
The Induced Pluripotent Stem Cells in Articular Cartilage Regeneration and Disease Modelling: Are We Ready for Their Clinical Use?

 , , , and
, , , and
Abstract
:1. Introduction
2. The Surgical Methods in OA Treatment
3. iPS Cells Definition
4. Comparison of iPSC and MSC
5. iChondrocytes Cell Generation—Protocols
5.1. Differentiation Methods Using Embryoid Bodies (EB)
5.2. Direct Methods for Obtaining Chondrocytes from iPSCs
6. The Future—Clinical Use
7. Disease Modelling from Chondrogenic iPSC
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barbero, A.; Grogan, S.; Schäfer, D.; Heberer, M.; Mainil-Varlet, P.; Martin, I. Age related changes in human articular chondrocyte yield, proliferation and post-expansion chondrogenic capacity. Osteoarthr. Cartil. 2004, 12, 476–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jämsen, E.; Nevalainen, P.; Eskelinen, A.; Huotari, K.; Kalliovalkama, J.; Moilanen, T. Obesity, Diabetes, and Preoperative Hyperglycemia as Predictors of Periprosthetic Joint Infection. J. Bone Jt. Surg. 2012, 94, e101. [Google Scholar] [CrossRef] [PubMed]
- Manek, N.J.; Hart, D.; Spector, T.D.; MacGregor, A.J. The association of body mass index and osteoarthritis of the knee joint: An examination of genetic and environmental influences. Arthritis Care Res. 2003, 48, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.D.; Brophy, R.H.; Siston, R.A.; Flanigan, D.C. Treatment of Chondral Defects in the Athlete’s Knee. Arthrosc. J. Arthrosc. Relat. Surg. 2010, 26, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Garstang, S.V.; Stitik, T.P. Osteoarthritis. Am. J. Phys. Med. Rehabil. 2006, 85, S2–S11. [Google Scholar] [CrossRef]
- Li, Y.P.; Wei, X.C.; Zhou, J.M.; Wei, L. The Age-Related Changes in Cartilage and Osteoarthritis. BioMed Res. Int. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pulsatelli, L.; Addimanda, O.; Brusi, V.; Pavloska, B.; Meliconi, R. New findings in osteoarthritis pathogenesis: Therapeutic implications. Ther. Adv. Chronic Dis. 2012, 4, 23–43. [Google Scholar] [CrossRef] [Green Version]
- Koelling, S.; Kruegel, J.; Irmer, M.; Path, J.R.; Sadowski, B.; Miro, X.; Miosge, N. Migratory Chondrogenic Progenitor Cells from Repair Tissue during the Later Stages of Human Osteoarthritis. Cell Stem Cell 2009, 4, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, J.; Cromme, C.; Umlauf, D.; Frank, S.; Pap, T. Molecular mechanisms of cartilage remodelling in osteoarthritis. Int. J. Biochem. Cell Biol. 2010, 42, 1594–1601. [Google Scholar] [CrossRef]
- Wang, Y.; Wluka, A.; Jones, G.; Ding, C.; Cicuttini, F.M. Use magnetic resonance imaging to assess articular cartilage. Ther. Adv. Musculoskelet. Dis. 2011, 4, 77–97. [Google Scholar] [CrossRef] [Green Version]
- Jungmann, P.M.; Baum, T.; Bauer, J.S.; Karampinos, D.; Erdle, B.; Link, T.M.; Li, X.; Trattnig, S.; Rummeny, E.J.; Woertler, K.; et al. Cartilage Repair Surgery: Outcome Evaluation by Using Noninvasive Cartilage Biomarkers Based on Quantitative MRI Techniques? BioMed Res. Int. 2014, 2014, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wieland, H.A.; Michaelis, M.; Kirschbaum, B.J.; Rudolphi, K.A. Osteoarthritis—An untreatable disease? Nat. Rev. Drug Discov. 2005, 4, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Oldershaw, R.A. Cell sources for the regeneration of articular cartilage: The past, the horizon and the future. Int. J. Exp. Pathol. 2012, 93, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lee, S.; Jo, Y.; Lee, K.; Nemeno, J.; Nam, B.; Kim, B.; Jang, I.; Kim, H.; Takebe, T.; et al. Effects of Natural Cartilaginous Extracellular Matrix on Chondrogenic Potential for Cartilage Cell Transplantation. Transplant. Proc. 2014, 46, 1247–1250. [Google Scholar] [CrossRef]
- Vynios, D.H. Metabolism of Cartilage Proteoglycans in Health and Disease. BioMed Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Vinatier, C.; Mrugala, D.; Jorgensen, C.; Guicheux, J.; Noël, D. Cartilage engineering: A crucial combination of cells, biomaterials and biofactors. Trends Biotechnol. 2009, 27, 307–314. [Google Scholar] [CrossRef]
- Gomoll, A.H.; Minas, T. The quality of healing: Articular cartilage. Wound Repair Regen. 2014, 22, 30–38. [Google Scholar] [CrossRef]
- Steele, J.; McCullen, S.; Callanan, A.; Autefage, H.; Accardi, M.; Dini, D.; Stevens, M. Combinatorial scaffold morphologies for zonal articular cartilage engineering. Acta Biomater. 2014, 10, 2065–2075. [Google Scholar] [CrossRef] [Green Version]
- Klein, T.; Malda, J.; Sah, R.L.; Hutmacher, D.W. Tissue Engineering of Articular Cartilage with Biomimetic Zones. Tissue Eng. Part B Rev. 2009, 15, 143–157. [Google Scholar] [CrossRef]
- Perez, R.; Won, J.-E.; Knowles, J.C.; Kim, H.-W. Naturally and synthetic smart composite biomaterials for tissue regeneration. Adv. Drug Deliv. Rev. 2013, 65, 471–496. [Google Scholar] [CrossRef]
- Gomes, S.; Leonor, I.B.; Mano, J.F.; Reis, R.L.; Kaplan, D.L. Natural and genetically engineered proteins for tissue engineering. Prog. Polym. Sci. 2012, 37, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Demoor, M.; Ollitrault, D.; Gomez-Leduc, T.; Bouyoucef, M.; Hervieu, M.; Fabre, H.; Lafont, J.; Denoix, J.-M.; Audigié, F.; Mallein-Gerin, F.; et al. Cartilage tissue engineering: Molecular control of chondrocyte differentiation for proper cartilage matrix reconstruction. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2014, 1840, 2414–2440. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, E.V.; Grebenik, E.A.; Gornostaeva, S.N.; Telpuhov, V.I.; Lychagin, A.V.; Timashev, P.S.; Chagin, A.S. Repair of Damaged Articular Cartilage: Current Approaches and Future Directions. Int. J. Mol. Sci. 2018, 19, 2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nehrer, S.; Brix, M. Long-Term Outcomes of Chondrocyte-Based Cartilage Repair. Oper. Tech. Orthop. 2014, 24, 48–53. [Google Scholar] [CrossRef]
- De Windt, T.S.; Vonk, L.A.; Brittberg, M.; Saris, D.B. Treatment and Prevention of (Early) Osteoarthritis Using Articular Cartilage Repair—Fact or Fiction? A Systematic Review. Cartilage 2013, 4, 5S–12S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmal, H.; Pestka, J.M.; Salzmann, G.; Strohm, P.C.; Südkamp, N.P.; Niemeyer, P. Autologous chondrocyte implantation in children and adolescents. Knee Surg. Sports Traumatol. Arthrosc. 2012, 21, 671–677. [Google Scholar] [CrossRef]
