
Drug Resistance and Endoplasmic Reticulum Stress in Hepatocellular Carcinoma



Abstract
:1. Introduction
2. Endoplasmic Reticulum Stress in Health and Disease
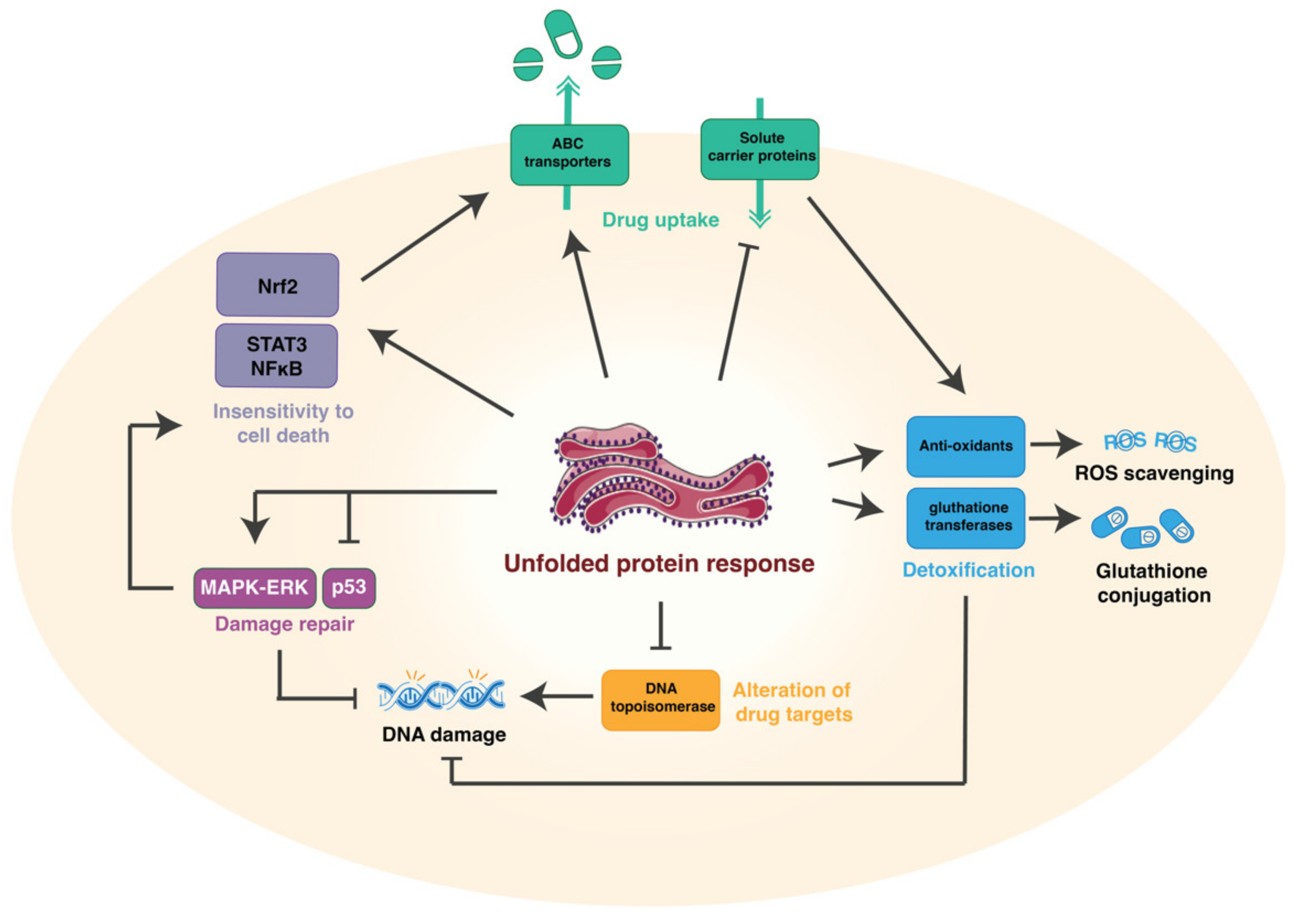
3. Mechanisms of Drug Resistance
3.1. Reduction of Drug Uptake and Enhanced Efflux
3.2. Alteration of the Drug Targets
3.3. Induction of Drug-Detoxifying Mechanisms
3.4. Repair Mechanisms
3.5. Insensitivity to Drug-Induced Cell Death
4. Role of the Tumor Microenvironment
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Mak, L.Y.; Cruz-Ramón, V.; Chinchilla-López, P.; Torres, H.A.; LoConte, N.K.; Rice, J.P.; Foxhall, L.E.; Sturgis, E.M.; Merrill, J.K.; Bailey, H.H.; et al. Global Epidemiology, Prevention, and Management of Hepatocellular Carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 262–279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, M. Current Options and Future Directions for NAFLD and NASH Treatment. Int. J. Mol. Sci. 2021, 22, 7571. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Ro, S.W. MAPK/ERK Signaling Pathway in Hepatocellular Carcinoma. Cancers 2021, 13, 3026. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Xiao, H. Metformin Actions on the Liver: Protection Mechanisms Emerging in Hepatocytes and Immune Cells against NASH-Related HCC. Int. J. Mol. Sci. 2021, 22, 5016. [Google Scholar] [CrossRef]
- Heindryckx, F.; Gerwins, P. Targeting the tumor stroma in hepatocellular carcinoma. World J. Hepatol. 2015, 7, 165–176. [Google Scholar] [CrossRef]
- Pocha, C.; Xie, C. Hepatocellular carcinoma in alcoholic and non-alcoholic fatty liver disease-one of a kind or two different enemies? Transl. Gastroenterol. Hepatol. 2019, 4, 72. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Walczak, A.; Gradzik, K.; Kabzinski, J.; Przybylowska-Sygut, K.; Majsterek, I. The Role of the ER-Induced UPR Pathway and the Efficacy of Its Inhibitors and Inducers in the Inhibition of Tumor Progression. Oxidative Med. Cell. Longev. 2019, 2019, 5729710. [Google Scholar] [CrossRef] [Green Version]
- Al-Rawashdeh, F.Y.; Scriven, P.; Cameron, I.C.; Vergani, P.V.; Wyld, L. Unfolded protein response activation contributes to chemoresistance in hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2010, 22, 1099–1105. [Google Scholar] [CrossRef]
- Sovolyova, N.; Healy, S.; Samali, A.; Logue, S.E. Stressed to death—Mechanisms of ER stress-induced cell death. Biol. Chem. 2014, 395, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Riaz, T.A.; Junjappa, R.P.; Handigund, M.; Ferdous, J.; Kim, H.R.; Chae, H.J. Role of Endoplasmic Reticulum Stress Sensor IRE1alpha in Cellular Physiology, Calcium, ROS Signaling, and Metaflammation. Cells 2020, 9, 1160. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.H.; Wong, C.C. Hypoxia, Metabolic Reprogramming, and Drug Resistance in Liver Cancer. Cells 2021, 10, 1715. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Duan, B.; Huang, C.; Bai, J.; Zhang, Y.L.; Wang, X.; Yang, J.; Li, J. Multidrug Resistance in Hepatocellular Carcinoma. In Hepatocellular Carcinoma; Tirnitz-Parker, J.E.E., Ed.; Codon Publications: Brisbane, Australia, 2019. [Google Scholar]
- Marin, J.J.G.; Macias, R.I.R.; Monte, M.J.; Romero, M.R.; Asensio, M.; Sanchez-Martin, A.; Cives-Losada, C.; Temprano, A.G.; Espinosa-Escudero, R.; Reviejo, M.; et al. Molecular Bases of Drug Resistance in Hepatocellular Carcinoma. Cancers 2020, 12, 1663. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Balgoma, D.; Kullenberg, F.; Calitz, C.; Kopsida, M.; Heindryckx, F.; Lennernas, H.; Hedeland, M. Anthracyclins Increase PUFAs: Potential Implications in ER Stress and Cell Death. Cells 2021, 10, 1163. [Google Scholar] [CrossRef]
