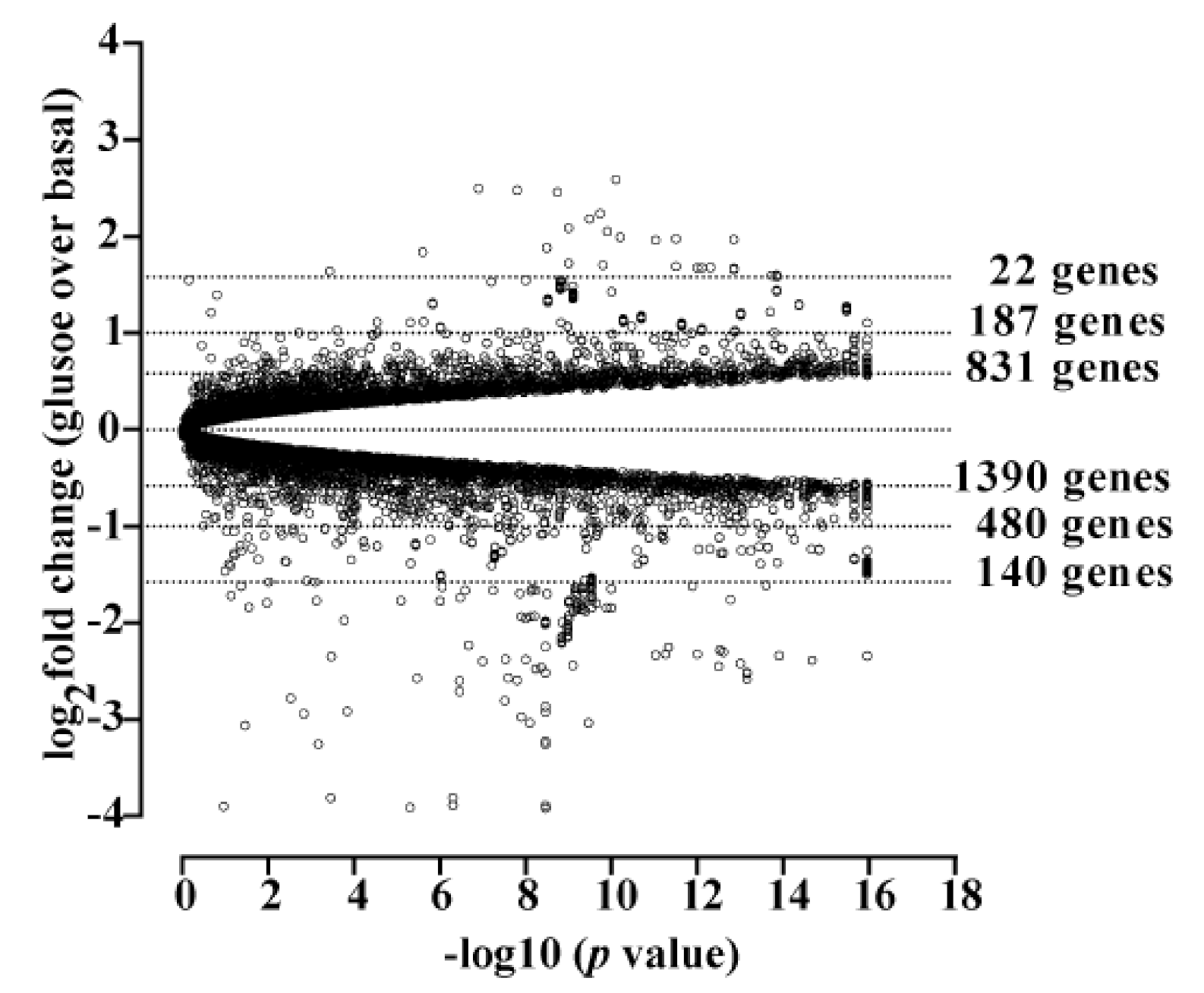
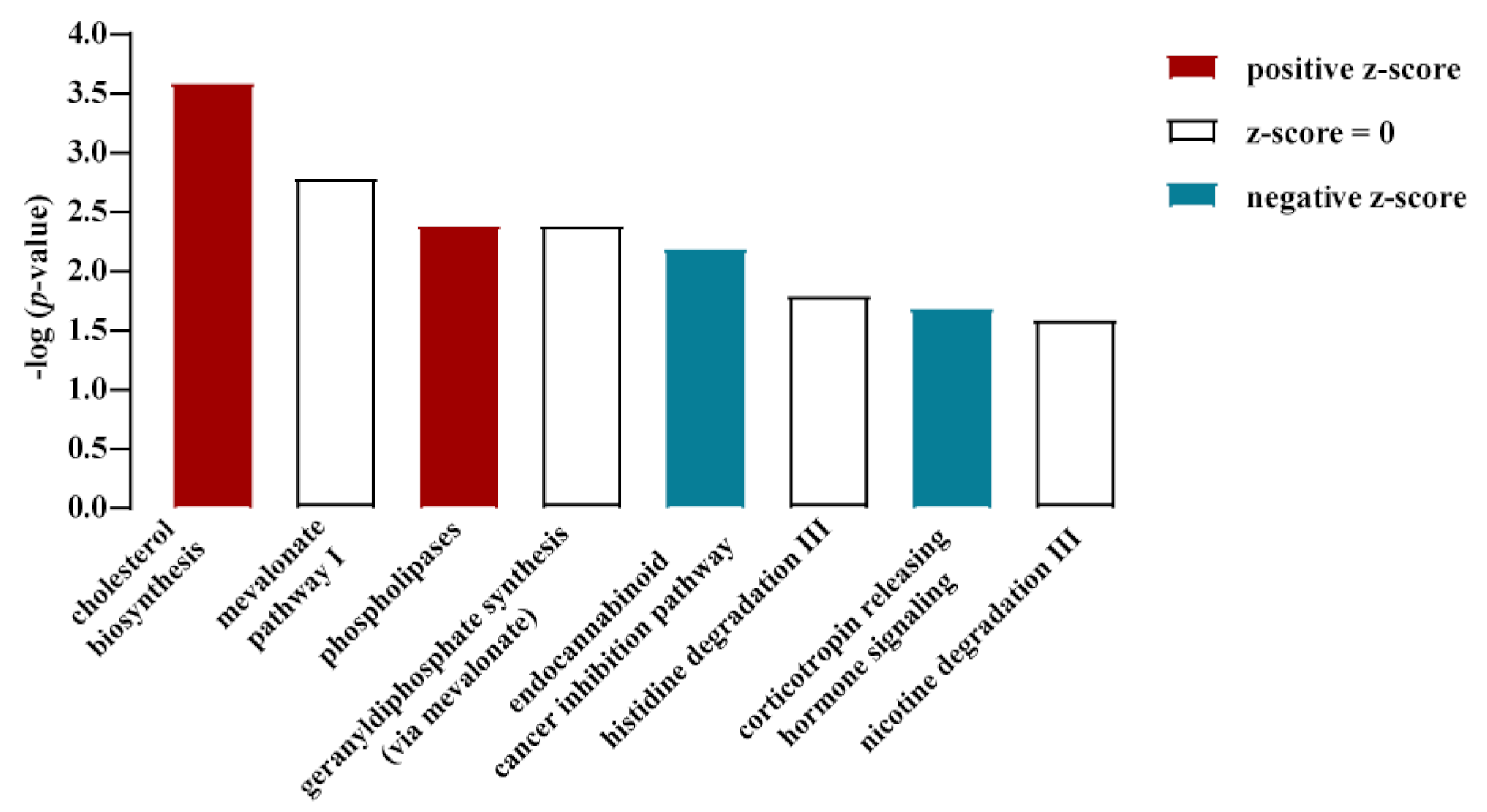
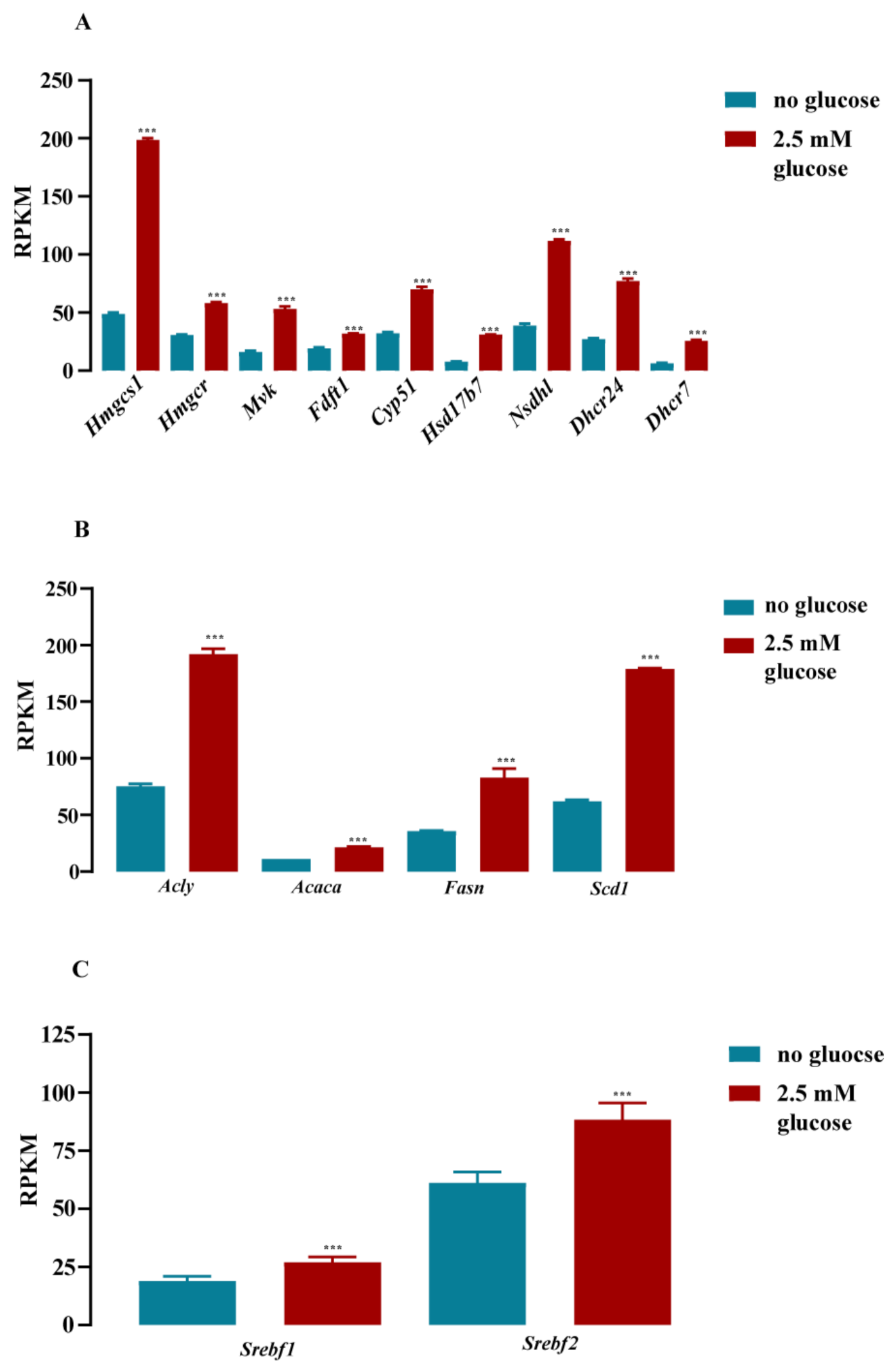
3.1. Cholesterol Biosynthesis Was the Major Target of Glucose-Dependent Gene Expression
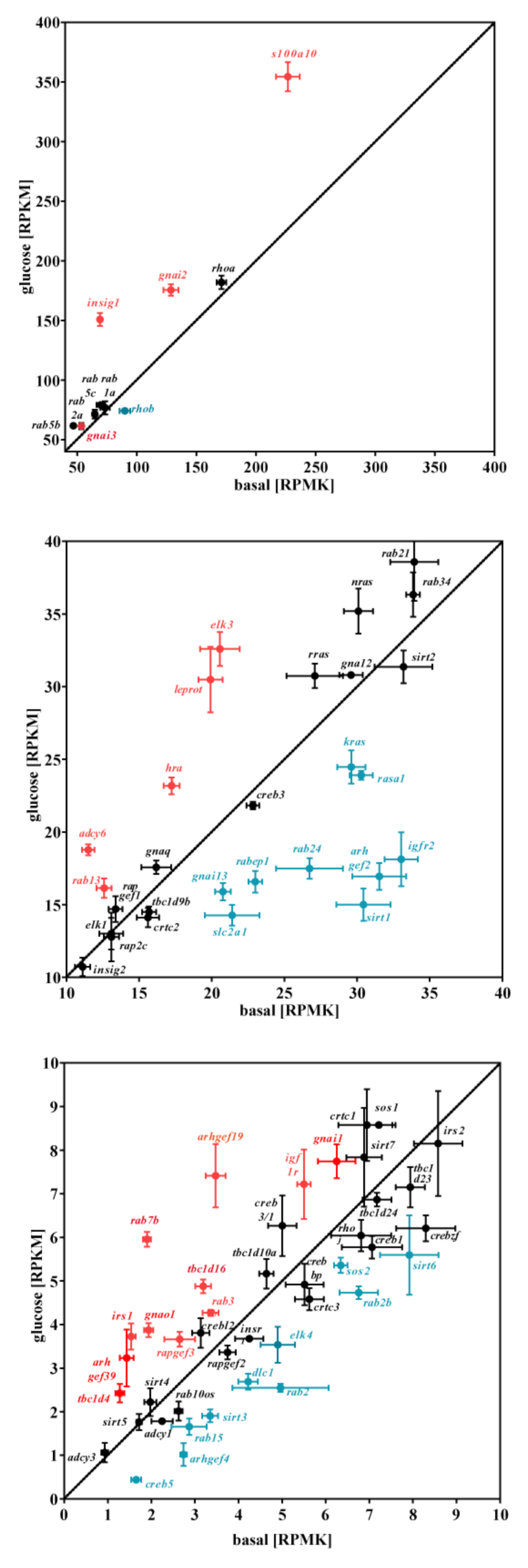
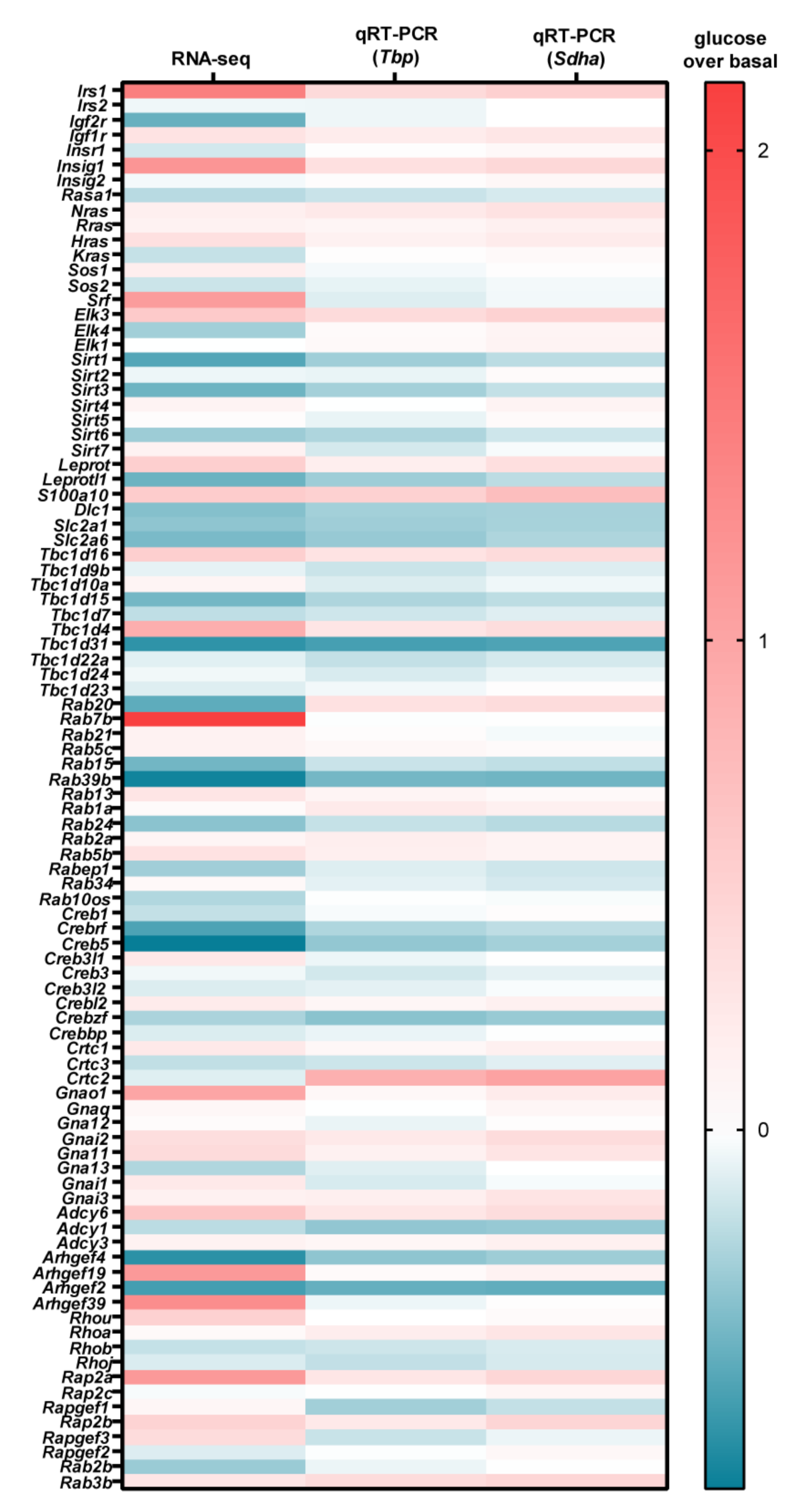
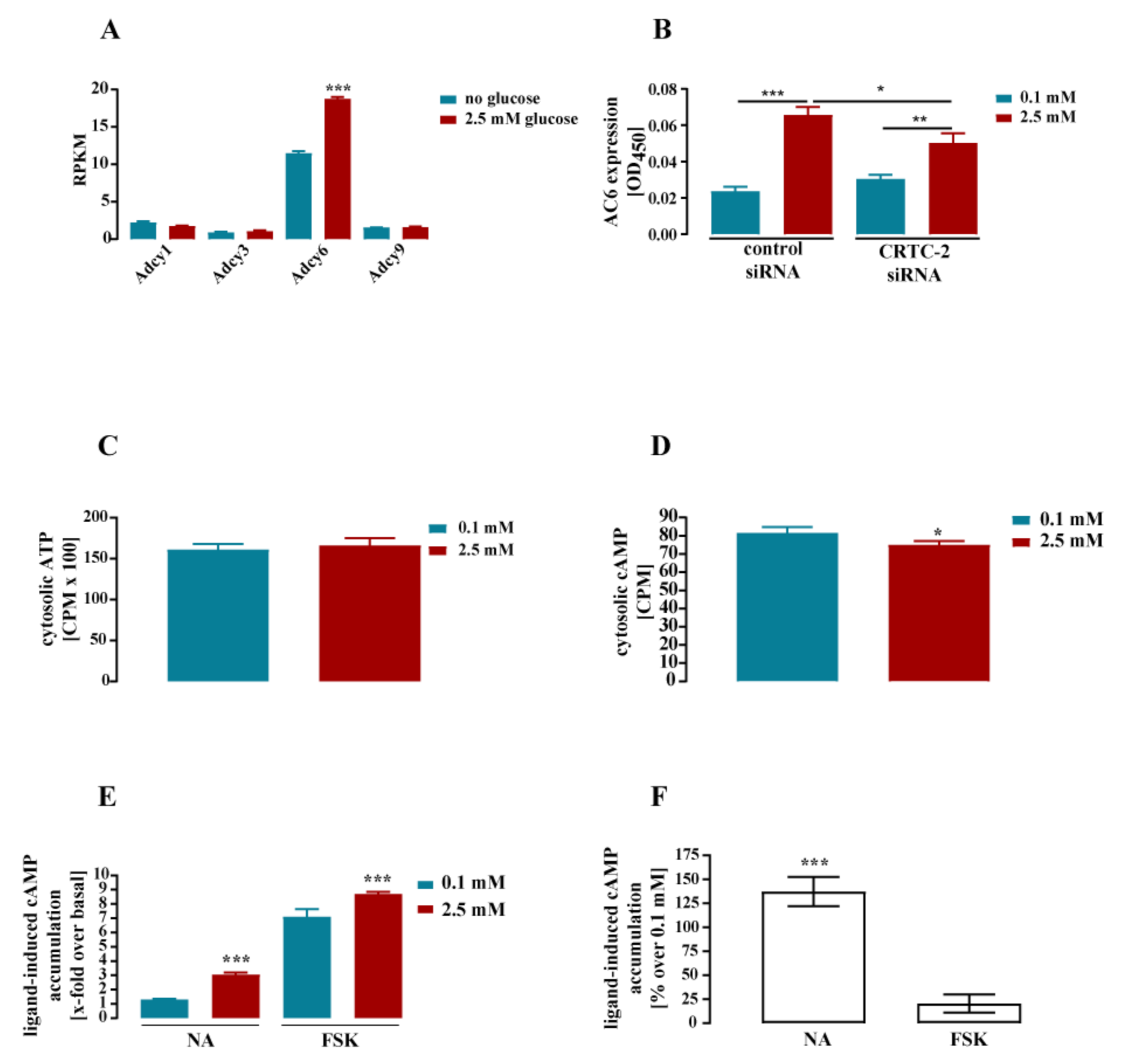
3.2. Glucose-Dependent Gene Expression Altered Signaling Pathways of RTK and GPCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Criego, A.B.; Tkac, I.; Kumar, A.; Thomas, W.; Gruetter, R.; Seaquist, E.R. Brain glucose concentrations in patients with type 1 diabetes and hypoglycemia unawareness. J. Neurosci. Res. 2005, 79, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Danaei, G.; Finucane, M.M.; Lu, Y.; Singh, G.M.; Cowan, M.J.; Paciorek, C.J.; Lin, J.K.; Farzadfar, F.; Khang, Y.H.; Stevens, G.A.; et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: Systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet 2011, 378, 31–40. [Google Scholar] [CrossRef]
- Cersosimo, E.; Triplitt, C.; Solis-Herrera, C.; Mandarino, L.J.; DeFronzo, R.A. Pathogenesis of Type 2 Diabetes Mellitus. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; Endotext: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Castillo, M.; Nemery, A.; Verdin, E.; Lefebvre, P.J.; Luyckx, A.S. Circadian profiles of blood glucose and plasma free insulin during treatment with semisynthetic and biosynthetic human insulin, and comparison with conventional monocomponent preparations. Eur. J. Clin. Pharmacol. 1983, 25, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Gruetter, R.; Novotny, E.J.; Boulware, S.D.; Rothman, D.L.; Mason, G.F.; Shulman, G.I.; Shulman, R.G.; Tamborlane, W.V. Direct measurement of brain glucose concentrations in humans by 13C NMR spectroscopy. Proc. Natl. Acad. Sci. USA 1992, 89, 1109–1112. [Google Scholar] [CrossRef]
- Silver, I.A.; Erecinska, M. Extracellular glucose concentration in mammalian brain: Continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J. Neurosci. 1994, 14, 5068–5076. [Google Scholar] [CrossRef]
- Seaquist, E.R.; Tkac, I.; Damberg, G.; Thomas, W.; Gruetter, R. Brain glucose concentrations in poorly controlled diabetes mellitus as measured by high-field magnetic resonance spectroscopy. Metabolism 2005, 54, 1008–1013. [Google Scholar] [CrossRef]
