
The Biogenesis of miRNAs and Their Role in the Development of Amyotrophic Lateral Sclerosis

 ,
,  , and
, and
Abstract
1. Introduction
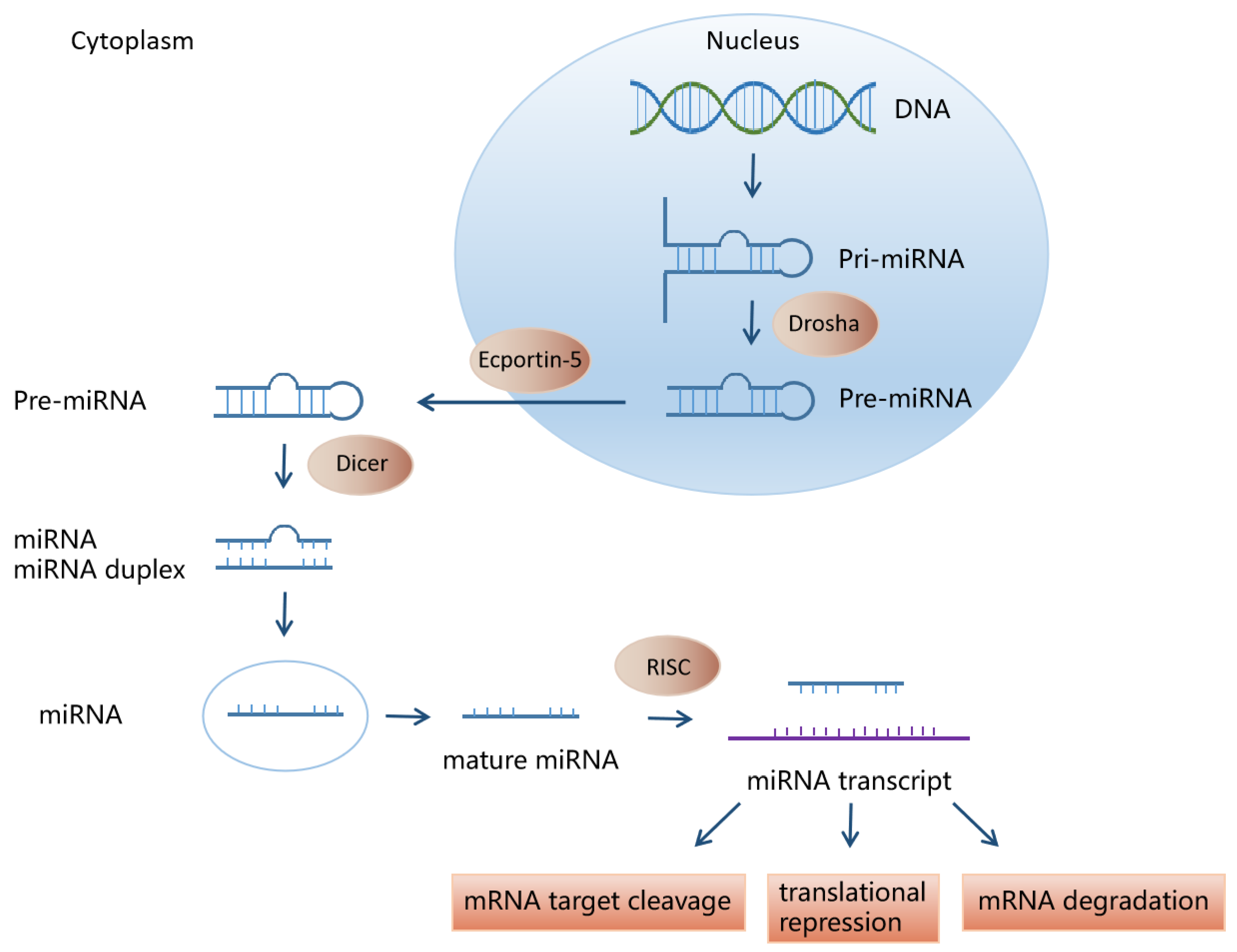
2. miRNA Biosynthesis and Function
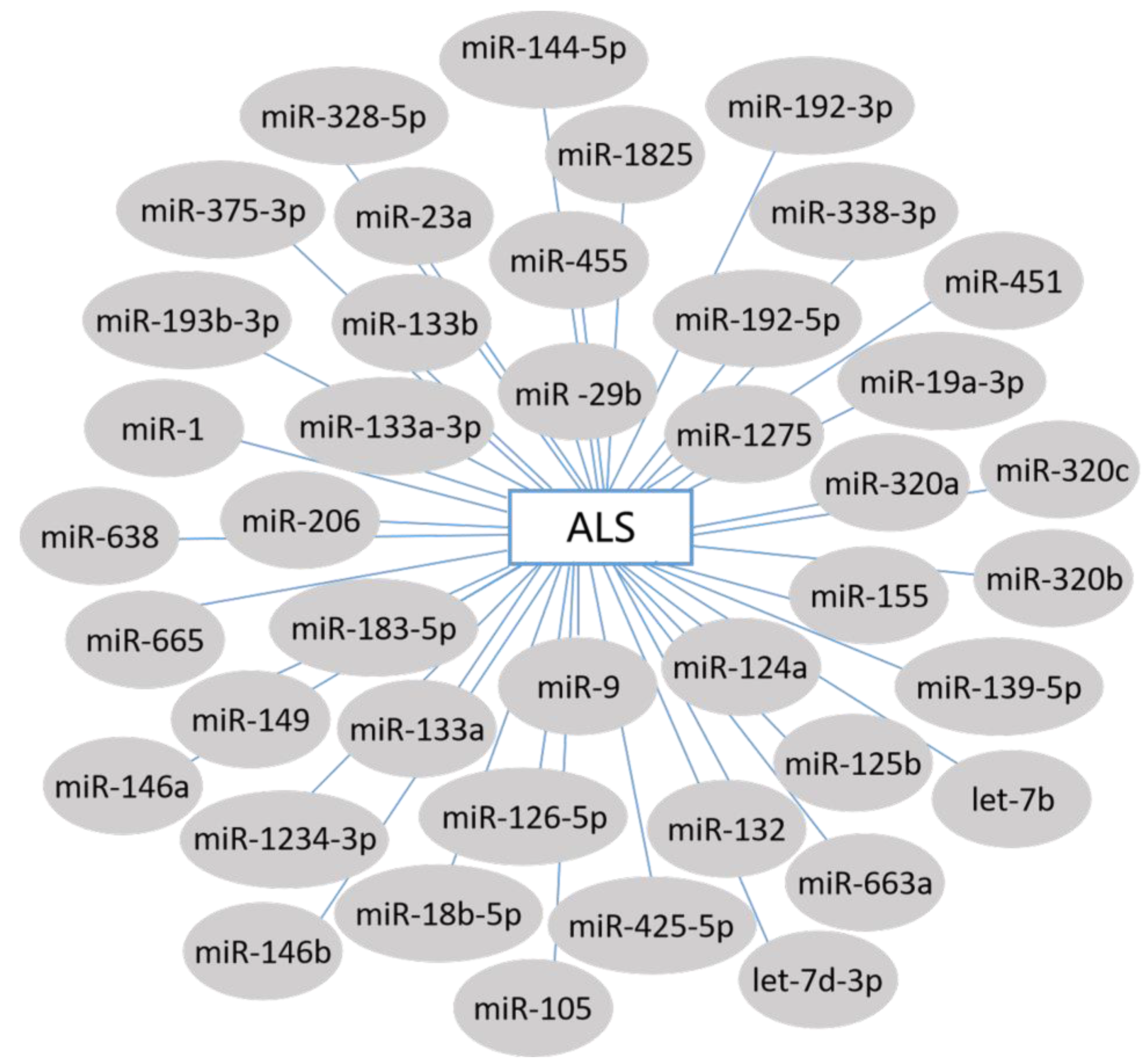
3. miRNAs and ALS
3.1. Circulating miRNA Biomarkers in ALS
3.1.1. Serum miRNAs in ALS
3.1.2. Muscle-Specific miRNAs in ALS
3.1.3. Leukocyte miRNAs in ALS
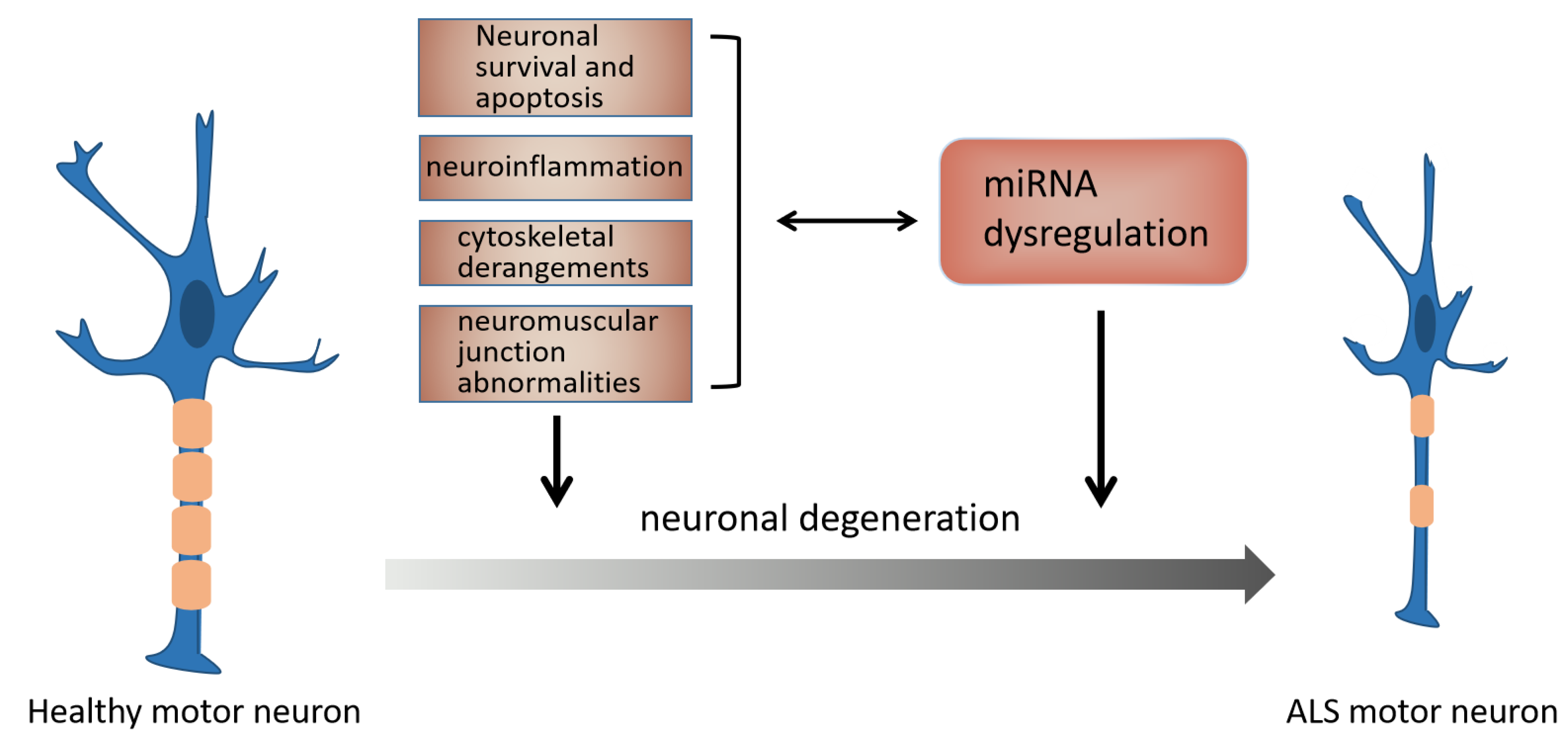
3.2. The Mechanism of miRNAs in ALS
3.2.1. miRNA and NMJ Abnormalities in ALS
3.2.2. miRNA and Neuroinflammation in ALS
3.2.3. miRNA and Neuronal Survival and Apoptosis in ALS
3.2.4. miRNA and Cytoskeletal Derangements in ALS
3.3. miRNA Treatment in ALS
3.3.1. miRNA Therapeutic Approaches
3.3.2. miRNA Therapy for ALS
4. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| miRNAs | microRNAs |
| MNs | motor neurons |
| ncRNA | noncoding RNA |
| miRbase | miRNA database |
| pri-miRNA | primary miRNA |
| precursor miRNA | pre-miRNA |
| Nt | nucleotides |
| snoRNAs | small nucleolar RNAs |
| miRISC | miRNA-induced silencing complexes |
| AD | Alzheimer’s disease |
| myo-miRNAs | muscle-specific miRNAs |
| ND | neurogenic disease |
| HDAC4 | histone deacetylase 4 |
| FGFBP1 | fibroblast growth factor binding protein 1 |
| NSC-34 cells | hybrid mouse motor neuron-like cells |
| TSC1 | tuberous sclerosis 1 |
| mTORC1 | mechanistic target of rapamycin complex 1 |
| NDRG2 | N-myc downstream-regulated gene 2 |
| WT | wild type |
| NMJ | neuromuscular junction |
| Sema3A | type 3 Semaphorins |
| TBCB | tubulin-folding cofactor b |
| A20 | TNF alpha-induced protein 3, TNFAIP3 |
| AMOs | anti-miRNA oligonucleotides |
| CNS | central nervous system |
| amiRSOD1 | hSOD1-specific amiR |
| ICV | intracerebral ventricular |
| rAAV | recombination adeno-associated virus |
| IT | intrathecal |
| ESC | embryonic stem cell |
| LMC-MNs | spinal lateral motor column MNs |
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Amado, D.A.; Davidson, B.L. Gene therapy for ALS: A review. Mol. Ther. 2021, 29, 3345–3358. [Google Scholar] [CrossRef] [PubMed]
- Sebastiao, A.M.; Rei, N.; Ribeiro, J.A. Amyotrophic Lateral Sclerosis (ALS) and Adenosine Receptors. Front. Pharm. 2018, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- Bucchia, M.; Ramirez, A.; Parente, V.; Simone, C.; Nizzardo, M.; Magri, F.; Dametti, S.; Corti, S. Therapeutic development in amyotrophic lateral sclerosis. Clin. Ther. 2015, 37, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; van den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Zhu, Y.; Fotinos, A.; Mao, L.L.; Atassi, N.; Zhou, E.W.; Ahmad, S.; Guan, Y.; Berry, J.D.; Cudkowicz, M.E.; Wang, X. Neuroprotective agents target molecular mechanisms of disease in ALS. Drug Discov. Today 2015, 20, 65–75. [Google Scholar] [CrossRef]
- Jiang, X.; Guan, Y.; Zhao, Z.; Meng, F.; Wang, X.; Gao, X.; Liu, J.; Chen, Y.; Zhou, F.; Zhou, S.; et al. Potential Roles of the WNT Signaling Pathway in Amyotrophic Lateral Sclerosis. Cells 2021, 10, 839. [Google Scholar] [CrossRef]
- Lattante, S.; Ciura, S.; Rouleau, G.A.; Kabashi, E. Defining the genetic connection linking amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD). Trends Genet. 2015, 31, 263–273. [Google Scholar] [CrossRef]
- Brenner, D.; Weishaupt, J.H. Update on amyotrophic lateral sclerosis genetics. Curr. Opin. Neurol. 2019, 32, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Karnati, H.K.; Panigrahi, M.K.; Gutti, R.K.; Greig, N.H.; Tamargo, I.A. miRNAs: Key Players in Neurodegenerative Disorders and Epilepsy. J. Alzheimers Dis. 2015, 48, 563–580. [Google Scholar] [CrossRef]
- Qiu, L.; Tan, E.K.; Zeng, L. microRNAs and Neurodegenerative Diseases. Adv. Exp. Med. Biol. 2015, 888, 85–105. [Google Scholar] [CrossRef]