- Jin, T.; Lin, J.; Gong, Y.; Bi, X.; Hu, S.; Lv, Q.; Chen, J.; Li, X.; Chen, J.; Zhang, W.; et al. iPLA2beta Contributes to ER Stress-Induced Apoptosis during Myocardial Ischemia/Reperfusion Injury. Cells 2021, 10, 1446. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Pavlovic, N.; Calitz, C.; Thanapirom, K.; Mazza, G.; Rombouts, K.; Gerwins, P.; Heindryckx, F. Inhibiting IRE1alpha-endonuclease activity decreases tumor burden in a mouse model for hepatocellular carcinoma. eLife 2020, 9, e55865. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, N.; Heindryckx, F. Exploring the Role of Endoplasmic Reticulum Stress in Hepatocellular Carcinoma through mining of the Human Protein Atlas. Biology 2021, 10, 640. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, N.; Heindryckx, F. Targeting ER stress in the hepatic tumor microenvironment. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kokott-Vuong, A.; Jung, J.; Fehr, A.T.; Kirschfink, N.; Noristani, R.; Voigt, A.; Reich, A.; Schulz, J.B.; Huber, M.; Habib, P. Increased Post-Hypoxic Oxidative Stress and Activation of the PERK Branch of the UPR in Trap1-Deficient Drosophila melanogaster Is Abrogated by Metformin. Int. J. Mol. Sci. 2021, 22, 11586. [Google Scholar] [CrossRef]
- Hou, J.; Zhang, H.; Sun, B.; Karin, M. The immunobiology of hepatocellular carcinoma in humans and mice: Basic concepts and therapeutic implications. J. Hepatol. 2020, 72, 167–182. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Marin, J.J.G.; Perez-Silva, L.; Macias, R.I.R.; Asensio, M.; Peleteiro-Vigil, A.; Sanchez-Martin, A.; Cives-Losada, C.; Sanchon-Sanchez, P.; Sanchez De Blas, B.; Herraez, E.; et al. Molecular Bases of Mechanisms Accounting for Drug Resistance in Gastric Adenocarcinoma. Cancers 2020, 12, 2116. [Google Scholar] [CrossRef]
- Khunweeraphong, N.; Kuchler, K. Multidrug Resistance in Mammals and Fungi-From MDR to PDR: A Rocky Road from Atomic Structures to Transport Mechanisms. Int. J. Mol. Sci. 2021, 22, 4806. [Google Scholar] [CrossRef]
- Serra, M.; Hattinger, C.M.; Pasello, M.; Casotti, C.; Fantoni, L.; Riganti, C.; Manara, M.C. Impact of ABC Transporters in Osteosarcoma and Ewing’s Sarcoma: Which Are Involved in Chemoresistance and Which Are Not? Cells 2021, 10, 2461. [Google Scholar] [CrossRef]
- He, J.; Fortunati, E.; Liu, D.X.; Li, Y. Pleiotropic Roles of ABC Transporters in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 3199. [Google Scholar] [CrossRef]
- Juan-Carlos, P.M.; Perla-Lidia, P.P.; Stephanie-Talia, M.M.; Monica-Griselda, A.M.; Luz-Maria, T.E. ABC transporter superfamily. An updated overview, relevance in cancer multidrug resistance and perspectives with personalized medicine. Mol. Biol. Rep. 2021, 48, 1883–1901. [Google Scholar] [CrossRef] [PubMed]
- Mitchell-White, J.I.; Stockner, T.; Holliday, N.; Briddon, S.J.; Kerr, I.D. Analysis of Sequence Divergence in Mammalian ABCGs Predicts a Structural Network of Residues That Underlies Functional Divergence. Int. J. Mol. Sci. 2021, 22, 3012. [Google Scholar] [CrossRef] [PubMed]
- Theile, D.; Wizgall, P. Acquired ABC-transporter overexpression in cancer cells: Transcriptional induction or Darwinian selection? Naunyn Schmiedebergs Arch. Pharmacol. 2021, 394, 1621–1632. [Google Scholar] [CrossRef]
- Gao, L.; Morine, Y.; Yamada, S.; Saito, Y.; Ikemoto, T.; Tokuda, K.; Takasu, C.; Miyazaki, K.; Shimada, M. Nrf2 signaling promotes cancer stemness, migration, and expression of ABC transporter genes in sorafenib-resistant hepatocellular carcinoma cells. PLoS ONE 2021, 16, e0256755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, X.; Li, J.; Song, Z. Systematic analysis of the ABC transporter family in hepatocellular carcinoma reveals the importance of ABCB6 in regulating ferroptosis. Life Sci. 2020, 257, 118131. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef]
- Leung, H.W.; Lau, E.Y.T.; Leung, C.O.N.; Lei, M.M.L.; Mok, E.H.K.; Ma, V.W.S.; Cho, W.C.S.; Ng, I.O.L.; Yun, J.P.; Cai, S.H.; et al. NRF2/SHH signaling cascade promotes tumor-initiating cell lineage and drug resistance in hepatocellular carcinoma. Cancer Lett. 2020, 476, 48–56. [Google Scholar] [CrossRef]
- Singh, A.; Wu, H.; Zhang, P.; Happel, C.; Ma, J.; Biswal, S. Expression of ABCG2 (BCRP) is regulated by Nrf2 in cancer cells that confers side population and chemoresistance phenotype. Mol. Cancer Ther. 2010, 9, 2365–2376. [Google Scholar] [CrossRef] [Green Version]