- Jacob, R.J.; Fan, X.; Evans, M.L.; Dziura, J.; Sherwin, R.S. Brain glucose levels are elevated in chronically hyperglycemic diabetic rats: No evidence for protective adaptation by the blood brain barrier. Metabolism 2002, 51, 1522–1524. [Google Scholar] [CrossRef]
- Mayer, C.H.; Fink, H.; Rex, A.; Voigt, J.P. Changes in extracellular hypothalamic glucose in relation to feeding. Eur. J. Neurosci. 2006, 24, 1695–1701. [Google Scholar] [CrossRef]
- de Andrade, I.S.; Zemdegs, J.C.; de Souza, A.P.; Watanabe, R.L.; Telles, M.M.; Nascimento, C.M.; Oyama, L.M.; Ribeiro, E.B. Diet-induced obesity impairs hypothalamic glucose sensing but not glucose hypothalamic extracellular levels, as measured by microdialysis. Nutr. Diabetes 2015, 5, e162. [Google Scholar] [CrossRef]
- Liu, C.C.; Hu, J.; Tsai, C.W.; Yue, M.; Melrose, H.L.; Kanekiyo, T.; Bu, G. Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J. Neurosci. 2015, 35, 5851–5859. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef]
- Solis, E., Jr.; Cameron-Burr, K.T.; Shaham, Y.; Kiyatkin, E.A. Intravenous Heroin Induces Rapid Brain Hypoxia and Hyperglycemia that Precede Brain Metabolic Response. eNeuro 2017, 4. [Google Scholar] [CrossRef]
- Amin, A.; Dhillo, W.S.; Murphy, K.G. The central effects of thyroid hormones on appetite. J. Thyroid Res. 2011, 2011, 306510. [Google Scholar] [CrossRef]
- Nillni, E.A. Regulation of the hypothalamic thyrotropin releasing hormone (TRH) neuron by neuronal and peripheral inputs. Front. Neuroendocrinol. 2010, 31, 134–156. [Google Scholar] [CrossRef]
- Celani, M.F.; Bonati, M.E.; Stucci, N. Prevalence of abnormal thyrotropin concentrations measured by a sensitive assay in patients with type 2 diabetes mellitus. Diabetes Res. 1994, 27, 15–25. [Google Scholar]
- Wilber, J.F.; Banerji, A.; Prasad, C.; Mori, M. Alterations in hypothalamic-pituitary-thyroid regulation produced by diabetes mellitus. Life Sci. 1981, 28, 1757–1763. [Google Scholar] [CrossRef]
- Hage, M.; Zantout, M.S.; Azar, S.T. Thyroid disorders and diabetes mellitus. J. Thyroid Res. 2011, 2011, 439463. [Google Scholar] [CrossRef]