- Paez-Colasante, X.; Figueroa-Romero, C.; Sakowski, S.A.; Goutman, S.A.; Feldman, E.L. Amyotrophic lateral sclerosis: Mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurol. 2015, 11, 266–279. [Google Scholar] [CrossRef]
- Mandrioli, J.; Mediani, L.; Alberti, S.; Carra, S. ALS and FTD: Where RNA metabolism meets protein quality control. Semin Cell Dev. Biol. 2020, 99, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, V.K.; Kerman, A.; Laister, R.C.; Sharda, P.R.; Arslan, P.E.; Chakrabartty, A. Early steps in oxidation-induced SOD1 misfolding: Implications for non-amyloid protein aggregation in familial ALS. J. Mol. Biol. 2012, 421, 631–652. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Armstrong, G.A.B.; Drapeau, P. Neuromuscular junction abnormalities in a zebrafish loss-of-function model of TDP-43. J. Neurophysiol. 2019, 121, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: Mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Chou, C.C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef]
- Mitra, J.; Hegde, M.L. A Commentary on TDP-43 and DNA Damage Response in Amyotrophic Lateral Sclerosis. J. Exp. Neurosci. 2019, 13, 1179069519880166. [Google Scholar] [CrossRef]
- Kawahara, Y.; Kwak, S. Excitotoxicity and ALS: What is unique about the AMPA receptors expressed on spinal motor neurons? Amyotroph. Lateral Scler. 2005, 6, 131–144. [Google Scholar] [CrossRef]
- Cozzolino, M.; Carri, M.T. Mitochondrial dysfunction in ALS. Prog. Neurobiol. 2012, 97, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Theunissen, F.; West, P.K.; Brennan, S.; Petrovic, B.; Hooshmand, K.; Akkari, P.A.; Keon, M.; Guennewig, B. New perspectives on cytoskeletal dysregulation and mitochondrial mislocalization in amyotrophic lateral sclerosis. Transl. Neurodegener. 2021, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Marinkovic, P.; Reuter, M.S.; Brill, M.S.; Godinho, L.; Kerschensteiner, M.; Misgeld, T. Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 4296–4301. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, S.; Boccazzi, M.; Fumagalli, M. Oligodendrocyte Dysfunction in Amyotrophic Lateral Sclerosis: Mechanisms and Therapeutic Perspectives. Cells 2021, 10, 565. [Google Scholar] [CrossRef]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front. Immunol. 2018, 9, 1024. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207. [Google Scholar] [CrossRef]
- Correia de Sousa, M.; Gjorgjieva, M.; Dolicka, D.; Sobolewski, C.; Foti, M. Deciphering miRNAs’ Action through miRNA Editing. Int. J. Mol. Sci. 2019, 20, 6249. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Lenart, J.; Salinska, E. MicroRNA in Brain pathology: Neurodegeneration the Other Side of the Brain Cancer. Non-Coding RNA 2019, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Gascon, E.; Gao, F.B. Cause or Effect: Misregulation of microRNA Pathways in Neurodegeneration. Front. Neurosci. 2012, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Herranz, H.; Cohen, S.M. MicroRNAs and gene regulatory networks: Managing the impact of noise in biological systems. Genes Dev. 2010, 24, 1339–1344. [Google Scholar] [CrossRef]
- Gershoni-Emek, N.; Altman, T.; Ionescu, A.; Costa, C.J.; Gradus-Pery, T.; Willis, D.E.; Perlson, E. Localization of RNAi Machinery to Axonal Branch Points and Growth Cones Is Facilitated by Mitochondria and Is Disrupted in ALS. Front. Mol. Neurosci. 2018, 11, 311. [Google Scholar] [CrossRef]
- Haramati, S.; Chapnik, E.; Sztainberg, Y.; Eilam, R.; Zwang, R.; Gershoni, N.; McGlinn, E.; Heiser, P.W.; Wills, A.M.; Wirguin, I.; et al. miRNA malfunction causes spinal motor neuron disease. Proc. Natl. Acad. Sci. USA 2010, 107, 13111–13116. [Google Scholar] [CrossRef]