- Minas, T.; Gomoll, A.H.; Rosenberger, R.; Royce, R.O.; Bryant, T. Increased Failure Rate of Autologous Chondrocyte Implantation after Previous Treatment with Marrow Stimulation Techniques. Am. J. Sports Med. 2009, 37, 902–908. [Google Scholar] [CrossRef]
- Siclari, A.; Mascaro, G.; Gentili, C.; Kaps, C.; Cancedda, R.; Boux, E. Cartilage repair in the knee with subchondral drilling augmented with a platelet-rich plasma-immersed polymer-based implant. Knee Surg. Sports Traumatol. Arthrosc. 2014, 22, 1225–1234. [Google Scholar] [CrossRef]
- Kreuz, P.; Steinwachs, M.; Erggelet, C.; Krause, S.; Konrad, G.; Uhl, M.; Südkamp, N. Results after microfracture of full-thickness chondral defects in different compartments in the knee. Osteoarthr. Cartil. 2006, 14, 1119–1125. [Google Scholar] [CrossRef] [Green Version]
- Trattnig, S.; Millington, S.A.; Szomolanyi, P.; Marlovits, S. MR imaging of osteochondral grafts and autologous chondrocyte implantation. Eur. Radiol. 2006, 17, 103–118. [Google Scholar] [CrossRef] [Green Version]
- Cole, B.J.; Pascual-Garrido, C.; Grumet, R.C. Surgical management of articular cartilage defects in the knee. J. Bone Jt. Surg. 2009, 91, 1778–1790. [Google Scholar]
- Hindle, P.; Hendry, J.L.; Keating, J.F.; Biant, L.C. Autologous osteochondral mosaicplasty or TruFit™ plugs for cartilage repair. Knee Surg. Sports Traumatol. Arthrosc. 2013, 22, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Heir, S.; Årøen, A.; Løken, S.; Holme, I.; Engebretsen, L.; Reinholt, F.P. Cartilage repair in the rabbit knee: Mosaic plasty resulted in higher degree of tissue filling but affected subchondral bone more than microfracture technique. Knee Surg. Sports Traumatol. Arthrosc. 2011, 20, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Pareek, A.; Reardon, P.J.; Macalena, J.A.; Levy, B.A.; Stuart, M.J.; Williams, R.J.; Krych, A.J. Osteochondral Autograft Transfer Versus Microfracture in the Knee: A Meta-analysis of Prospective Comparative Studies at Midterm. Arthrosc. J. Arthrosc. Relat. Surg. 2016, 32, 2118–2130. [Google Scholar] [CrossRef] [PubMed]
- Li, X.A.; Iyer, S.; Cross, M.B.; Figgie, M.P. Total joint replacement in adolescents. Curr. Opin. Pediatr. 2012, 24, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Ando, W.; Yasui, H.; Yamamoto, K.; Oinuma, K.; Tokunaga, H.; Inaba, Y.; Kobayashi, N.; Aihara, M.; Nakanishi, R.; Ohzono, K. A comparison of the effect of large and small metal-on-metal bearings in total hip arthroplasty on metal ion levels and the incidence of pseudotumour. Bone Jt. J. 2018, 100-B, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Reito, A.; Parkkinen, J.; Puolakka, T.; Pajamäki, J.; Eskelinen, A. Diagnostic utility of joint fluid metal ion measurement for histopathological findings in metal-on-metal hip replacements. BMC Musculoskelet. Disord. 2015, 16, 1–12. [Google Scholar] [CrossRef]
- Gomoll, A.H.; Filardo, G.; DE Girolamo, L.; Esprequeira-Mendes, J.; Marcacci, M.; Rodkey, W.G.; Steadman, R.J.; Zaffagnini, S.; Kon, E. Surgical treatment for early osteoarthritis. Part I: Cartilage repair procedures. Knee Surg. Sports Traumatol. Arthrosc. 2011, 20, 450–466. [Google Scholar] [CrossRef]
- Wang, C.-C.; Yang, K.-C.; Lin, K.-H.; Liu, H.-C.; Lin, F.-H. A highly organized three-dimensional alginate scaffold for cartilage tissue engineering prepared by microfluidic technology. Biomaterials 2011, 32, 7118–7126. [Google Scholar] [CrossRef]
- Toh, W.S.; Guo, X.-M.; Choo, A.B.; Lu, K.; Lee, E.H.; Cao, T. Differentiation and enrichment of expandable chondrogenic cells from human embryonic stem cells in vitro. J. Cell. Mol. Med. 2009, 13, 3570–3590. [Google Scholar] [CrossRef] [Green Version]
- Heywood, H.K.; Nalesso, G.; Lee, D.A.; Dell’Accio, F. Culture Expansion in Low-Glucose Conditions Preserves Chondrocyte Differentiation and Enhances Their Subsequent Capacity to Form Cartilage Tissue in Three-Dimensional Culture. BioResearch Open Access 2014, 3, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-L.; Duan, L.; Zhu, W.; Xiong, J.; Wang, D. Extracellular matrix production in vitro in cartilage tissue engineering. J. Transl. Med. 2014, 12, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saris, D.B.F.; Vanlauwe, J.; Victor, J.; Haspl, M.; Bohnsack, M.; Fortems, Y.; Vandekerckhove, B.; Almqvist, K.F.; Claes, T.; Handelberg, F.; et al. Characterized Chondrocyte Implantation Results in Better Structural Repair when Treating Symptomatic Cartilage Defects of the Knee in a Randomized Controlled Trial versus Microfracture. Am. J. Sports Med. 2008, 36, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Kamarul, T.; Selvaratnam, L.; Masjuddin, T.; Ab-Rahim, S.; Ng, C.; Chan, K.; Ahmad, T.S. Autologous Chondrocyte Transplantation in the Repair of Full-Thickness Focal Cartilage Damage in Rabbits. J. Orthop. Surg. 2008, 16, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Ozbey, O.; Sahin, Z.; Acar, N.; Ozcelik, F.T.; Ozenci, A.M.; Koksoy, S.; Ustunel, I. Characterization of colony-forming cells in adult human articular cartilage. Acta Histochem. 2014, 116, 763–770. [Google Scholar] [CrossRef]
- Lahner, M.; Duif, C.; Ficklscherer, A.; Kaps, C.; Kalwa, L.; Seidl, T. Arthroscopic Fixation of Cell Free Polymer-Based Cartilage Implants with a Bioinspired Polymer Surface on the Hip Joint: A Cadaveric Pilot Study. BioMed Res. Int. 2014, 2014, 1–6. [Google Scholar] [CrossRef]
- Schrock, J.B.; Kraeutler, M.J.; Houck, D.A.; McQueen, M.B.; Mccarty, E.C. A Cost-Effectiveness Analysis of Surgical Treatment Modalities for Chondral Lesions of the Knee: Microfracture, Osteochondral Autograft Transplantation, and Autologous Chondrocyte Implantation. Orthop. J. Sports Med. 2017, 5, 232596711770463. [Google Scholar] [CrossRef]
- Inderhaug, E.; Solheim, E. Osteochondral Autograft Transplant (Mosaicplasty) for Knee Articular Cartilage Defects. JBJS Essent. Surg. Tech. 2019, 9, e34. [Google Scholar] [CrossRef]