- Nascimento-Saba, C.C.; Breitenbach, M.M.; Rosenthal, D. Pituitary-thyroid axis in short- and long-term experimental diabetes mellitus. Braz. J. Med. Biol. Res. 1997, 30, 269–274. [Google Scholar] [CrossRef][Green Version]
- Breit, A.; Miek, L.; Schredelseker, J.; Geibel, M.; Merrow, M.; Gudermann, T. Insulin-like growth factor-1 acts as a zeitgeber on hypothalamic circadian clock gene expression via glycogen synthase kinase-3beta signaling. J. Biol. Chem. 2018, 293, 17278–17290. [Google Scholar] [CrossRef]
- Little, K.Y.; Duncan, G.E.; Breese, G.R.; Stumpf, W.E. Beta-adrenergic receptor binding in human and rat hypothalamus. Biol. Psychiatry 1992, 32, 512–522. [Google Scholar] [CrossRef]
- Grimm, Y.; Reichlin, S. Thyrotropin releasing hormone (TRH): Neurotransmitter regulation of secretion by mouse hypothalamic tissue in vitro. Endocrinology 1973, 93, 626–631. [Google Scholar] [CrossRef]
- Daftary, S.S.; Gore, A.C. IGF-1 in the brain as a regulator of reproductive neuroendocrine function. Exp. Biol. Med. 2005, 230, 292–306. [Google Scholar] [CrossRef]
- Daftary, S.S.; Gore, A.C. The hypothalamic insulin-like growth factor-1 receptor and its relationship to gonadotropin-releasing hormones neurones during postnatal development. J. Neuroendocrinol. 2004, 16, 160–169. [Google Scholar] [CrossRef]
- Daftary, S.S.; Gore, A.C. Developmental changes in hypothalamic insulin-like growth factor-1: Relationship to gonadotropin-releasing hormone neurons. Endocrinology 2003, 144, 2034–2045. [Google Scholar] [CrossRef][Green Version]
- Routh, V.H. Glucose sensing neurons in the ventromedial hypothalamus. Sensors 2010, 10, 9002–9025. [Google Scholar] [CrossRef]
- Melnick, I.V.; Price, C.J.; Colmers, W.F. Glucosensing in parvocellular neurons of the rat hypothalamic paraventricular nucleus. Eur. J. Neurosci. 2011, 34, 272–282. [Google Scholar] [CrossRef]
- Burdakov, D.; Luckman, S.M.; Verkhratsky, A. Glucose-sensing neurons of the hypothalamus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2227–2235. [Google Scholar] [CrossRef]
- Song, Z.; Routh, V.H. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 2005, 54, 15–22. [Google Scholar] [CrossRef]