- Wu, D.; Raafat, A.; Pak, E.; Clemens, S.; Murashov, A.K. Dicer-microRNA pathway is critical for peripheral nerve regeneration and functional recovery in vivo and regenerative axonogenesis in vitro. Exp. Neurol. 2012, 233, 555–565. [Google Scholar] [CrossRef]
- Otaegi, G.; Pollock, A.; Sun, T. An Optimized Sponge for microRNA miR-9 Affects Spinal Motor Neuron Development in vivo. Front. Neurosci. 2011, 5, 146. [Google Scholar] [CrossRef][Green Version]
- Gagliardi, D.; Comi, G.P.; Bresolin, N.; Corti, S. MicroRNAs as regulators of cell death mechanisms in amyotrophic lateral sclerosis. J. Cell Mol. Med. 2019, 23, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Klatt, C.L.; Theis, V.; Hahn, S.; Theiss, C.; Matschke, V. Deregulated miR-29b-3p Correlates with Tissue-Specific Activation of Intrinsic Apoptosis in An Animal Model of Amyotrophic Lateral Sclerosis. Cells 2019, 8, 1077. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhang, C.; Guan, Y.; Chen, Y.; Lu, Q.; Jie, L.; Gao, H.; Du, H.; Zhang, H.; Liu, Y.; et al. Screening the expression characteristics of several miRNAs in G93A-SOD1 transgenic mouse: Altered expression of miRNA-124 is associated with astrocyte differentiation by targeting Sox2 and Sox9. J. Neurochem. 2018, 145, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Kye, M.J.; Goncalves Ido, C. The role of miRNA in motor neuron disease. Front. Cell Neurosci. 2014, 8, 15. [Google Scholar] [CrossRef]
- Singh, T.; Yadav, S. Role of microRNAs in neurodegeneration induced by environmental neurotoxicants and aging. Ageing Res. Rev. 2020, 60, 101068. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Michlewski, G.; Caceres, J.F. Post-transcriptional control of miRNA biogenesis. RNA 2019, 25, 1–16. [Google Scholar] [CrossRef]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef]
- Pu, M.; Chen, J.; Tao, Z.; Miao, L.; Qi, X.; Wang, Y.; Ren, J. Regulatory network of miRNA on its target: Coordination between transcriptional and post-transcriptional regulation of gene expression. Cell Mol. Life Sci. 2019, 76, 441–451. [Google Scholar] [CrossRef]
- Chen, L.; Heikkinen, L.; Wang, C.; Yang, Y.; Sun, H.; Wong, G. Trends in the development of miRNA bioinformatics tools. Brief. Bioinform. 2019, 20, 1836–1852. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Vishnoi, A.; Rani, S. MiRNA Biogenesis and Regulation of Diseases: An Overview. Methods Mol. Biol. 2017, 1509, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, L. MicroRNAs in amyotrophic lateral sclerosis: From pathogenetic involvement to diagnostic biomarker and therapeutic agent development. Neurol. Sci. 2020, 41, 3569–3577. [Google Scholar] [CrossRef] [PubMed]
- Goodall, E.F.; Heath, P.R.; Bandmann, O.; Kirby, J.; Shaw, P.J. Neuronal dark matter: The emerging role of microRNAs in neurodegeneration. Front. Cell Neurosci. 2013, 7, 178. [Google Scholar] [CrossRef] [PubMed]
- Juzwik, C.A.; Drake, S.S.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Douglas, C.; Morquette, B.; Moore, C.S.; Fournier, A.E. microRNA dysregulation in neurodegenerative diseases: A systematic review. Prog. Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef] [PubMed]
- Ravnik-Glavac, M.; Glavac, D. Circulating RNAs as Potential Biomarkers in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 1714. [Google Scholar] [CrossRef]
- Dardiotis, E.; Aloizou, A.M.; Siokas, V.; Patrinos, G.P.; Deretzi, G.; Mitsias, P.; Aschner, M.; Tsatsakis, A. The Role of MicroRNAs in Patients with Amyotrophic Lateral Sclerosis. J. Mol. Neurosci. 2018, 66, 617–628. [Google Scholar] [CrossRef]
- Hamzeiy, H.; Suluyayla, R.; Brinkrolf, C.; Janowski, S.J.; Hofestädt, R.; Allmer, J. Visualization and Analysis of miRNAs Implicated in Amyotrophic Lateral Sclerosis Within Gene Regulatory Pathways. Stud. Health Technol. Inf. 2018, 253, 183–187. [Google Scholar] [CrossRef]