- Raghunath, A.; Sundarraj, K.; Arfuso, F.; Sethi, G.; Perumal, E. Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance. Cancers 2018, 10, 481. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.C.; Yang, P.M.; Liu, T.P. PERK/ATF4-Dependent ZFAS1 Upregulation Is Associated with Sorafenib Resistance in Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2021, 22, 5848. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Rivera, D.; Delvaeye, T.; Roelandt, R.; Nerinckx, W.; Augustyns, K.; Vandenabeele, P.; Bertrand, M.J.M. When PERK inhibitors turn out to be new potent RIPK1 inhibitors: Critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 2017, 24, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Li, X.X.; Xu, Y.M.; Zhang, J.Z.; Rong, S.D.; Qin, Y.Q.; Fang, J. IRE1alpha-targeting downregulates ABC transporters and overcomes drug resistance of colon cancer cells. Cancer Lett. 2020, 476, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Chevet, E. IRE1-mediated miRNA maturation in macrophage phosphoinositide signaling. EMBO Rep. 2020, 21, e51929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, H.; Song, Z.; Jiang, Y.; Kim, H.; Samavati, L.; Nguyen, H.M.; Yang, Z.Q. The UPR Transducer IRE1 Promotes Breast Cancer Malignancy by Degrading Tumor Suppressor microRNAs. iScience 2020, 23, 101503. [Google Scholar] [CrossRef]
- Heindryckx, F.; Binet, F.; Ponticos, M.; Rombouts, K.; Lau, J.; Kreuger, J.; Gerwins, P. Endoplasmic reticulum stress enhances fibrosis through IRE1alpha-mediated degradation of miR-150 and XBP-1 splicing. EMBO Mol. Med. 2016, 8, 729–744. [Google Scholar] [CrossRef]
- Kim, T.; Croce, C.M. MicroRNA and ER stress in cancer. Semin. Cancer Biol. 2021, 75, 3–14. [Google Scholar] [CrossRef]
- Shi, L.; Wu, L.; Chen, Z.; Yang, J.; Chen, X.; Yu, F.; Zheng, F.; Lin, X. MiR-141 Activates Nrf2-Dependent Antioxidant Pathway via Down-Regulating the Expression of Keap1 Conferring the Resistance of Hepatocellular Carcinoma Cells to 5-Fluorouracil. Cell Physiol. Biochem. 2015, 35, 2333–2348. [Google Scholar] [CrossRef] [Green Version]
- Kishikawa, T.; Otsuka, M.; Tan, P.S.; Ohno, M.; Sun, X.; Yoshikawa, T.; Shibata, C.; Takata, A.; Kojima, K.; Takehana, K.; et al. Decreased miR122 in hepatocellular carcinoma leads to chemoresistance with increased arginine. Oncotarget 2015, 6, 8339–8352. [Google Scholar] [CrossRef]
- Gao, R.; Kalathur, R.K.R.; Coto-Llerena, M.; Ercan, C.; Buechel, D.; Shuang, S.; Piscuoglio, S.; Dill, M.T.; Camargo, F.D.; Christofori, G.; et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol. Med. 2021, 13, e14351. [Google Scholar] [CrossRef]
- Baglini, E.; Salerno, S.; Barresi, E.; Robello, M.; Da Settimo, F.; Taliani, S.; Marini, A.M. Multiple Topoisomerase I (TopoI), Topoisomerase II (TopoII) and Tyrosyl-DNA Phosphodiesterase (TDP) inhibitors in the development of anticancer drugs. Eur. J. Pharm. Sci. 2021, 156, 105594. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; Shen, J.W.; Subjeck, J.R. Resistance to etoposide induced by three glucose-regulated stresses in Chinese hamster ovary cells. Cancer Res. 1989, 49, 4452–4454. [Google Scholar] [PubMed]
- Gray, M.D.; Mann, M.; Nitiss, J.L.; Hendershot, L.M. Activation of the unfolded protein response is necessary and sufficient for reducing topoisomerase IIalpha protein levels and decreasing sensitivity to topoisomerase-targeted drugs. Mol. Pharmacol. 2005, 68, 1699–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufey, E.; Bravo-San Pedro, J.M.; Eggers, C.; Gonzalez-Quiroz, M.; Urra, H.; Sagredo, A.I.; Sepulveda, D.; Pihan, P.; Carreras-Sureda, A.; Hazari, Y.; et al. Genotoxic stress triggers the activation of IRE1alpha-dependent RNA decay to modulate the DNA damage response. Nat. Commun. 2020, 11, 2401. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.J.; Pereira, E.R.; Liao, N.; Hendershot, L.M. UPR-induced resistance to etoposide is downstream of PERK and independent of changes in topoisomerase IIalpha levels. PLoS ONE 2012, 7, e47931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.T.; Song, K.; Zhou, J.; Shi, Y.H.; Liu, W.R.; Tian, M.X.; Jin, L.; Shi, G.M.; Gao, Q.; Ding, Z.B.; et al. Autophagy activation contributes to glutathione transferase Mu 1-mediated chemoresistance in hepatocellular carcinoma. Oncol. Lett. 2018, 16, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [Green Version]