- Wang, R.; Liu, X.; Hentges, S.T.; Dunn-Meynell, A.A.; Levin, B.E.; Wang, W.; Routh, V.H. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 2004, 53, 1959–1965. [Google Scholar] [CrossRef]
- Lerner, R.G.; Depatie, C.; Rutter, G.A.; Screaton, R.A.; Balthasar, N. A role for the CREB co-activator CRTC2 in the hypothalamic mechanisms linking glucose sensing with gene regulation. EMBO Rep. 2009, 10, 1175–1181. [Google Scholar] [CrossRef]
- Damm, E.; Buech, T.R.; Gudermann, T.; Breit, A. Melanocortin-induced PKA activation inhibits AMPK activity via ERK-1/2 and LKB-1 in hypothalamic GT1-7 cells. Mol. Endocrinol. 2012, 26, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Guan, T.; Zhang, H.; Xia, Y.; Liu, F.; Zhang, Y. Inhibitory crosstalk between ERK and AMPK in the growth and proliferation of cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2008, 368, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Havula, E.; Hietakangas, V. Glucose sensing by ChREBP/MondoA-Mlx transcription factors. Semin. Cell Dev. Biol. 2012, 23, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Meugnier, E.; Rome, S.; Vidal, H. Regulation of gene expression by glucose. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 518–522. [Google Scholar] [CrossRef]
- Vaulont, S.; Vasseur-Cognet, M.; Kahn, A. Glucose regulation of gene transcription. J. Biol. Chem. 2000, 275, 31555–31558. [Google Scholar] [CrossRef]
- Breit, A.; Wicht, K.; Boekhoff, I.; Glas, E.; Lauffer, L.; Muckter, H.; Gudermann, T. Glucose Enhances Basal or Melanocortin-Induced cAMP-Response Element Activity in Hypothalamic Cells. Mol. Endocrinol. 2016, 30, 748–762. [Google Scholar] [CrossRef][Green Version]
- Belsham, D.D.; Fick, L.J.; Dalvi, P.S.; Centeno, M.L.; Chalmers, J.A.; Lee, P.K.; Wang, Y.; Drucker, D.J.; Koletar, M.M. Ciliary neurotrophic factor recruitment of glucagon-like peptide-1 mediates neurogenesis, allowing immortalization of adult murine hypothalamic neurons. FASEB J. 2009, 23, 4256–4265. [Google Scholar] [CrossRef]
- Breit, A.; Besik, V.; Solinski, H.J.; Muehlich, S.; Glas, E.; Yarwood, S.J.; Gudermann, T. Serine-727 phosphorylation activates hypothalamic STAT-3 independently from tyrosine-705 phosphorylation. Mol. Endocrinol. 2015, 29, 445–459. [Google Scholar] [CrossRef]