- Gudas, R.; Gudaitė, A.; Pocius, A.; Gudienė, A.; Čekanauskas, E.; Monastyreckienė, E.; Basevičius, A. Ten-Year Follow-up of a Prospective, Randomized Clinical Study of Mosaic Osteochondral Autologous Transplantation Versus Microfracture for the Treatment of Osteochondral Defects in the Knee Joint of Athletes. Am. J. Sports Med. 2012, 40, 2499–2508. [Google Scholar] [CrossRef]
- Patil, S.; Tapasvi, S.R. Osteochondral autografts. Curr. Rev. Musculoskelet. Med. 2015, 8, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.-D.; Xu, W.; Tian, M.; Cheng, Q.; Huang, W. Cup-on-cup technique to manage severe protrusio acetabular defects. Ann. R. Coll. Surg. Engl. 2018, 100, e181–e184. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitalipov, S.; Wolf, D. Totipotency, Pluripotency and Nuclear Reprogramming. Eng. Stem Cells 2009, 114, 185–199. [Google Scholar]
- Mikkelsen, T.S.; Hanna, J.; Zhang, X.; Ku, M.; Wernig, M.; Schorderet, P.; Bernstein, B.E.; Jaenisch, R.; Lander, E.S.; Meissner, A. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008, 454, 49–55. [Google Scholar] [CrossRef]
- Stadtfeld, M.; Maherali, N.; Breault, D.T.; Hochedlinger, K. Defining Molecular Cornerstones during Fibroblast to iPS Cell Reprogramming in Mouse. Cell Stem Cell 2008, 2, 230–240. [Google Scholar] [CrossRef] [Green Version]
- Brambrink, T.; Foreman, R.; Welstead, G.G.; Lengner, C.; Wernig, M.; Suh, H.; Jaenisch, R. Sequential Expression of Pluripotency Markers during Direct Reprogramming of Mouse Somatic Cells. Cell Stem Cell 2008, 2, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Omole, A.E.; Fakoya, A.O.J. Ten years of progress and promise of induced pluripotent stem cells: Historical origins, characteristics, mechanisms, limitations, and potential applications. PeerJ 2018, 6, e4370. [Google Scholar] [CrossRef] [Green Version]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced Pluripotent Stem Cells Generated Without Viral Integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [Green Version]
- Turinetto, V.; Orlando, L.; Giachino, C. Induced Pluripotent Stem Cells: Advances in the Quest for Genetic Stability during Reprogramming Process. Int. J. Mol. Sci. 2017, 18, 1952. [Google Scholar] [CrossRef] [Green Version]
- Lyssiotis, C.; Foreman, R.K.; Staerk, J.; Garcia, M.; Mathur, D.; Markoulaki, S.; Hanna, J.H.; Lairson, L.; Charette, B.D.; Bouchez, L.C.; et al. Reprogramming of murine fibroblasts to induced pluripotent stem cells with chemical complementation of Klf4. Proc. Natl. Acad. Sci. USA 2009, 106, 8912–8917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.B.; Greber, B.; Araúzo-Bravo, M.J.; Meyer, J.; Park, K.I.; Zaehres, H.; Schöler, H. Direct reprogramming of human neural stem cells by OCT4. Nature 2009, 461, 649–653. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Lowe, S.W. The promises and perils of p53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelcer, E.; Kulcenty, K.; Rucinski, M.; Jopek, K.; Richter, M.; Trzeciak, T.; Suchorska, W.M. Forced differentiation in vitro leads to stress-induced activation of DNA damage response in hiPSC-derived chondrocyte-like cells. PLoS ONE 2018, 13, e0198079. [Google Scholar] [CrossRef] [PubMed]
- Oleksiewicz, U.; Gładych, M.; Raman, A.T.; Heyn, H.; Mereu, E.; Chlebanowska, P.; Andrzejewska, A.; Sozańska, B.; Samant, N.; Fąk, K.; et al. TRIM28 and Interacting KRAB-ZNFs Control Self-Renewal of Human Pluripotent Stem Cells through Epigenetic Repression of Pro-differentiation Genes. Stem Cell Rep. 2017, 9, 2065–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Ling, T.; Xie, W.; Sun, H.; Zhou, Y.; Zhu, Q.; Shen, M.; Zong, L.; Lyu, G.; Zhao, Y.; et al. NuRD Blocks Reprogramming of Mouse Somatic Cells into Pluripotent Stem Cells. Stem Cells 2013, 31, 1278–1286. [Google Scholar] [CrossRef]
- Liu, X.; Li, W.; Fu, X.; Xu, Y. The Immunogenicity and Immune Tolerance of Pluripotent Stem Cell Derivatives. Front. Immunol. 2017, 8, 645. [Google Scholar] [CrossRef] [Green Version]
- Eguizabal, C.; Aran, B.; Lopes, S.M.C.D.S.; Geens, M.; Heindryckx, B.; Panula, S.; Popovic, M.; Vassena, R.; Veiga, A. Two decades of embryonic stem cells: A historical overview. Hum. Reprod. Open 2019, 2019, hoy024. [Google Scholar] [CrossRef] [Green Version]
- Garreta, E.; Sanchez, S.; Lajara, J.; Montserrat, N.; Belmonte, J.C.I. Roadblocks in the Path of iPSC to the Clinic. Curr. Transplant. Rep. 2018, 5, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Li, X.; Lu, X.; Zhang, C.; Yu, H.; Zhao, T. Cells derived from iPSC can be immunogenic — Yes or No? Protein Cell 2014, 5, 1–3. [Google Scholar] [CrossRef]
- Kaneko, S.; Yamanaka, S. To Be Immunogenic, or Not to Be: That’s the iPSC Question. Cell Stem Cell 2013, 12, 385–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Zhang, Z.-N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nature 2011, 474, 212–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Chen, S.; Zhou, Y.; Zhao, Y.; Liu, P.; Cai, J. Application of induced pluripotent stem cell transplants: Autologous or allogeneic? Life Sci. 2018, 212, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, L.; Stephenson, E.; Collins, R.; Patel, H.; Trussler, J.; Al-Bedaery, R.; Renwick, P.; Ogilvie, C.; Vaughan, R.; Ilic, D. Strategy for the creation of clinical grade hESC line banks that HLA-match a target population. EMBO Mol. Med. 2012, 5, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Peacock, S.; Chaudhry, A.N.; Bradley, J.A.; Bolton, E. Generating an iPSC Bank for HLA-Matched Tissue Transplantation Based on Known Donor and Recipient HLA Types. Cell Stem Cell 2012, 11, 147–152. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, P.E.; Ransohoff, J.D.; Nahid, A.; Wu, J.C. Immunogenicity of Pluripotent Stem Cells and Their Derivatives. Circ. Res. 2013, 112, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Caplan, A.I. Mesenchymal Stem Cells: Time to Change the Name! STEM CELLS Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, S.; Shi, Y.; Galipeau, J.; Krampera, M.; Leblanc, K.; Martin, I.; Nolta, J.; Phinney, D.G.; Sensebe, L. Mesenchymal stem versus stromal cells: International Society for Cell & Gene Therapy (ISCT®) Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy 2019, 21, 1019–1024. [Google Scholar] [CrossRef]