- Robin, S.K.D.; Ansari, M.; Uppugunduri, C.R.S. Spectrophotometric Screening for Potential Inhibitors of Cytosolic Glutathione S-Transferases. J. Vis. Exp. 2020, 164, e61347. [Google Scholar] [CrossRef]
- Shi, Y.H.; Ding, Z.B.; Zhou, J.; Hui, B.; Shi, G.M.; Ke, A.W.; Wang, X.Y.; Dai, Z.; Peng, Y.F.; Gu, C.Y.; et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011, 7, 1159–1172. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.W.; Chen, W.; Culpepper, J.; Jiang, H.; Ball, L.E.; Mehrotra, S.; Blumental-Perry, A.; Tew, K.D.; Townsend, D.M. Altered redox regulation and S-glutathionylation of BiP contribute to bortezomib resistance in multiple myeloma. Free Radic. Biol. Med. 2020, 160, 755–767. [Google Scholar] [CrossRef]
- Dubbelboer, I.R.; Pavlovic, N.; Heindryckx, F.; Sjogren, E.; Lennernas, H. Liver Cancer Cell Lines Treated with Doxorubicin under Normoxia and Hypoxia: Cell Viability and Oncologic Protein Profile. Cancers 2019, 11, 1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshi, M.; Kim, T.H.; Tokumaru, Y.; Yan, L.; Matsuyama, R.; Endo, I.; Cherkassky, L.; Takabe, K. Enhanced DNA Repair Pathway is Associated with Cell Proliferation and Worse Survival in Hepatocellular Carcinoma (HCC). Cancers 2021, 13, 323. [Google Scholar] [CrossRef] [PubMed]
- Pavan, I.C.B.; Peres de Oliveira, A.; Dias, P.R.F.; Basei, F.L.; Issayama, L.K.; Ferezin, C.C.; Silva, F.R.; Rodrigues de Oliveira, A.L.; Alves Dos Reis Moura, L.; Martins, M.B.; et al. On Broken Ne(c)ks and Broken DNA: The Role of Human NEKs in the DNA Damage Response. Cells 2021, 10, 507. [Google Scholar] [CrossRef] [PubMed]
- Sakthivel, K.M.; Hariharan, S. Regulatory players of DNA damage repair mechanisms: Role in Cancer Chemoresistance. Biomed. Pharmacother. 2017, 93, 1238–1245. [Google Scholar] [CrossRef]
- Yuan, H.; Lu, Y.; Chan, Y.-T.; Zhang, C.; Wang, N.; Feng, Y. The Role of Protein SUMOylation in Human Hepatocellular Carcinoma: A Potential Target of New Drug Discovery and Development. Cancers 2021, 13, 5700. [Google Scholar] [CrossRef]
- Zhang, S.; Fu, Y.; Wang, D.; Wang, J. Icotinib enhances lung cancer cell radiosensitivity in vitro and in vivo by inhibiting MAPK/ERK and AKT activation. Clin. Exp. Pharmacol. Physiol. 2018, 45, 969–977. [Google Scholar] [CrossRef]
- Che, J.; Lu, Y.W.; Sun, K.K.; Feng, C.; Dong, A.J.; Jiao, Y. Overexpression of TOB1 confers radioprotection to bronchial epithelial cells through the MAPK/ERK pathway. Oncol. Rep. 2013, 30, 637–642. [Google Scholar] [CrossRef]
- Marza, E.; Taouji, S.; Barroso, K.; Raymond, A.A.; Guignard, L.; Bonneu, M.; Pallares-Lupon, N.; Dupuy, J.W.; Fernandez-Zapico, M.E.; Rosenbaum, J.; et al. Genome-wide screen identifies a novel p97/CDC-48-dependent pathway regulating ER-stress-induced gene transcription. EMBO Rep. 2015, 16, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Pantazi, V.; Berzsenyi, I.; Borsos, B.N.; Pankotai, T. Visualizing and Quantifying Endonuclease-Based Site-Specific DNA Damage. J. Vis. Exp. 2021, 174, e62175. [Google Scholar] [CrossRef]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Bravo-San Pedro, J.M.; Galluzzi, L. Novel function of cytoplasmic p53 at the interface between mitochondria and the endoplasmic reticulum. Cell Death Dis. 2015, 6, e1698. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Chuang, Y.C.; Chang, Y.S.; Lai, M.D.; Teng, Y.N.; Su, I.J.; Wang, C.C.; Lee, K.H.; Hung, J.H. Endoplasmic reticulum stress stimulates p53 expression through NF-kappaB activation. PLoS ONE 2012, 7, e39120. [Google Scholar]
- Pluquet, O.; Qu, L.K.; Baltzis, D.; Koromilas, A.E. Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3beta. Mol. Cell Biol. 2005, 25, 9392–9405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, L.; Huang, S.; Baltzis, D.; Rivas-Estilla, A.M.; Pluquet, O.; Hatzoglou, M.; Koumenis, C.; Taya, Y.; Yoshimura, A.; Koromilas, A.E. Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes Dev. 2004, 18, 261–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Li, J.; Lee, A.S. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007, 67, 3734–3740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Hamanaka, R.B.; Bobrovnikova-Marjon, E.; Gordan, J.D.; Dai, M.S.; Lu, H.; Simon, M.C.; Diehl, J.A. Ribosomal stress couples the unfolded protein response to p53-dependent cell cycle arrest. J. Biol. Chem. 2006, 281, 30036–30045. [Google Scholar] [CrossRef] [Green Version]