- Laneve, P.; Tollis, P.; Caffarelli, E. RNA Deregulation in Amyotrophic Lateral Sclerosis: The Noncoding Perspective. Int. J. Mol. Sci. 2021, 22, 285. [Google Scholar] [CrossRef]
- Freischmidt, A.; Muller, K.; Zondler, L.; Weydt, P.; Volk, A.E.; Bozic, A.L.; Walter, M.; Bonin, M.; Mayer, B.; von Arnim, C.A.; et al. Serum microRNAs in patients with genetic amyotrophic lateral sclerosis and pre-manifest mutation carriers. Brain 2014, 137, 2938–2950. [Google Scholar] [CrossRef]
- Freischmidt, A.; Muller, K.; Zondler, L.; Weydt, P.; Mayer, B.; von Arnim, C.A.; Hubers, A.; Dorst, J.; Otto, M.; Holzmann, K.; et al. Serum microRNAs in sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, 2660.e15–2660.e20. [Google Scholar] [CrossRef] [PubMed]
- Raheja, R.; Regev, K.; Healy, B.C.; Mazzola, M.A.; Beynon, V.; Von Glehn, F.; Paul, A.; Diaz-Cruz, C.; Gholipour, T.; Glanz, B.I.; et al. Correlating serum micrornas and clinical parameters in amyotrophic lateral sclerosis. Muscle Nerve 2018, 58, 261–269. [Google Scholar] [CrossRef]
- Pegoraro, V.; Marozzo, R.; Angelini, C. MicroRNAs and HDAC4 protein expression in the skeletal muscle of ALS patients. Clin. Neuropathol. 2020, 39, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.P.; Wada, S.; Vergani, L.; Hock, M.B.; Lamon, S.; Leger, B.; Ushida, T.; Cartoni, R.; Wadley, G.D.; Hespel, P.; et al. Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 49, 107–117. [Google Scholar] [CrossRef]
- De Felice, B.; Guida, M.; Guida, M.; Coppola, C.; De Mieri, G.; Cotrufo, R. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene 2012, 508, 35–40. [Google Scholar] [CrossRef]
- Shioya, M.; Obayashi, S.; Tabunoki, H.; Arima, K.; Saito, Y.; Ishida, T.; Satoh, J. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol. Appl. Neurobiol. 2010, 36, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Vrabec, K.; Bostjancic, E.; Koritnik, B.; Leonardis, L.; Dolenc Groselj, L.; Zidar, J.; Rogelj, B.; Glavac, D.; Ravnik-Glavac, M. Differential Expression of Several miRNAs and the Host Genes AATK and DNM2 in Leukocytes of Sporadic ALS Patients. Front. Mol. Neurosci. 2018, 11, 106. [Google Scholar] [CrossRef]
- Joilin, G.; Leigh, P.N.; Newbury, S.F.; Hafezparast, M. An Overview of MicroRNAs as Biomarkers of ALS. Front. Neurol. 2019, 10, 186. [Google Scholar] [CrossRef]
- Gomes, C.; Cunha, C.; Nascimento, F.; Ribeiro, J.A.; Vaz, A.R.; Brites, D. Cortical Neurotoxic Astrocytes with Early ALS Pathology and miR-146a Deficit Replicate Gliosis Markers of Symptomatic SOD1G93A Mouse Model. Mol. Neurobiol. 2019, 56, 2137–2158. [Google Scholar] [CrossRef]
- Gomes, C.; Sequeira, C.; Barbosa, M.; Cunha, C.; Vaz, A.R.; Brites, D. Astrocyte regional diversity in ALS includes distinct aberrant phenotypes with common and causal pathological processes. Exp. Cell Res. 2020, 395, 112209. [Google Scholar] [CrossRef]
- Barbosa, M.; Gomes, C.; Sequeira, C.; Goncalves-Ribeiro, J.; Pina, C.C.; Carvalho, L.A.; Moreira, R.; Vaz, S.H.; Vaz, A.R.; Brites, D. Recovery of Depleted miR-146a in ALS Cortical Astrocytes Reverts Cell Aberrancies and Prevents Paracrine Pathogenicity on Microglia and Motor Neurons. Front. Cell Dev. Biol. 2021, 9, 634355. [Google Scholar] [CrossRef]
- Sison, S.L.; Patitucci, T.N.; Seminary, E.R.; Villalon, E.; Lorson, C.L.; Ebert, A.D. Astrocyte-produced miR-146a as a mediator of motor neuron loss in spinal muscular atrophy. Hum. Mol. Genet. 2017, 26, 3409–3420. [Google Scholar] [CrossRef]
- Parisi, C.; Arisi, I.; D’Ambrosi, N.; Storti, A.E.; Brandi, R.; D’Onofrio, M.; Volonte, C. Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation. Cell Death Dis. 2013, 4, e959. [Google Scholar] [CrossRef] [PubMed]