- Beane, O.S.; Darling, E.M. Isolation, Characterization, and Differentiation of Stem Cells for Cartilage Regeneration. Ann. Biomed. Eng. 2012, 40, 2079–2097. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkova, A.V.; Vakhrushev, I.V.; Basok, Y.B.; Grigor’Ev, A.M.; Kirsanova, L.A.; Lupatov, A.Y.; Sevastianov, V.I.; Yarygin, K.N. Chondrogeneic Potential of MSC from Different Sources in Spheroid Culture. Bull. Exp. Biol. Med. 2021, 170, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Jiang, J.; Gu, Z.; Zhang, J.; Chen, Y.; Liu, X. Mesenchymal stromal cell therapies: Immunomodulatory properties and clinical progress. Stem Cell Res. Ther. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, C.; Ritacco, C.; Hannon, M.; Seidel, L.; Delens, L.; Belle, L.; Dubois, S.; Vériter, S.; Lechanteur, C.; Briquet, A.; et al. Comparison of Mesenchymal Stromal Cells From Different Origins for the Treatment of Graft-vs.-Host-Disease in a Humanized Mouse Model. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, Y.; Vackova, I.; Kekulova, K.; Chudickova, M.; Koci, Z.; Turnovcova, K.; Skalnikova, H.K.; Vodicka, P.; Kubinova, S. A Comparative Analysis of Multipotent Mesenchymal Stromal Cells derived from Different Sources, with a Focus on Neuroregenerative Potential. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mohamed-Ahmed, S.; Fristad, I.; Lie, S.A.; Suliman, S.; Mustafa, K.; Vindenes, H.; Idris, S.B. Adipose-derived and bone marrow mesenchymal stem cells: A donor-matched comparison. Stem Cell Res. Ther. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pievani, A.; Scagliotti, V.; Russo, F.M.; Azario, I.; Rambaldi, B.; Sacchetti, B.; Marzorati, S.; Erba, E.; Giudici, G.; Riminucci, M.; et al. Comparative analysis of multilineage properties of mesenchymal stromal cells derived from fetal sources shows an advantage of mesenchymal stromal cells isolated from cord blood in chondrogenic differentiation potential. Cytotherapy 2014, 16, 893–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na Yang, H.; Park, J.S.; Woo, D.G.; Jeon, S.Y.; Do, H.-J.; Lim, H.-Y.; Kim, S.W.; Kim, J.-H.; Park, K.-H. Chondrogenesis of mesenchymal stem cells and dedifferentiated chondrocytes by transfection with SOX Trio genes. Biomaterials 2011, 32, 7695–7704. [Google Scholar] [CrossRef]
- Brown, P.T.; Squire, M.W.; Li, W.-J. Characterization and evaluation of mesenchymal stem cells derived from human embryonic stem cells and bone marrow. Cell Tissue Res. 2014, 358, 149–164. [Google Scholar] [CrossRef] [Green Version]
- Abujarour, R.; Valamehr, B.; Robinson, M.; Rezner, B.; Vranceanu, F.; Flynn, P. Optimized Surface Markers for the Prospective Isolation of High-Quality hiPSCs using Flow Cytometry Selection. Sci. Rep. 2013, 3, srep01179. [Google Scholar] [CrossRef] [Green Version]
- Priester, C.; Macdonald, A.; Dhar, M.; Bow, A. Examining the Characteristics and Applications of Mesenchymal, Induced Pluripotent, and Embryonic Stem Cells for Tissue Engineering Approaches across the Germ Layers. Pharmaceuticals 2020, 13, 344. [Google Scholar] [CrossRef]
- Yilmazer, A.; De Lázaro, I.; Bussy, C.; Kostarelos, K. In vivo reprogramming of adult somatic cells to pluripotency by overexpression of Yamanaka factors. J. Vis. Exp. 2013, e50837, e50837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Aranda, I.; Ramos-Mejia, V.; Bueno, C.; Munoz-Lopez, M.; Real, P.J.; Mácia, A.; Sanchez, L.; Ligero, G.; Garcia-Parez, J.L.; Menendez, P. Human Induced Pluripotent Stem Cells Develop Teratoma More Efficiently and Faster Than Human Embryonic Stem Cells Regardless the Site of Injection. Stem Cells 2010, 28, 1568–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-David, U.; Gan, Q.-F.; Golan-Lev, T.; Arora, P.; Yanuka, O.; Oren, Y.S.; Leikin-Frenkel, A.; Graf, M.; Garippa, R.; Boehringer, M.; et al. Selective Elimination of Human Pluripotent Stem Cells by an Oleate Synthesis Inhibitor Discovered in a High-Throughput Screen. Cell Stem Cell 2013, 12, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, T.; Hanamatsu, H.; Xu, L.; Onodera, T.; Furukawa, J.-I.; Homan, K.; Baba, R.; Kawasaki, T.; Iwasaki, N. Evaluation of Residual Human-Induced Pluripotent Stem Cells in Human Chondrocytes by Cell Type-Specific Glycosphingolipid Glycome Analysis Based on the Aminolysis-SALSA Technique. Int. J. Mol. Sci. 2019, 21, 231. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Xu, W.; Zhang, H.; Wang, Q.; Yu, J.; Zhang, R.; Chen, Y.; Xia, Y.; Wang, J.; Wang, D. Transplantation of human induced pluripotent stem cell-derived cardiomyocytes improves myocardial function and reverses ventricular remodeling in infarcted rat hearts. Stem Cell Res. Ther. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lotz, M.K. New developments in osteoarthritis: Posttraumatic osteoarthritis: Pathogenesis and pharmacological treatment options. Arthritis Res. Ther. 2010, 12, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ng, F.; Boucher, S.; Koh, S.; Sastry, K.S.R.; Chase, L.; Lakshmipathy, U.; Choong, C.; Yang, Z.; Vemuri, M.C.; Rao, M.S.; et al. PDGF, TGF-β, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): Transcriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood 2008, 112, 295–307. [Google Scholar] [CrossRef]
- Hanada, K.; Solchaga, L.A.; Caplan, A.I.; Hering, T.M.; Goldberg, V.M.; Yoo, J.U.; Johnstone, B. BMP-2 induction and TGF-beta 1 modulation of rat periosteal cell chondrogenesis. J. Cell Biochem. 2001, 81, 284–294. [Google Scholar] [CrossRef]
- Oldershaw, A.R.; Baxter, A.M.; Lowe, E.T.; Bates, N.; Grady, L.M.; Soncin, F.; Brison, D.R.; E Hardingham, T.; Kimber, S.J. Directed differentiation of human embryonic stem cells toward chondrocytes. Nat. Biotechnol. 2010, 28, 1187–1194. [Google Scholar] [CrossRef]
- Steinert, A.F.; Nöth, U.; Tuan, R.S. Concepts in gene therapy for cartilage repair. Injury 2008, 39, 97–113. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Caldwell, K.; Lu, Q.; Feng, Y.; Barnthouse, N.; Miller, A. NFAT1 deficiency provokes hypertrophic repair of articular cartilage defects and progression of posttraumatic osteoarthritis. Osteoarthr. Cartil. 2016, 24, S19. [Google Scholar] [CrossRef]