- Mihailidou, C.; Papazian, I.; Papavassiliou, A.G.; Kiaris, H. CHOP-dependent regulation of p21/waf1 during ER stress. Cell Physiol. Biochem. 2010, 25, 761–766. [Google Scholar] [CrossRef]
- Inoue, Y.; Kawachi, S.; Ohkubo, T.; Nagasaka, M.; Ito, S.; Fukuura, K.; Itoh, Y.; Ohoka, N.; Morishita, D.; Hayashi, H. The CDK inhibitor p21 is a novel target gene of ATF4 and contributes to cell survival under ER stress. FEBS Lett. 2017, 591, 3682–3691. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, P.; Zarb, Y.; Noborn, F.; Kreuger, J. Modeling the structural implications of an alternatively spliced Exoc3l2, a paralog of the tunneling nanotube-forming M-Sec. PLoS ONE 2018, 13, e0201557. [Google Scholar] [CrossRef]
- Fusee, L.T.S.; Marin, M.; Fahraeus, R.; Lopez, I. Alternative Mechanisms of p53 Action During the Unfolded Protein Response. Cancers 2020, 12, 401. [Google Scholar] [CrossRef] [Green Version]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.; Yu, X.; Yuan, M.; Lv, W.; Feng, T.; Bai, R.; Zhong, H. Activation of the PERK-ATF4 pathway promotes chemo-resistance in colon cancer cells. Sci. Rep. 2019, 9, 3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Yun, G.J.; Kim, S.E. Metabolic Regulation of Ferroptosis in Cancer. Biology 2021, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Stieger, B.; Steiger, J.; Locher, K.P. Membrane lipids and transporter function. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166079. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Lv, X.; Guo, X.; Ruan, B.; Liu, D.; Ding, R.; Gao, Y.; Ding, J.; Dou, K.; Chen, Y. RACK1 modulates apoptosis induced by sorafenib in HCC cells by interfering with the IRE1/XBP1 axis. Oncol. Rep. 2015, 33, 3006–3014. [Google Scholar] [CrossRef] [Green Version]
- Orru, C.; Giordano, S.; Columbano, A. Nrf2 in Neoplastic and Non-Neoplastic Liver Diseases. Cancers 2020, 12, 2932. [Google Scholar] [CrossRef]
- Zhou, S.; Ye, W.; Zhang, Y.; Yu, D.; Shao, Q.; Liang, J.; Zhang, M. miR-144 reverses chemoresistance of hepatocellular carcinoma cell lines by targeting Nrf2-dependent antioxidant pathway. Am. J. Transl. Res. 2016, 8, 2992–3002. [Google Scholar]
- Gonzalez-Sanchez, E.; Marin, J.J.; Perez, M.J. The expression of genes involved in hepatocellular carcinoma chemoresistance is affected by mitochondrial genome depletion. Mol. Pharm. 2014, 11, 1856–1868. [Google Scholar] [CrossRef]
- Bronner, D.N.; O’Riordan, M.X. Measurement of Mitochondrial DNA Release in Response to ER Stress. Bio-Protocol 2016, 6, e1839. [Google Scholar] [CrossRef]
- Souza-Neto, F.V.; Jimenez-Gonzalez, S.; Delgado-Valero, B.; Jurado-Lopez, R.; Genty, M.; Romero-Miranda, A.; Rodriguez, C.; Nieto, M.L.; Martinez-Martinez, E.; Cachofeiro, V. The Interplay of Mitochondrial Oxidative Stress and Endoplasmic Reticulum Stress in Cardiovascular Fibrosis in Obese Rats. Antioxidants 2021, 10, 1274. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Maity, S. ER Stress-Sensor Proteins and ER-Mitochondrial Crosstalk-Signaling Beyond (ER) Stress Response. Biomolecules 2021, 11, 173. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Tyagi, A.K.; Agboke, F.A.; Mathur, R.; Pokharel, N.; Jordan, V.C. Modulation of nuclear factor-kappa B activation by the endoplasmic reticulum stress sensor PERK to mediate estrogen-induced apoptosis in breast cancer cells. Cell Death Discov. 2018, 4, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makhov, P.; Naito, S.; Haifler, M.; Kutikov, A.; Boumber, Y.; Uzzo, R.G.; Kolenko, V.M. The convergent roles of NF-kappaB and ER stress in sunitinib-mediated expression of pro-tumorigenic cytokines and refractory phenotype in renal cell carcinoma. Cell Death Dis. 2018, 9, 374. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, M.L.; Shaban, M.S.; Albert, B.V.; Gokcen, A.; Kracht, M. The Crosstalk of Endoplasmic Reticulum (ER) Stress Pathways with NF-kappaB: Complex Mechanisms Relevant for Cancer, Inflammation and Infection. Biomedicines 2018, 6, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.T.; Jia, L.; Wang, P.; Wang, H.; Farren, T.W.; Agrawal, S.G. STAT3 and NF-kappaB cooperatively control in vitro spontaneous apoptosis and poor chemo-responsiveness in patients with chronic lymphocytic leukemia. Oncotarget 2016, 7, 32031–32045. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.; Min, H.Y.; Pei, H.; Wei, X.; Sim, J.Y.; Park, S.H.; Hwang, S.J.; Lee, H.J.; Hong, S.; Shin, Y.K.; et al. The ATF6-EGF Pathway Mediates the Awakening of Slow-Cycling Chemoresistant Cells and Tumor Recurrence by Stimulating Tumor Angiogenesis. Cancers 2020, 12, 1772. [Google Scholar] [CrossRef]
- Van Huizen, R.; Martindale, J.L.; Gorospe, M.; Holbrook, N.J. P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J. Biol. Chem. 2003, 278, 15558–15564. [Google Scholar] [CrossRef] [Green Version]
- DeZwaan-McCabe, D.; Riordan, J.D.; Arensdorf, A.M.; Icardi, M.S.; Dupuy, A.J.; Rutkowski, D.T. The stress-regulated transcription factor CHOP promotes hepatic inflammatory gene expression, fibrosis, and oncogenesis. PLoS Genet. 2013, 9, e1003937. [Google Scholar] [CrossRef] [Green Version]
- Scaiewicz, V.; Nahmias, A.; Chung, R.T.; Mueller, T.; Tirosh, B.; Shibolet, O. CCAAT/enhancer-binding protein homologous (CHOP) protein promotes carcinogenesis in the DEN-induced hepatocellular carcinoma model. PLoS ONE 2013, 8, e81065. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogaerts, E.; Heindryckx, F.; Devisscher, L.; Paridaens, A.; Vandewynckel, Y.P.; Van den Bussche, A.; Verhelst, X.; Libbrecht, L.; van Grunsven, L.A.; Geerts, A.; et al. Time-dependent effect of hypoxia on tumor progression and liver progenitor cell markers in primary liver tumors. PLoS ONE 2015, 10, e0119555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heindryckx, F.; Coulon, S.; Terrie, E.; Casteleyn, C.; Stassen, J.M.; Geerts, A.; Libbrecht, L.; Allemeersch, J.; Carmeliet, P.; Colle, I.; et al. The placental growth factor as a target against hepatocellular carcinoma in a diethylnitrosamine-induced mouse model. J. Hepatol. 2013, 58, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Heindryckx, F.; Kuchnio, A.; Casteleyn, C.; Coulon, S.; Olievier, K.; Colle, I.; Geerts, A.; Libbrecht, L.; Carmeliet, P.; Van Vlierberghe, H. Effect of prolyl hydroxylase domain-2 haplodeficiency on the hepatocarcinogenesis in mice. J. Hepatol. 2012, 57, 61–68. [Google Scholar] [CrossRef]
- Vu, N.B.; Nguyen, T.T.; Tran, L.C.; Do, C.D.; Nguyen, B.H.; Phan, N.K.; Pham, P.V. Doxorubicin and 5-fluorouracil resistant hepatic cancer cells demonstrate stem-like properties. Cytotechnology 2013, 65, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Pi, L.; Li, X.; Song, Q.; Shen, Y.; Lu, X.; Di, B. Knockdown of glucose-regulated protein 78 abrogates chemoresistance of hypopharyngeal carcinoma cells to cisplatin induced by unfolded protein in response to severe hypoxia. Oncol. Lett. 2014, 7, 685–692. [Google Scholar] [CrossRef] [Green Version]
- You, Y.; Zheng, Q.; Dong, Y.; Xie, X.; Wang, Y.; Wu, S.; Zhang, L.; Wang, Y.; Xue, T.; Wang, Z.; et al. Matrix stiffness-mediated effects on stemness characteristics occurring in HCC cells. Oncotarget 2016, 7, 32221–32231. [Google Scholar] [CrossRef] [Green Version]
- Tameire, F.; Verginadis, I.I.; Koumenis, C. Cell intrinsic and extrinsic activators of the unfolded protein response in cancer: Mechanisms and targets for therapy. Semin. Cancer Biol. 2015, 33, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Ozkan, A.; Stolley, D.L.; Cressman, E.N.K.; McMillin, M.; DeMorrow, S.; Yankeelov, T.E.; Rylander, M.N. Tumor Microenvironment Alters Chemoresistance of Hepatocellular Carcinoma Through CYP3A4 Metabolic Activity. Front. Oncol. 2021, 11, 662135. [Google Scholar] [CrossRef]