- Peplow, P.; Martinez, B. MicroRNA expression in animal models of amyotrophic lateral sclerosis and potential therapeutic approaches. Neural Regen. Res. 2022, 17, 728. [Google Scholar] [CrossRef]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef]
- de Andrade, H.M.; de Albuquerque, M.; Avansini, S.H.; de, S.R.C.; Dogini, D.B.; Nucci, A.; Carvalho, B.; Lopes-Cendes, I.; Franca, M.C., Jr. MicroRNAs-424 and 206 are potential prognostic markers in spinal onset amyotrophic lateral sclerosis. J. Neurol. Sci. 2016, 368, 19–24. [Google Scholar] [CrossRef]
- Waller, R.; Goodall, E.F.; Milo, M.; Cooper-Knock, J.; Da Costa, M.; Hobson, E.; Kazoka, M.; Wollff, H.; Heath, P.R.; Shaw, P.J.; et al. Serum miRNAs miR-206, 143–3p and 374b-5p as potential biomarkers for amyotrophic lateral sclerosis (ALS). Neurobiol. Aging 2017, 55, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.; Santos, C.; Gomes, C.; Fernandes, A.; Correia, A.M.; Sebastiao, A.M.; Vaz, A.R.; Brites, D. Downregulated Glia Interplay and Increased miRNA-155 as Promising Markers to Track ALS at an Early Stage. Mol. Neurobiol. 2018, 55, 4207–4224. [Google Scholar] [CrossRef] [PubMed]
- Koval, E.D.; Shaner, C.; Zhang, P.; du Maine, X.; Fischer, K.; Tay, J.; Chau, B.N.; Wu, G.F.; Miller, T.M. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 2013, 22, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef]
- Parisi, C.; Napoli, G.; Amadio, S.; Spalloni, A.; Apolloni, S.; Longone, P.; Volonte, C. MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ. 2016, 23, 531–541. [Google Scholar] [CrossRef]
- Li, C.; Chen, Y.; Chen, X.; Wei, Q.; Cao, B.; Shang, H. Downregulation of MicroRNA-193b-3p Promotes Autophagy and Cell Survival by Targeting TSC1/mTOR Signaling in NSC-34 Cells. Front. Mol. Neurosci. 2017, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Rohm, M.; May, C.; Marcus, K.; Steinbach, S.; Theis, V.; Theiss, C.; Matschke, V. The microRNA miR-375-3p and the Tumor Suppressor NDRG2 are Involved in Sporadic Amyotrophic Lateral Sclerosis. Cell Physiol. Biochem. 2019, 52, 1412–1426. [Google Scholar] [CrossRef]
- Kim, K.Y.; Kim, Y.R.; Choi, K.W.; Lee, M.; Lee, S.; Im, W.; Shin, J.Y.; Kim, J.Y.; Hong, Y.H.; Kim, M.; et al. Downregulated miR-18b-5p triggers apoptosis by inhibition of calcium signaling and neuronal cell differentiation in transgenic SOD1 (G93A) mice and SOD1 (G17S and G86S) ALS patients. Transl. Neurodegener. 2020, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, Y.; Chen, X.; Wei, Q.; Ou, R.; Gu, X.; Cao, B.; Shang, H. MicroRNA-183-5p is stress-inducible and protects neurons against cell death in amyotrophic lateral sclerosis. J. Cell Mol. Med. 2020, 24, 8614–8622. [Google Scholar] [CrossRef]
- Vaz, A.R.; Vizinha, D.; Morais, H.; Colaco, A.R.; Loch-Neckel, G.; Barbosa, M.; Brites, D. Overexpression of miR-124 in Motor Neurons Plays a Key Role in ALS Pathological Processes. Int. J. Mol. Sci. 2021, 22, 6128. [Google Scholar] [CrossRef]
- Hawley, Z.C.E.; Campos-Melo, D.; Strong, M.J. MiR-105 and miR-9 regulate the mRNA stability of neuronal intermediate filaments. Implications for the pathogenesis of amyotrophic lateral sclerosis (ALS). Brain Res. 2019, 1706, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.B.; de Winter, F.; Verhaagen, J. ALS as a distal axonopathy: Molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 2014, 8, 252. [Google Scholar] [CrossRef] [PubMed]
- Van Harten, A.C.M.; Phatnani, H.; Przedborski, S. Non-cell-autonomous pathogenic mechanisms in amyotrophic lateral sclerosis. Trends Neurosci. 2021, 44, 658–668. [Google Scholar] [CrossRef]