- Ko, J.-Y.; Kim, K.-I.; Park, S.; Im, G.-I. In vitro chondrogenesis and in vivo repair of osteochondral defect with human induced pluripotent stem cells. Biomaterials 2014, 35, 3571–3581. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wu, X.; Liang, Y.; Gu, H.; Song, K.; Zou, X.; Zhou, G. Repair of cartilage defects in osteoarthritis rats with induced pluripotent stem cell derived chondrocytes. BMC Biotechnol. 2016, 16, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, T.; Van Halm-Lutterodt, N.; Chen, J.; Su, Q.; Hai, Y. Reprogramming of blood cells into induced pluripotent stem cells as a new cell source for cartilage repair. Stem Cell Res. Ther. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rim, Y.A.; Nam, Y.; Park, N.; Jung, H.; Lee, K.; Lee, J.; Ju, J.H. Chondrogenic Differentiation from Induced Pluripotent Stem Cells Using Non-Viral Minicircle Vectors. Cells 2020, 9, 582. [Google Scholar] [CrossRef]
- Koyama, N.; Miura, M.; Nakao, K.; Kondo, E.; Fujii, T.; Taura, D.; Kanamoto, N.; Sone, M.; Yasoda, A.; Arai, H.; et al. Human Induced Pluripotent Stem Cells Differentiated into Chondrogenic LineageViaGeneration of Mesenchymal Progenitor Cells. Stem Cells Dev. 2013, 22, 102–113. [Google Scholar] [CrossRef]
- Craft, A.M.; Rockel, J.S.; Nartiss, Y.; A Kandel, R.; A Alman, B.; Keller, G. Generation of articular chondrocytes from human pluripotent stem cells. Nat. Biotechnol. 2015, 33, 638–645. [Google Scholar] [CrossRef]
- Lee, J.; Taylor, S.E.B.; Smeriglio, P.; Lai, J.; Maloney, W.J.; Yang, F.; Bhutani, N. Early induction of a prechondrogenic population allows efficient generation of stable chondrocytes from human induced pluripotent stem cells. FASEB J. 2015, 29, 3399–3410. [Google Scholar] [CrossRef] [Green Version]
- Boreström, C.; Simonsson, S.; Enochson, L.; Bigdeli, N.; Brantsing, C.; Ellerström, C.; Hyllner, J.; Lindahl, A. Footprint-Free Human Induced Pluripotent Stem Cells From Articular Cartilage With Redifferentiation Capacity: A First Step Toward a Clinical-Grade Cell Source. STEM CELLS Transl. Med. 2014, 3, 433–447. [Google Scholar] [CrossRef]
- Yang, S.-L.; Harnish, E.; Leeuw, T.; Dietz, U.; Batchelder, E.; Wright, P.S.; Peppard, J.; August, P.; Volle-Challier, C.; Bono, F.; et al. Compound screening platform using human induced pluripotent stem cells to identify small molecules that promote chondrogenesis. Protein Cell 2012, 3, 934–942. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Yano, F.; Mori, D.; Kawata, M.; Hoshi, K.; Takato, T.; Masaki, H.; Otsu, M.; Eto, K.; Nakauchi, H.; et al. Hyaline cartilage formation and tumorigenesis of implanted tissues derived from human induced pluripotent stem cells. Biomed. Res. 2015, 36, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A.; Morioka, M.; Yahara, Y.; Okada, M.; Kobayashi, T.; Kuriyama, S.; Matsuda, S.; Tsumaki, N. Generation of Scaffoldless Hyaline Cartilaginous Tissue from Human iPSCs. Stem Cell Rep. 2015, 4, 404–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawata, M.; Mori, D.; Kanke, K.; Hojo, H.; Ohba, S.; Chung, U.-I.; Yano, F.; Masaki, H.; Otsu, M.; Nakauchi, H.; et al. Simple and Robust Differentiation of Human Pluripotent Stem Cells toward Chondrocytes by Two Small-Molecule Compounds. Stem Cell Rep. 2019, 13, 530–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuser, U.; Buchert, J.; Haase, A.; Richter, W.; Diederichs, S. Initial WNT/β-Catenin Activation Enhanced Mesoderm Commitment, Extracellular Matrix Expression, Cell Aggregation and Cartilage Tissue Yield From Induced Pluripotent Stem Cells. Front. Cell Dev. Biol. 2020, 8, 581331. [Google Scholar] [CrossRef] [PubMed]
- Adkar, S.S.; Wu, C.-L.; Willard, V.P.; Dicks, A.; Ettyreddy, A.; Steward, N.; Bhutani, N.; Gersbach, C.A.; Guilak, F. Step-Wise Chondrogenesis of Human Induced Pluripotent Stem Cells and Purification Via a Reporter Allele Generated by CRISPR-Cas9 Genome Editing. Stem Cells 2018, 37, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzzo, R.M.; Gibson, J.; Xu, R.-H.; Lee, F.Y.; Drissi, H. Efficient differentiation of human iPSC-derived mesenchymal stem cells to chondroprogenitor cells. J. Cell. Biochem. 2012, 114, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Nejadnik, H.; Diecke, S.; Lenkov, O.D.; Chapelin, F.; Donig, J.; Tong, X.; Derugin, N.; Chan, R.C.F.; Gaur, A.; Yang, F.; et al. Improved Approach for Chondrogenic Differentiation of Human Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2015, 11, 242–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diederichs, S.; Gabler, J.; Autenrieth, J.; Kynast, K.L.; Merle, C.; Walles, H.; Utikal, J.; Richter, W. Differential Regulation of SOX9 Protein During Chondrogenesis of Induced Pluripotent Stem Cells Versus Mesenchymal Stromal Cells: A Shortcoming for Cartilage Formation. Stem Cells Dev. 2016, 25, 598–609. [Google Scholar] [CrossRef]
- Diederichs, S.; Klampfleuthner, F.A.M.; Moradi, B.; Richter, W. Chondral Differentiation of Induced Pluripotent Stem Cells Without Progression Into the Endochondral Pathway. Front. Cell Dev. Biol. 2019, 7, 270. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-H.; Wu, K.-C.; Ding, D.-C. Induced Pluripotent Stem Cell-Differentiated Chondrocytes Repair Cartilage Defect in a Rabbit Osteoarthritis Model. Stem Cells Int. 2020, 2020, 1–16. [Google Scholar] [CrossRef]
- Aisenbrey, E.A.; Bilousova, G.; Payne, K.; Bryant, S.J. Dynamic mechanical loading and growth factors influence chondrogenesis of induced pluripotent mesenchymal progenitor cells in a cartilage-mimetic hydrogel. Biomater. Sci. 2019, 7, 5388–5403. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, L.; Gao, Y.; Cheng, P.; Yang, L.; Wu, C.; Jie, Q. A lithium-containing biomaterial promotes chondrogenic differentiation of induced pluripotent stem cells with reducing hypertrophy. Stem Cell Res. Ther. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, C.; Puttonen, K.; Lindeberg, H.; Ruponen, M.; Hovatta, O.; Koistinaho, J.; Lammi, M. Chondrogenic differentiation of human pluripotent stem cells in chondrocyte co-culture. Int. J. Biochem. Cell Biol. 2013, 45, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zeng, W.; Wan, R.; Wang, J.; Zhou, Q.; Qiu, S.; Singh, S. Chondrogenic differentiation of induced pluripotent stem cells from osteoarthritic chondrocytes in alginate matrix. Eur. Cells Mater. 2012, 23, 1–12. [Google Scholar] [CrossRef]