- Lauffer, L.; Glas, E.; Gudermann, T.; Breit, A. Endogenous 5-HT2C Receptors Phosphorylate the cAMP Response Element Binding Protein via Protein Kinase C-Promoted Activation of Extracellular-Regulated Kinases-1/2 in Hypothalamic mHypoA-2/10 Cells. J. Pharmacol. Exp. Ther. 2016, 358, 39–49. [Google Scholar] [CrossRef]
- Faro, D.; Boekhoff, I.; Gudermann, T.; Breit, A. Physiological Temperature Changes Fine-Tune beta 2-Adrenergic Receptor-Induced Cytosolic cAMP Accumulation. Mol. Pharmacol. 2021, 100, 203–216. [Google Scholar] [CrossRef]
- Khong, K.; Kurtz, S.E.; Sykes, R.L.; Cone, R.D. Expression of functional melanocortin-4 receptor in the hypothalamic GT1-1 cell line. Neuroendocrinology 2001, 74, 193–201. [Google Scholar] [CrossRef]
- Anderson, R.A.; Zwain, I.H.; Arroyo, A.; Mellon, P.L.; Yen, S.S. The insulin-like growth factor system in the GT1-7 GnRH neuronal cell line. Neuroendocrinology 1999, 70, 353–359. [Google Scholar] [CrossRef]
- Dunn-Meynell, A.A.; Sanders, N.M.; Compton, D.; Becker, T.C.; Eiki, J.; Zhang, B.B.; Levin, B.E. Relationship among brain and blood glucose levels and spontaneous and glucoprivic feeding. J. Neurosci. 2009, 29, 7015–7022. [Google Scholar] [CrossRef]
- DeVries, J.H.; Snoek, F.J.; Kostense, P.J.; Heine, R.J. Improved glycaemic control in type 1 diabetes patients following participation per se in a clinical trial—Mechanisms and implications. Diabetes Metab. Res. Rev. 2003, 19, 357–362. [Google Scholar] [CrossRef]
- Diggs-Andrews, K.A.; Zhang, X.; Song, Z.; Daphna-Iken, D.; Routh, V.H.; Fisher, S.J. Brain insulin action regulates hypothalamic glucose sensing and the counterregulatory response to hypoglycemia. Diabetes 2010, 59, 2271–2280. [Google Scholar] [CrossRef]
- Lavoie, C.; Mercier, J.F.; Salahpour, A.; Umapathy, D.; Breit, A.; Villeneuve, L.R.; Zhu, W.Z.; Xiao, R.P.; Lakatta, E.G.; Bouvier, M.; et al. Beta 1/beta 2-adrenergic receptor heterodimerization regulates beta 2-adrenergic receptor internalization and ERK signaling efficacy. J. Biol. Chem. 2002, 277, 35402–35410. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef]