- Maimon, R.; Ionescu, A.; Bonnie, A.; Sweetat, S.; Wald-Altman, S.; Inbar, S.; Gradus, T.; Trotti, D.; Weil, M.; Behar, O.; et al. miR126-5p Downregulation Facilitates Axon Degeneration and NMJ Disruption via a Non-Cell-Autonomous Mechanism in ALS. J. Neurosci. 2018, 38, 5478–5494. [Google Scholar] [CrossRef]
- Helferich, A.M.; Brockmann, S.J.; Reinders, J.; Deshpande, D.; Holzmann, K.; Brenner, D.; Andersen, P.M.; Petri, S.; Thal, D.R.; Michaelis, J.; et al. Dysregulation of a novel miR-1825/TBCB/TUBA4A pathway in sporadic and familial ALS. Cell Mol. Life Sci. 2018, 75, 4301–4319. [Google Scholar] [CrossRef]
- Wahid, F.; Khan, T.; Kim, Y.Y. MicroRNA and diseases: Therapeutic potential as new generation of drugs. Biochimie 2014, 104, 12–26. [Google Scholar] [CrossRef]
- Akbari Dilmaghani, N.; Hussen, B.M.; Nateghinia, S.; Taheri, M.; Ghafouri-Fard, S. Emerging role of microRNAs in the pathogenesis of amyotrophic lateral sclerosis. Metab. Brain Dis. 2021, 36, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Wei, J.; Yu, C.; Han, X.; Qin, X.; Zhang, C.; Liao, W.; Li, L.; Huang, W. Non-viral nanocarriers for intracellular delivery of microRNA therapeutics. J. Mater. Chem. B 2019, 7, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci. Adv. 2017, 3, eaar3952. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.W.; Gapinske, M.; Brooks, A.K.; Woods, W.S.; Powell, J.E.; Zeballos, C.M.; Winter, J.; Perez-Pinera, P.; Gaj, T. Treatment of a Mouse Model of ALS by In Vivo Base Editing. Mol. Ther. 2020, 28, 1177–1189. [Google Scholar] [CrossRef]
- Yang, N. An overview of viral and nonviral delivery systems for microRNA. Int J. Pharm. Investig. 2015, 5. [Google Scholar] [CrossRef]
- Aigner, A. Cellular delivery in vivo of siRNA-based therapeutics. Curr. Pharm. Des. 2008, 14, 3603–3619. [Google Scholar] [CrossRef]
- van Zundert, B.; Brown, R.H., Jr. Silencing strategies for therapy of SOD1-mediated ALS. Neurosci. Lett. 2017, 636, 32–39. [Google Scholar] [CrossRef]
- Nolan, K.; Mitchem, M.R.; Jimenez-Mateos, E.M.; Henshall, D.C.; Concannon, C.G.; Prehn, J.H. Increased expression of microRNA-29a in ALS mice: Functional analysis of its inhibition. J. Mol. Neurosci. 2014, 53, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Zerah, M.; Piguet, F.; Colle, M.A.; Raoul, S.; Deschamps, J.Y.; Deniaud, J.; Gautier, B.; Toulgoat, F.; Bieche, I.; Laurendeau, I.; et al. Intracerebral Gene Therapy Using AAVrh.10-hARSA Recombinant Vector to Treat Patients with Early-Onset Forms of Metachromatic Leukodystrophy: Preclinical Feasibility and Safety Assessments in Nonhuman Primates. Hum. Gene Ther. Clin. Dev. 2015, 26, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Borel, F.; Gernoux, G.; Cardozo, B.; Metterville, J.P.; Toro Cabrera, G.C.; Song, L.; Su, Q.; Gao, G.P.; Elmallah, M.K.; Brown, R.H., Jr.; et al. Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1(G93A) Mice and Nonhuman Primates. Hum. Gene Ther. 2016, 27, 19–31. [Google Scholar] [CrossRef]
- Stoica, L.; Todeasa, S.H.; Cabrera, G.T.; Salameh, J.S.; ElMallah, M.K.; Mueller, C.; Brown, R.H., Jr.; Sena-Esteves, M. AAV delivered artificial microRNA extends survival and delays paralysis in an Amyotrophic Lateral Sclerosis mouse model. Ann. Neurol. 2016, 79, 687–700. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.; Yang, C.; Wang, D.; Wu, D.; Qi, Y.; Su, Q.; Gao, G.; Xu, Z.; Guo, Y. Slow Intrathecal Injection of rAAVrh10 Enhances its Transduction of Spinal Cord and Therapeutic Efficacy in a Mutant SOD1 Model of ALS. Neuroscience 2017, 365, 192–205. [Google Scholar] [CrossRef]
- Tung, Y.T.; Lu, Y.L.; Peng, K.C.; Yen, Y.P.; Chang, M.; Li, J.; Jung, H.; Thams, S.; Huang, Y.P.; Hung, J.H.; et al. Mir-17 approximately 92 Governs Motor Neuron Subtype Survival by Mediating Nuclear PTEN. Cell Rep. 2015, 11, 1305–1318. [Google Scholar] [CrossRef]