- Calitz, C.; Pavlovic, N.; Rosenquist, J.; Zagami, C.; Samanta, A.; Heindryckx, F. A Biomimetic Model for Liver Cancer to Study Tumor-Stroma Interactions in a 3D Environment with Tunable Bio-Physical Properties. J. Vis. Exp. 2020, 162, e61606. [Google Scholar] [CrossRef]
- Samarelli, A.V.; Masciale, V.; Aramini, B.; Coló, G.P.; Tonelli, R.; Marchioni, A.; Bruzzi, G.; Gozzi, F.; Andrisani, D.; Castaniere, I.; et al. Molecular Mechanisms and Cellular Contribution from Lung Fibrosis to Lung Cancer Development. Int. J. Mol. Sci. 2021, 22, 12179. [Google Scholar] [CrossRef] [PubMed]
- Kaps, L.; Schuppan, D. Targeting Cancer Associated Fibroblasts in Liver Fibrosis and Liver Cancer Using Nanocarriers. Cells 2020, 9, 2027. [Google Scholar] [CrossRef]
- Maiers, J.L.; Kostallari, E.; Mushref, M.; de Assuncao, T.M.; Li, H.; Jalan-Sakrikar, N.; Huebert, R.C.; Cao, S.; Malhi, H.; Shah, V.H. The unfolded protein response mediates fibrogenesis and collagen I secretion through regulating TANGO1 in mice. Hepatology 2017, 65, 983–998. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, C.; Kang, N.; Malhi, H.; Shah, V.H.; Maiers, J.L. Transforming growth factor beta (TGFbeta) cross-talk with the unfolded protein response is critical for hepatic stellate cell activation. J. Biol. Chem. 2019, 294, 3137–3151. [Google Scholar] [CrossRef] [Green Version]
- Jia, C.; Wang, G.; Wang, T.; Fu, B.; Zhang, Y.; Huang, L.; Deng, Y.; Chen, G.; Wu, X.; Chen, J.; et al. Cancer-associated Fibroblasts induce epithelial-mesenchymal transition via the Transglutaminase 2-dependent IL-6/IL6R/STAT3 axis in Hepatocellular Carcinoma. Int. J. Biol. Sci. 2020, 16, 2542–2558. [Google Scholar] [CrossRef] [PubMed]
- Palamaris, K.; Felekouras, E.; Sakellariou, S. Epithelial to Mesenchymal Transition: Key Regulator of Pancreatic Ductal Adenocarcinoma Progression and Chemoresistance. Cancers 2021, 13, 5532. [Google Scholar] [CrossRef]
- Hirao, A.; Sato, Y.; Tanaka, H.; Nishida, K.; Tomonari, T.; Hirata, M.; Bando, M.; Kida, Y.; Tanaka, T.; Kawaguchi, T.; et al. MiR-125b-5p Is Involved in Sorafenib Resistance through Ataxin-1-Mediated Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma. Cancers 2021, 13, 4917. [Google Scholar] [CrossRef] [PubMed]
- Gibert-Ramos, A.; Sanfeliu-Redondo, D.; Aristu-Zabalza, P.; Martínez-Alcocer, A.; Gracia-Sancho, J.; Guixé-Muntet, S.; Fernández-Iglesias, A. The Hepatic Sinusoid in Chronic Liver Disease: The Optimal Milieu for Cancer. Cancers 2021, 13, 5719. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Koletsa, T.; Mitroulis, I.; Germanidis, G. Tumor-Associated Macrophages in Hepatocellular Carcinoma Pathogenesis, Prognosis and Therapy. Cancers 2022, 14, 226. [Google Scholar] [CrossRef]
- Fu, X.T.; Song, K.; Zhou, J.; Shi, Y.H.; Liu, W.R.; Shi, G.M.; Gao, Q.; Wang, X.Y.; Ding, Z.B.; Fan, J. Tumor-associated macrophages modulate resistance to oxaliplatin via inducing autophagy in hepatocellular carcinoma. Cancer Cell Int. 2019, 19, 71. [Google Scholar] [CrossRef] [Green Version]
- Pavlovic, N.; Kopsida, M.; Gerwins, P.; Heindryckx, F. Inhibiting P2Y12 in Macrophages Induces Endoplasmic Reticulum Stress and Promotes an Anti-Tumoral Phenotype. Int. J. Mol. Sci. 2020, 21, 8177. [Google Scholar] [CrossRef]
- Yang, F.; Liu, Y.; Ren, H.; Zhou, G.; Yuan, X.; Shi, X. ER-stress regulates macrophage polarization through pancreatic EIF-2alpha kinase. Cell Immunol. 2019, 336, 40–47. [Google Scholar] [CrossRef]
- Suzuki, T.; Gao, J.; Ishigaki, Y.; Kondo, K.; Sawada, S.; Izumi, T.; Uno, K.; Kaneko, K.; Tsukita, S.; Takahashi, K.; et al. ER Stress Protein CHOP Mediates Insulin Resistance by Modulating Adipose Tissue Macrophage Polarity. Cell Rep. 2017, 18, 2045–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, D.; Nakao, Y.; Mauer, A.S.; Thompson, J.M.; Sehrawat, T.S.; Liao, C.Y.; Krishnan, A.; Lucien, F.; Guo, Q.; Liu, M.; et al. IRE1A Stimulates Hepatocyte-Derived Extracellular Vesicles That Promote Inflammation in Mice With Steatohepatitis. Gastroenterology 2020, 159, 1487–1503.e17. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.Z.; Lourie, R.; Das, I.; Chen, A.C.; McGuckin, M.A. The interplay between endoplasmic reticulum stress and inflammation. Immunol. Cell Biol. 2012, 90, 260–270. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Mitroulis, I.; Germanidis, G. Tumor-Associated Neutrophils in Hepatocellular Carcinoma Pathogenesis, Prognosis, and Therapy. Cancers 2021, 13, 2899. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Navas, R.; Gajate, C.; Mollinedo, F. Neutrophils drive endoplasmic reticulum stress-mediated apoptosis in cancer cells through arginase-1 release. Sci. Rep. 2021, 11, 12574. [Google Scholar] [CrossRef] [PubMed]