- Zhu, L.; Du, W.; Liu, Y.; Cheng, M.; Wang, X.; Zhang, C.; Lv, X.; Li, F.; Zhao, S.; Hao, J. Prolonged high-glucose exposure decreased SREBP-1/FASN/ACC in Schwann cells of diabetic mice via blocking PI3K/Akt pathway. J. Cell. Biochem. 2019, 120, 5777–5789. [Google Scholar] [CrossRef]
- Xu, M.; Jiang, F.; Li, B.; Zhang, Z. 1alpha,25(OH)2 D3 alleviates high glucose-induced lipid accumulation in rat renal tubular epithelial cells by inhibiting SREBPs. J. Cell. Biochem. 2019, 120, 15211–15221. [Google Scholar] [CrossRef]
- Han, J.; Li, E.; Chen, L.; Zhang, Y.; Wei, F.; Liu, J.; Deng, H.; Wang, Y. The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature 2015, 524, 243–246. [Google Scholar] [CrossRef]
- Mellon, P.L.; Windle, J.J.; Goldsmith, P.C.; Padula, C.A.; Roberts, J.L.; Weiner, R.I. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 1990, 5, 1–10. [Google Scholar] [CrossRef]
- Muehlich, S.; Wang, R.; Lee, S.M.; Lewis, T.C.; Dai, C.; Prywes, R. Serum-induced phosphorylation of the serum response factor coactivator MKL1 by the extracellular signal-regulated kinase 1/2 pathway inhibits its nuclear localization. Mol. Cell. Biol. 2008, 28, 6302–6313. [Google Scholar] [CrossRef]
- Ge, Q.; Feng, F.; Liu, L.; Chen, L.; Lv, P.; Ma, S.; Chen, K.; Yao, Q. RNA-Seq analysis of the pathogenesis of STZ-induced male diabetic mouse liver. J. Diabetes Complicat. 2020, 34, 107444. [Google Scholar] [CrossRef]
- Ottosson-Laakso, E.; Krus, U.; Storm, P.; Prasad, R.B.; Oskolkov, N.; Ahlqvist, E.; Fadista, J.; Hansson, O.; Groop, L.; Vikman, P. Glucose-Induced Changes in Gene Expression in Human Pancreatic Islets: Causes or Consequences of Chronic Hyperglycemia. Diabetes 2017, 66, 3013–3028. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Shu, J.; Lee, C.T. Whole-genome expression analyses of type 2 diabetes in human skin reveal altered immune function and burden of infection. Oncotarget 2017, 8, 34601–34609. [Google Scholar] [CrossRef]
- Nishitani, K.; Hayakawa, K.; Tanaka, S. Extracellular glucose levels in cultures of undifferentiated mouse trophoblast stem cells affect gene expression during subsequent differentiation with replicable cell line-dependent variation. J. Reprod. Dev. 2019, 65, 19–27. [Google Scholar] [CrossRef]