- Kurosawa, H. Methods for inducing embryoid body formation: In vitro differentiation system of embryonic stem cells. J. Biosci. Bioeng. 2007, 103, 389–398. [Google Scholar] [CrossRef]
- Schukur, L.; Zorlutuna, P.; Cha, J.M.; Bae, H.; Khademhosseini, A. Directed Differentiation of Size-Controlled Embryoid Bodies Towards Endothelial and Cardiac Lineages in RGD-Modified Poly(Ethylene Glycol) Hydrogels. Adv. Heal. Mater. 2013, 2, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Kim, C.; Yang, J.Y.; Lee, H.; Ahn, B.; Xu, L.; Kang, J.Y.; Oh, K.W. Gravity-oriented microfluidic device for uniform and massive cell spheroid formation. Biomicrofluidics 2012, 6, 014114–141147. [Google Scholar] [CrossRef] [Green Version]
- Dias, A.D.; Unser, A.M.; Xie, Y.; Chrisey, D.B.; Corr, D.T. Generating size-controlled embryoid bodies using laser direct-write. Biofabrication 2014, 6, 025007. [Google Scholar] [CrossRef] [Green Version]
- Sa, S. Round-bottomed Honeycomb Microwells: Embryoid body shape correlates with stem cell fate. J. Dev. Biol. Tissue Eng. 2012, 4, 12–22. [Google Scholar] [CrossRef]
- Choi, Y.Y.; Chung, B.G.; Lee, D.H.; Khademhosseini, A.; Kim, J.-H.; Lee, S.-H. Controlled-size embryoid body formation in concave microwell arrays. Biomaterials 2010, 31, 4296–4303. [Google Scholar] [CrossRef]
- Hwang, Y.-S.; Chung, B.G.; Ortmann, D.; Hattori, N.; Moeller, H.-C.; Khademhosseini, A. Microwell-mediated control of embryoid body size regulates embryonic stem cell fate via differential expression of WNT5a and WNT11. Proc. Natl. Acad. Sci. USA 2009, 106, 16978–16983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, J.M.; Bae, H.; Sadr, N.; Manoucheri, S.; Edalat, F.; Kim, K.; Kim, S.B.; Kwon, I.K.; Hwang, Y.-S.; Khademhosseini, A. Embryoid body size-mediated differential endodermal and mesodermal differentiation using polyethylene glycol (PEG) microwell array. Macromol. Res. 2015, 23, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Van Winkle, A.P.; Gates, I.; Kallos, M. Mass Transfer Limitations in Embryoid Bodies during Human Embryonic Stem Cell Differentiation. Cells Tissues Organs 2012, 196, 34–47. [Google Scholar] [CrossRef]
- Dahlin, R.L.; Ni, M.; Meretoja, V.; Kasper, F.; Mikos, A.G. TGF-β3-induced chondrogenesis in co-cultures of chondrocytes and mesenchymal stem cells on biodegradable scaffolds. Biomaterials 2014, 35, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rim, Y.A.; Nam, Y.; Park, N.; Jung, H.; Jang, Y.; Lee, J.; Ju, J.H. Different Chondrogenic Potential among Human Induced Pluripotent Stem Cells from Diverse Origin Primary Cells. Stem Cells Int. 2018, 2018, 9432616. [Google Scholar] [CrossRef] [Green Version]
- Nam, Y.; Rim, Y.A.; Jung, S.M.; Ju, J.H. Cord blood cell-derived iPSCs as a new candidate for chondrogenic differentiation and cartilage regeneration. Stem Cell Res. Ther. 2017, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, B.; Wei, A.; Tao, H.; Diwan, A.D.; Ma, D.D. BMP-2 Enhances TGF-β3–Mediated Chondrogenic Differentiation of Human Bone Marrow Multipotent Mesenchymal Stromal Cells in Alginate Bead Culture. Tissue Eng. Part A 2009, 15, 1311–1320. [Google Scholar] [CrossRef]
- Lee, H.-H.; Chang, C.-C.; Shieh, M.-J.; Wang, J.-P.; Chen, Y.-T.; Young, T.-H.; Hung, S.-C. Hypoxia Enhances Chondrogenesis and Prevents Terminal Differentiation through PI3K/Akt/FoxO Dependent Anti-Apoptotic Effect. Sci. Rep. 2013, 3, 2683. [Google Scholar] [CrossRef] [Green Version]
- Carroll, S.; Buckley, C.; Kelly, D. Cyclic hydrostatic pressure promotes a stable cartilage phenotype and enhances the functional development of cartilaginous grafts engineered using multipotent stromal cells isolated from bone marrow and infrapatellar fat pad. J. Biomech. 2014, 47, 2115–2121. [Google Scholar] [CrossRef]
- Allendorph, G.P.; Read, J.D.; Kawakami, Y.; Kelber, J.; Isaacs, M.J.; Choe, S. Designer TGFβ Superfamily Ligands with Diversified Functionality. PLoS ONE 2011, 6, e26402. [Google Scholar] [CrossRef]
- Chen, X.; Yamashita, A.; Morioka, M.; Senba, T.; Kamatani, T.; Watanabe, A.; Kosai, A.; Tsumaki, N. Integration Capacity of Human Induced Pluripotent Stem Cell-Derived Cartilage. Tissue Eng. Part A 2019, 25, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, D.F.; Langille, R.M.; Dress, V.; Solursh, M. Selective stimulation of in vitro limb-bud chondrogenesis by retinoic acid. Differentiation 1988, 39, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Diederichs, S.; Tuan, R.S. Functional Comparison of Human-Induced Pluripotent Stem Cell-Derived Mesenchymal Cells and Bone Marrow-Derived Mesenchymal Stromal Cells from the Same Donor. Stem Cells Dev. 2014, 23, 1594–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narcisi, R.; Arikan, O.H.; Lehmann, J.; Berge, D.T.; van Osch, G.J. Differential Effects of Small Molecule WNT Agonists on the Multilineage Differentiation Capacity of Human Mesenchymal Stem Cells. Tissue Eng. Part A 2016, 22, 1264–1273. [Google Scholar] [CrossRef]
- Thompson, C.L.; Wiles, A.; Poole, C.A.; Knight, M.M. Lithium chloride modulates chondrocyte primary cilia and inhibits Hedgehog signaling. FASEB J. 2016, 30, 716–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustyniak, E.; Trzeciak, T.; Richter, M.; Kaczmarczyk, J.; Suchorska, W. The role of growth factors in stem cell-directed chondrogenesis: A real hope for damaged cartilage regeneration. Int. Orthop. 2015, 39, 995–1003. [Google Scholar] [CrossRef] [Green Version]
- Bian, L.; Zhai, D.Y.; Tous, E.; Rai, R.; Mauck, R.; Burdick, J.A. Enhanced MSC chondrogenesis following delivery of TGF-β3 from alginate microspheres within hyaluronic acid hydrogels in vitro and in vivo. Biomater. 2011, 32, 6425–6434. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Lapcevich, R.K.; Underhill, C.B.; Han, Z.; Gao, F.; Swartz, G.; Plum, S.M.; Zhang, L.; Green, S.J. Metastatin: A hyaluronan-binding complex from cartilage that inhibits tumor growth. Cancer Res. 2001, 61, 1022–1028. [Google Scholar]