- Kanning, K.C.; Kaplan, A.; Henderson, C.E. Motor neuron diversity in development and disease. Annu. Rev. Neurosci. 2010, 33, 409–440. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef]
- Tung, Y.T.; Peng, K.C.; Chen, Y.C.; Yen, Y.P.; Chang, M.; Thams, S.; Chen, J.A. Mir-17~92 Confers Motor Neuron Subtype Differential Resistance to ALS-Associated Degeneration. Cell Stem Cell 2019, 25, 193–209.e197. [Google Scholar] [CrossRef]
- Martier, R.; Liefhebber, J.M.; Garcia-Osta, A.; Miniarikova, J.; Cuadrado-Tejedor, M.; Espelosin, M.; Ursua, S.; Petry, H.; van Deventer, S.J.; Evers, M.M.; et al. Targeting RNA-Mediated Toxicity in C9orf72 ALS and/or FTD by RNAi-Based Gene Therapy. Mol. Ther. Nucleic Acids 2019, 16, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Lind, L.A.; Andel, E.M.; McCall, A.L.; Dhindsa, J.S.; Johnson, K.A.; Stricklin, O.E.; Mueller, C.; ElMallah, M.K.; Lever, T.E.; Nichols, N.L. Intralingual Administration of AAVrh10-miR(SOD1) Improves Respiratory But Not Swallowing Function in a Superoxide Dismutase-1 Mouse Model of Amyotrophic Lateral Sclerosis. Hum. Gene Ther. 2020, 31, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Berry, J.D.; McKenna-Yasek, D.M.; Gernoux, G.; Owegi, M.A.; Pothier, L.M.; Douthwright, C.L.; Gelevski, D.; Luppino, S.D.; Blackwood, M.; et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med. 2020, 383, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Musaro, A. Understanding ALS: New therapeutic approaches. FEBS J. 2013, 280, 4315–4322. [Google Scholar] [CrossRef]
- Worringer, K.A.; Rand, T.A.; Hayashi, Y.; Sami, S.; Takahashi, K.; Tanabe, K.; Narita, M.; Srivastava, D.; Yamanaka, S. The let-7/LIN-41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell Stem Cell 2014, 14, 40–52. [Google Scholar] [CrossRef]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef]
- Foust, K.D.; Salazar, D.L.; Likhite, S.; Ferraiuolo, L.; Ditsworth, D.; Ilieva, H.; Meyer, K.; Schmelzer, L.; Braun, L.; Cleveland, D.W.; et al. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol. Ther. 2013, 21, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, V.; Richardson, R.M. Gene Therapy for Neurodegenerative Diseases. Neurotherapeutics 2019, 16, 166–175. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| miRNA | Model | Change | Target/Signaling Pathway | Functions |
|---|---|---|---|---|
| miR-206 [75] | SOD1G93A mice | ↑ | HDAC4 FGFBP1 | miR-206 slowed progression of ALS by sensing MN damage and promoting compensatory regeneration of neuromuscular synapses. |
| miR-155 [80] | SOD1G93A mice | ↑ | APOE pathway | Genetic ablation of miR-155 reversed the proinflammatory signature of both peripheral tissues. |
| miR-125b [81] | SOD1G93A mice | ↑ | NF-κB pathway A20 | miR-125b inhibition through A20 protein protects MNs from death induced by activating G93A microglia. |
| miR-193b-3p [82] | SOD1G93A mice NSC-34 cell | ↓ | mTOR TSC1 | Downregulation of miR-193b-3p is essential for cell survival by targeting TSC1–mTOR signaling in NSC-34 cells. |
| miR-375-3p [83] | wobbler mouse | P0: ↑ P20:↑ P40:↓ | p53 NDRG2 | After downregulating miR-375-3p expression, inefficient inhibition of p53 results in overexpression of NDRG2, increasing ROS generation and creating a vicious cycle. |
| miR-18b-5p [84] | fALS patient SOD1G93A mice NSC-34 cell | Hif1α Mef2c miR-206 Mctp1 Rarb | miR-18b-5p induced HIF1α, which increased the expression of Mef2c. Mef2c upregulated miR-206 as a transcription factor. Inhibition of mctp1 and RARB as miR-206 targets induced intracellular Ca2+ levels and reduced cell differentiation, respectively. | |
| miR-183-5p [85] | SOD1G93A mice | PDCD4 RIPK3 | miR-183-5p regulated cell apoptosis by targeting PDCD4 and necroptosis by RIPK3, respectively. | |