- Abdul-Rahman, O.; Sasvari-Szekely, M.; Ver, A.; Rosta, K.; Szasz, B.K.; Kereszturi, E.; Keszler, G. Altered gene expression profiles in the hippocampus and prefrontal cortex of type 2 diabetic rats. BMC Genom. 2012, 13, 81. [Google Scholar] [CrossRef]
- Nie, T.; Zhang, S.; Vazhoor Amarsingh, G.; Liu, H.; McCann, M.J.; Cooper, G.J.S. Altered metabolic gene expression in the brain of a triprolyl-human amylin transgenic mouse model of type 2 diabetes. Sci. Rep. 2019, 9, 14588. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Espenshade, P.J.; Wright, M.E.; Yabe, D.; Gong, Y.; Aebersold, R.; Goldstein, J.L.; Brown, M.S. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 2002, 110, 489–500. [Google Scholar] [CrossRef]
- Peng, Y.; Schwarz, E.J.; Lazar, M.A.; Genin, A.; Spinner, N.B.; Taub, R. Cloning, human chromosomal assignment, and adipose and hepatic expression of the CL-6/INSIG1 gene. Genomics 1997, 43, 278–284. [Google Scholar] [CrossRef]
- Suzuki, R.; Lee, K.; Jing, E.; Biddinger, S.B.; McDonald, J.G.; Montine, T.J.; Craft, S.; Kahn, C.R. Diabetes and insulin in regulation of brain cholesterol metabolism. Cell Metab. 2010, 12, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Ferris, H.A.; Chee, M.J.; Maratos-Flier, E.; Kahn, C.R. Reduction of the cholesterol sensor SCAP in the brains of mice causes impaired synaptic transmission and altered cognitive function. PLoS Biol. 2013, 11, e1001532. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.; Kerry, R.; Aviram, M.; Hayek, T. High glucose concentration increases macrophage cholesterol biosynthesis in diabetes through activation of the sterol regulatory element binding protein 1 (SREBP1): Inhibitory effect of insulin. J. Cardiovasc. Pharmacol. 2008, 52, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K.; Ferris, H.A.; Kahn, C.R. Effect of cholesterol reduction on receptor signaling in neurons. J. Biol. Chem. 2016, 291, 15910. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | RNA-seq | qRT-PCR | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | Function | Change (%) | Bon-ferroni-Corrected | tbp | Sdha | Mean Change (%) | ||||
| Change (%) | Sidak’s ΔΔCp ANOVA | ΔΔCp t-Test | Change (%) | Sidak’s ΔΔCp ANOVA | ΔΔCp t-Test | |||||