- Hongisto, H.; Ilmarinen, T.; Vattulainen, M.; Mikhailova, A.; Skottman, H. Xeno- and feeder-free differentiation of human pluripotent stem cells to two distinct ocular epithelial cell types using simple modifications of one method. Stem Cell Res. Ther. 2017, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell–Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef]
- Turner, M.; Leslie, S.; Martin, N.; Peschanski, M.; Rao, M.; Taylor, C.J.; Trounson, A.; Turner, D.; Yamanaka, S.; Wilmut, I. Toward the Development of a Global Induced Pluripotent Stem Cell Library. Cell Stem Cell 2013, 13, 382–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Valk, J.; Mellor, D.; Brands, R.; Fischer, R.; Gruber, F.; Gstraunthaler, G.; Hellebrekers, L.; Hyllner, J.; Jonker, F.; Prieto, P.; et al. The humane collection of fetal bovine serum and possibilities for serum-free cell and tissue culture. Toxicol. Vitr. 2004, 18, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, N.; Rambhia, P.; Gishto, A. Human embryonic stem cell cultivation: Historical perspective and evolution of xeno-free culture systems. Reprod. Biol. Endocrinol. 2015, 13, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, K.; Fujita, N.; Nakagawa, T.; Nishimura, R. Effect of Fibroblast Growth Factor-2 and Serum on Canine Mesenchymal Stem Cell Chondrogenesis. Tissue Eng. Part A 2019, 25, 901–910. [Google Scholar] [CrossRef]
- Bilgen, B.; Orsini, E.; Aaron, R.K.; Ciombor, D.M. FBS suppresses TGF-β1-induced chondrogenesis in synoviocyte pellet cultures while dexamethasone and dynamic stimuli are beneficial. J. Tissue Eng. Regen. Med. 2007, 1, 436–442. [Google Scholar] [CrossRef]
- Jing-Li, Z.; Wu, X.-Y.; Tong, J.-B.; Yang, X.-X.; Zhao, J.-L.; Zheng, Q.-F.; Zhao, G.-B.; Ma, Z.-J. Comparative analysis of human mesenchymal stem cells from bone marrow and adipose tissue under xeno-free conditions for cell therapy. Stem Cell Res. Ther. 2015, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Kapacee, Z.; Peng, J.; Lu, S.; Lucas, R.J.; Hardingham, T.E.; Kimber, S.J. Cartilage Repair Using Human Embryonic Stem Cell-Derived Chondroprogenitors. STEM CELLS Transl. Med. 2014, 3, 1287–1294. [Google Scholar] [CrossRef]
- Moskalewski, S.; Hyc, A.; Osiecka-Iwan, A. Immune response by host after allogeneic chondrocyte transplant to the cartilage. Microsc. Res. Tech. 2002, 58, 3–13. [Google Scholar] [CrossRef]
- Kimura, T.; Yamashita, A.; Ozono, K.; Tsumaki, N. Limited Immunogenicity of Human Induced Pluripotent Stem Cell-Derived Cartilages. Tissue Eng. Part A 2016, 22, 1367–1375. [Google Scholar] [CrossRef]
- Zimmermann, A.; Preynat-Seauve, O.; Tiercy, J.-M.; Krause, K.-H.; Villard, J. Haplotype-Based Banking of Human Pluripotent Stem Cells for Transplantation: Potential and Limitations. Stem Cells Dev. 2012, 21, 2364–2373. [Google Scholar] [CrossRef] [Green Version]
- Nakatsuji, N.; Nakajima, F.; Tokunaga, K. HLA-haplotype banking and iPS cells. Nat. Biotechnol. 2008, 26, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Liu, S.; Woltjen, K.; Thomas, B.; Meng, G.; Hotta, A.; Takahashi, K.; Ellis, J.; Yamanaka, S.; Rancourt, D.E. Cartilage tissue engineering identifies abnormal human induced pluripotent stem cells. Sci. Rep. 2013, 3, srep01978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, A.; Morioka, M.; Kishi, H.; Kimura, T.; Yahara, Y.; Okada, M.; Fujita, K.; Sawai, H.; Ikegawa, S.; Tsumaki, N. Statin treatment rescues FGFR3 skeletal dysplasia phenotypes. Nature 2014, 513, 507–511. [Google Scholar] [CrossRef]
- Goyal, D.; Goyal, A.; Keyhani, S.; Lee, E.H.; Hui, J.H. Evidence-Based Status of Second- and Third-Generation Autologous Chondrocyte Implantation Over First Generation: A Systematic Review of Level I and II Studies. Arthrosc. J. Arthrosc. Relat. Surg. 2013, 29, 1872–1878. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Liu, C.-L.; Ting, C.-Y.; Chiu, Y.-T.; Cheng, Y.-C.; Nicholson, M.W.; Hsieh, P.C.H. Human iPSC banking: Barriers and opportunities. J. Biomed. Sci. 2019, 26, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, S.J. Disease modelling using human iPSCs. Hum. Mol. Genet. 2016, 25, R173–R181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Yang, L.; Yu, F.; Wang, S.; Wu, C.; Qu, C.; Lammi, M.; Guo, X. The potential of induced pluripotent stem cells as a tool to study skeletal dysplasias and cartilage-related pathologic conditions. Osteoarthr. Cartil. 2017, 25, 616–624. [Google Scholar] [CrossRef]
- Kim, T.-W.; Che, J.-H.; Yun, J.-W. Use of stem cells as alternative methods to animal experimentation in predictive toxicology. Regul. Toxicol. Pharmacol. 2019, 105, 15–29. [Google Scholar] [CrossRef]
- Kimura, T.; Ozaki, T.; Fujita, K.; Yamashita, A.; Morioka, M.; Ozono, K.; Tsumaki, N. Proposal of patient-specific growth plate cartilage xenograft model for FGFR3 chondrodysplasia. Osteoarthr. Cartil. 2018, 26, 1551–1561. [Google Scholar] [CrossRef] [Green Version]
- Horie, N.; Hikita, A.; Nishizawa, S.; Uto, S.; Takato, T.; Hoshi, K. Impairment of the transition from proliferative stage to prehypertrophic stage in chondrogenic differentiation of human induced pluripotent stem cells harboring the causative mutation of achondroplasia in fibroblast growth factor receptor 3. Regen. Ther. 2017, 6, 15–20. [Google Scholar] [CrossRef]
- Pretemer, Y.; Kawai, S.; Nagata, S.; Nishio, M.; Watanabe, M.; Tamaki, S.; Alev, C.; Yamanaka, Y.; Xue, J.-Y.; Wang, Z.; et al. Differentiation of Hypertrophic Chondrocytes from Human iPSCs for the In Vitro Modeling of Chondrodysplasias. Stem Cell Rep. 2021, 16, 610–625. [Google Scholar] [CrossRef]