| miR-124 [86] | SOD1G93A mice NSC-34 cell | Upregulation of miR-124 is related to the degeneration of mSOD1 MNs, the deregulation of neuro-immune crosstalk and the imbalance of homeostasis. | ||
| miR-105 miR-9 [87] | sALS patient | ↓ | NEFL PRPH, INA | Downregulation of miR-9 and miR-105 in sALS might contribute to the loss of intermediate filament stoichiometry, ultimately leading to intermediate filament aggregation and eventually neuronal death. |
| miR-126-5p [90] | SOD1G93A mice | ↓ | Sema3A | Downregulation of miR-126-5p levels facilitated axon degeneration and NMJ disruption. |
| miR-1825 [91] | sALS patient | ↓ | TBCB CASP3 | Downregulation of miR-1825 caused translational upregulation of TBCB, which might lead to depolymerization and degradation of TUBA4A. |
| miRNA | Model | Approach Type | Therapeutic Efficacy |
|---|---|---|---|
| miR-155 [79] | SOD1G93A mice | anti-miR-155 | Inhibition of miR-155 extended survival for 10 days and disease duration for 15 days (38%). |
| miR-29a [101] | SOD1G93A mice | miR-29a-specific antagomir | miR-29a knockout did not show significant changes in disease progression or pathology of ALS, but there was a trend of prolonged lifespan and delayed disease onset in male mice. |
| miR-SOD1 [103] | SOD1G93A mice | rAAVrh10-miR-SOD1 | Silencing SOD1 significantly delayed the onset and death of SOD1G93A mice and preserved muscle strength as well as motor and respiratory function. |
| miR-SOD1 [104] | SOD1G93A mice | AAV9-amiRSOD1 vector | Treatment of AAV9-amiRSOD1 vector prolonged median survival by 50% and delayed hindlimb paralysis. |
| miR-SOD1 [105] | SOD1G93A mice | rAAVrh10-GFP-amiR-SOD1 | Slow injection of rAAVrh10-GFP-amiR-SOD1 to silence SOD1 has better therapeutic effect than rapid injection. |
| miR-17~92 [109] | SOD1G93A mice | AAV9- miR-17~92 | Overexpression of miR-17~92 in adult MNs can delay MN degeneration, enhance motor function and prolong lifespan in SOD1-linked ALS. |
| miR-C9orf72 [110] | Tg(C9orf72_3) Line 112 mice | AAV5-miC | After treatment, repeat-containing c9orf72 transcripts and RNA foci were significantly reduced. |
| miR-SOD1 [111] | SOD1G93A mice | AAVrh10-miR-SOD1 | Intralingual miR-SOD1 injection extends survival and improves respiratory function in mice. |
| miR-SOD1 [112] | SOD1 fALS patient | AAV-miR-SOD1 | Two patients with SOD1-mediated ALS were injected with AAV-miR-SOD1, which achieved short-term therapeutic effect. The results showed that intrathecal miRNA could be a potential treatment for SOD1-mediated ALS. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Zhou, F.; Guan, Y.; Meng, F.; Zhao, Z.; Su, Q.; Bao, W.; Wang, X.; Zhao, J.; Huo, Z.; et al. The Biogenesis of miRNAs and Their Role in the Development of Amyotrophic Lateral Sclerosis. Cells 2022, 11, 572. https://doi.org/10.3390/cells11030572
Liu J, Zhou F, Guan Y, Meng F, Zhao Z, Su Q, Bao W, Wang X, Zhao J, Huo Z, et al. The Biogenesis of miRNAs and Their Role in the Development of Amyotrophic Lateral Sclerosis. Cells. 2022; 11(3):572. https://doi.org/10.3390/cells11030572
Chicago/Turabian StyleLiu, Jinmeng, Fenghua Zhou, Yingjun Guan, Fandi Meng, Zhenhan Zhao, Qi Su, Weiwei Bao, Xuemei Wang, Jiantao Zhao, Zijun Huo, and et al. 2022. "The Biogenesis of miRNAs and Their Role in the Development of Amyotrophic Lateral Sclerosis" Cells 11, no. 3: 572. https://doi.org/10.3390/cells11030572
APA StyleLiu, J., Zhou, F., Guan, Y., Meng, F., Zhao, Z., Su, Q., Bao, W., Wang, X., Zhao, J., Huo, Z., Zhang, L., Zhou, S., Chen, Y., & Wang, X. (2022). The Biogenesis of miRNAs and Their Role in the Development of Amyotrophic Lateral Sclerosis. Cells, 11(3), 572. https://doi.org/10.3390/cells11030572