| Irs1 | adaptor protein for IR and IGFR | 123 | *** | 41 | *** | * | 52 | *** | * | 72 ± 26 |
| Igfr1 | IGF receptor | 34 | *** | 21 | ** | *** | 28 | ** | *** | 28 ± 4 |
| Adcy6 | cAMP production | 66 | *** | 28 | ** | *** | 37 | ** | ** | 44 ± 11 |
| Gnai1 | inhibition of cAMP production | 43 | *** | 15 | * | *** | 30 | * | ** | 29 ± 8 |
| Gnai2 | inhibition of cAMP production | 40 | *** | 24 | ** | *** | 39 | *** | *** | 35 ± 5 |
| Gnai3 | inhibition of cAMP production | 16 | * | 17 | * | ** | 30 | * | *** | 21 ± 5 |
| Gna11 | PLC activation/Ca2+ | 17 | * | 24 | * | ** | 30 | * | ** | 24 ± 4 |
| Hras | monomeric G protein/MAPK activation | 38 | *** | 15 | * | *** | 22 | * | ** | 25 ± 7 |
| Elk3 | transriptional activator/SRF/SRE | 61 | *** | 40 | *** | ** | 50 | *** | ** | 50 ± 6 |
| Insig1 | cholesterolsynthesis inhibitor | 108 | *** | 34 | *** | ** | 43 | *** | ** | 62 ± 23 |
| Rap2a | monomeric G protein/MAPK activation | 105 | *** | 27 | *** | ** | 45 | *** | ** | 59 ± 24 |
| Tbc1d1 | Rab inhibitor | 90 | *** | 29 | *** | ** | 38 | *** | ** | 52 ± 19 |
| S100a10 | calcium-binding protein | 59 | *** | 51 | *** | ** | 72 | *** | ** | 61 ± 6 |
| Arhgef2 | Rho activator | −58 | *** | −46 | *** | ** | −47 | *** | *** | −50 ± 4 |
| Tbc1d31 | Rab inhibitor | −61 | *** | −55 | *** | *** | −53 | *** | *** | −56 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webert, L.; Faro, D.; Zeitlmayr, S.; Gudermann, T.; Breit, A. Analysis of the Glucose-Dependent Transcriptome in Murine Hypothalamic Cells. Cells 2022, 11, 639. https://doi.org/10.3390/cells11040639
Webert L, Faro D, Zeitlmayr S, Gudermann T, Breit A. Analysis of the Glucose-Dependent Transcriptome in Murine Hypothalamic Cells. Cells. 2022; 11(4):639. https://doi.org/10.3390/cells11040639
Chicago/Turabian StyleWebert, Leonhard, Dennis Faro, Sarah Zeitlmayr, Thomas Gudermann, and Andreas Breit. 2022. "Analysis of the Glucose-Dependent Transcriptome in Murine Hypothalamic Cells" Cells 11, no. 4: 639. https://doi.org/10.3390/cells11040639
APA StyleWebert, L., Faro, D., Zeitlmayr, S., Gudermann, T., & Breit, A. (2022). Analysis of the Glucose-Dependent Transcriptome in Murine Hypothalamic Cells. Cells, 11(4), 639. https://doi.org/10.3390/cells11040639