- Setrerrahmane, S.; Xu, H. Tumor-related interleukins: Old validated targets for new anti-cancer drug development. Mol. Cancer 2017, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Wang, X.; Peng, R.; Zhang, B.; Han, Q.; Lin, J.; Wang, J.; Lin, J.; Jiang, M.; Liu, H.; et al. Induction of IL-6Ralpha by ATF3 enhances IL-6 mediated sorafenib and regorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2022, 524, 161–171. [Google Scholar] [CrossRef]
- Fang, P.; Xiang, L.; Huang, S.; Jin, L.; Zhou, G.; Zhuge, L.; Li, J.; Fan, H.; Zhou, L.; Pan, C.; et al. IRE1alpha-XBP1 signaling pathway regulates IL-6 expression and promotes progression of hepatocellular carcinoma. Oncol. Lett. 2018, 16, 4729–4736. [Google Scholar]
- Lohitesh, K.; Chowdhury, R.; Mukherjee, S. Resistance a major hindrance to chemotherapy in hepatocellular carcinoma: An insight. Cancer Cell Int. 2018, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| TMD Fold | Subfamily | Gene | Function | Upregulation in Cancer |
|---|---|---|---|---|
| Type IV | ABCB | ABCB1 (P-gp) | Drug efflux and regulator of lipids and steroids homeostasis in central and peripheral nervous system | Adrenocortical, breast, colorectal, leukemic, ovarian, and renal cancers |
| ABCB5 | GSH mediator | Leukemic, lung, melanoma, ovarian, renal, and thyroidal cancers | ||
| ABCB8 | Iron metabolism and homeostasis, OS protection | Head and neck, pancreatic, and renal cancers | ||
| ABCC | ABCC1 (MRP1) | Organic anion transporter and GSH mediator | Breast, endometrium, glioma, head and neck, lung, lymphoma, melanoma, ovarian, prostate, neuroblastoma, and thyroid cancers | |
| ABCC2 | Organic anion transporter | Colorectal, gastric, hepatic, and lung cancers | ||
| ABCC3 | Organic anion transporter | Breast, cervical, colorectal, gastric, hepatic, lung, ovarian pancreatic, renal, and thyroid cancers | ||
| ABCC4 | Nucleoside transporter | Breast, endometrial, gastric, head and neck, hepatic, lung, neuroblastoma, ovarian, prostate, andrenal cancers | ||
| ABCC5 | Nucleoside transporter | Breast, cervical, glioma hepatic, lung, pancreatic, renal, and urothelial cancers | ||
| ABCC6 | Putative biomineralization modulator | Liver cancer | ||
| ABCC10 | E(2)17βG transporter | Breast, colorectal, liver, lung, and prostate cancers | ||
| ABCC11 | Bile salts transporter | Breast cancer | ||
| ABCC12 | Unknown | Breast, colorectal, liver, lung, and prostate cancers | ||
| Type V | ABCG | ABCG2 | Toxin efflux, cell differentiation | Cervical, glioma, liver, ovarian, prostate, pulmonary, renal, and testicular cancers |
| ABCA | ABCA2 | Lipid transporter | Breast, colon, leukemia, and liver cancers | |
| ABCA8 | Lipophilic drugs transporter | Ovarian cancer | ||
| - | ABCF | ABCF2 | Inflammatory development | Breast cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khaled, J.; Kopsida, M.; Lennernäs, H.; Heindryckx, F. Drug Resistance and Endoplasmic Reticulum Stress in Hepatocellular Carcinoma. Cells 2022, 11, 632. https://doi.org/10.3390/cells11040632
Khaled J, Kopsida M, Lennernäs H, Heindryckx F. Drug Resistance and Endoplasmic Reticulum Stress in Hepatocellular Carcinoma. Cells. 2022; 11(4):632. https://doi.org/10.3390/cells11040632
Chicago/Turabian StyleKhaled, Jaafar, Maria Kopsida, Hans Lennernäs, and Femke Heindryckx. 2022. "Drug Resistance and Endoplasmic Reticulum Stress in Hepatocellular Carcinoma" Cells 11, no. 4: 632. https://doi.org/10.3390/cells11040632
APA StyleKhaled, J., Kopsida, M., Lennernäs, H., & Heindryckx, F. (2022). Drug Resistance and Endoplasmic Reticulum Stress in Hepatocellular Carcinoma. Cells, 11(4), 632. https://doi.org/10.3390/cells11040632