- Okada, M.; Ikegawa, S.; Morioka, M.; Yamashita, A.; Saito, A.; Sawai, H.; Murotsuki, J.; Ohashi, H.; Okamoto, T.; Nishimura, G.; et al. Modeling type II collagenopathy skeletal dysplasia by directed conversion and induced pluripotent stem cells. Hum. Mol. Genet. 2015, 24, 299–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Stattin, E.-L.; Shaw, G.; Heinegård, D.; Sullivan, G.; Wilmut, I.; Colman, A.; Önnerfjord, P.; Khabut, A.; Aspberg, A.; et al. Chondrocytes Derived From Mesenchymal Stromal Cells and Induced Pluripotent Cells of Patients With Familial Osteochondritis Dissecans Exhibit an Endoplasmic Reticulum Stress Response and Defective Matrix Assembly. STEM CELLS Transl. Med. 2016, 5, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Ikeya, M.; Umeda, K.; Oda, H.; Nodomi, S.; Nasu, A.; Matsumoto, Y.; Izawa, K.; Horigome, K.; Kusaka, T.; et al. Enhanced Chondrogenesis of Induced Pluripotent Stem Cells From Patients With Neonatal-Onset Multisystem Inflammatory Disease Occurs via the Caspase 1-Independent cAMP/Protein Kinase A/CREB Pathway. Arthritis Rheumatol. 2014, 67, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Saitta, B.; Passarini, J.; Sareen, D.; Ornelas, L.; Sahabian, A.; Argade, S.; Krakow, D.; Cohn, D.H.; Svendsen, C.N.; Rimoin, D.L. Patient-Derived Skeletal Dysplasia Induced Pluripotent Stem Cells Display Abnormal Chondrogenic Marker Expression and Regulation by BMP2 and TGFβ1. Stem Cells Dev. 2014, 23, 1464–1478. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Ikeya, M.; Hino, K.; Horigome, K.; Fukuta, M.; Watanabe, M.; Nagata, S.; Yamamoto, T.; Otsuka, T.; Toguchida, J. New Protocol to Optimize iPS Cells for Genome Analysis of Fibrodysplasia Ossificans Progressiva. Stem Cells 2015, 33, 1730–1742. [Google Scholar] [CrossRef] [PubMed]
- Castro-Viñuelas, R.; Sanjurjo-Rodríguez, C.; Piñeiro-Ramil, M.; Hermida-Gómez, T.; Rodríguez-Fernández, S.; Oreiro, N.; De Toro, J.; Fuentes, I.; Blanco, F.J.; Díaz-Prado, S. Generation and characterization of human induced pluripotent stem cells (iPSCs) from hand osteoarthritis patient-derived fibroblasts. Sci. Rep. 2020, 10, 4272. [Google Scholar] [CrossRef] [Green Version]
- Rim, Y.; Nam, Y.; Park, N.; Lee, K.; Jung, H.; Jung, S.; Lee, J.; Ju, J. Characterization of Early-Onset Finger Osteoarthritis-Like Condition Using Patient-Derived Induced Pluripotent Stem Cells. Cells 2021, 10, 317. [Google Scholar] [CrossRef]
- Lin, Z.; Li, Z.; Li, E.N.; Li, X.; Del Duke, C.J.; Shen, H.; Hao, T.; O’Donnell, B.; Bunnell, B.; Goodman, S.B.; et al. Osteochondral Tissue Chip Derived From iPSCs: Modeling OA Pathologies and Testing Drugs. Front. Bioeng. Biotechnol. 2019, 7, 411. [Google Scholar] [CrossRef]
- Zhou, Z.-Q.; Ota, S.; Deng, C.; Akiyama, H.; Hurlin, P.J. Mutant activated FGFR3 impairs endochondral bone growth by preventing SOX9 downregulation in differentiating chondrocytes. Hum. Mol. Genet. 2014, 24, 1764–1773. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Xie, Y.; Li, W.; Huang, J.; Wang, Z.; Tang, J.; Xu, W.; Sun, X.; Tan, Q.; Huang, S.; et al. Conditional Deletion of Fgfr3 in Chondrocytes leads to Osteoarthritis-like Defects in Temporomandibular Joint of Adult Mice. Sci. Rep. 2016, 6, 24039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, M.D.; Dennis, E.; Dietmar, H.F.; Pirog, K.A. New developments in chondrocyte ER-stress and related diseases. F1000Research 2020, 9, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkitie, O.; Susic, M.; Ward, L.; Barclay, C.; Glorieux, F.H.; Cole, W.G. Schmid type of metaphyseal chondrodysplasia andCOL10A1mutations-findings in 10 patients. Am. J. Med Genet. Part A 2005, 137A, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Mäkitie, O.; Mortier, G.; Czarny-Ratajczak, M.; Wright, M.J.; Suri, M.; Rogala, P.; Freund, M.; Jackson, G.C.; Jakkula, E.; Ala-Kokko, L.; et al. Clinical and radiographic findings in multiple epiphyseal dysplasia caused by MATN3 mutations: Description of 12 patients. Am. J. Med Genet. Part A 2004, 125A, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Muttigi, M.S.; Han, I.; Park, H.-K.; Park, H.; Lee, S.-H. Matrilin-3 Role in Cartilage Development and Osteoarthritis. Int. J. Mol. Sci. 2016, 17, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Wang, W.; Ludeman, M.; Cheng, K.; Hayami, T.; Lotz, J.C.; Kapila, S. Chondrogenic Differentiation of Human Mesenchymal Stem Cells in Three-Dimensional Alginate Gels. Tissue Eng. Part A 2008, 14, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Nenna, R.; Turchetti, A.; Mastrogiorgio, G.; Midulla, F. COL2A1 Gene Mutations: Mechanisms of Spondyloepiphyseal Dysplasia Congenita. Appl. Clin. Genet. 2019, ume 12, 235–238. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, G.; Haga, N.; Kitoh, H.; Tanaka, Y.; Sonoda, T.; Kitamura, M.; Shirahama, S.; Itoh, T.; Nakashima, E.; Ohashi, H.; et al. The phenotypic spectrum ofCOL2A1mutations. Hum. Mutat. 2005, 26, 36–43. [Google Scholar] [CrossRef]
- Fosang, A.J.; Beier, F. Emerging Frontiers in cartilage and chondrocyte biology. Best Pr. Res. Clin. Rheumatol. 2011, 25, 751–766. [Google Scholar] [CrossRef]
- Nikitovic, D.; Aggelidakis, J.; Young, M.F.; Iozzo, R.; Karamanos, N.; Tzanakakis, G. The Biology of Small Leucine-rich Proteoglycans in Bone Pathophysiology. J. Biol. Chem. 2012, 287, 33926–33933. [Google Scholar] [CrossRef] [Green Version]
- Jenei-Lanzl, Z.; Meurer, A.; Zaucke, F. Interleukin-1β signaling in osteoarthritis–chondrocytes in focus. Cell. Signal. 2019, 53, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonar, S.L.; Brydges, S.D.; Mueller, J.L.; McGeough, M.D.; Pena, C.; Chen, D.; Grimston, S.K.; Hickman-Brecks, C.L.; Ravindran, S.; McAlinden, A.; et al. Constitutively Activated NLRP3 Inflammasome Causes Inflammation and Abnormal Skeletal Development in Mice. PLoS ONE 2012, 7, e35979. [Google Scholar] [CrossRef] [PubMed]
- Everaerts, W.; Nilius, B.; Owsianik, G. The vanilloid transient receptor potential channel TRPV4: From structure to disease. Prog. Biophys. Mol. Biol. 2010, 103, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Geneviève, D.; Le Merrer, M.; Feingold, J.; Munnich, A.; Maroteaux, P.; Cormier-Daire, V. Revisiting metatropic dysplasia: Presentation of a series of 19 novel patients and review of the literature. Am. J. Med Genet. Part A 2008, 146A, 992–996. [Google Scholar] [CrossRef]
- Graversen, L.; Haagerup, A.; Andersen, B.N.; Petersen, K.K.; Gjørup, V.; Gudmundsdottir, G.; Vogel, I.; Gregersen, P.A. Novel TRPV4 variant causes a severe form of metatropic dysplasia. Clin. Case Rep. 2018, 6, 1774–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, Y.; Hayashi, Y.; Schlieve, C.R.; Ikeya, M.; Kim, H.; Nguyen, T.D.; Sami, S.; Baba, S.; Barruet, E.; Nasu, A.; et al. Induced pluripotent stem cells from patients with human fibrodysplasia ossificans progressiva show increased mineralization and cartilage formation. Orphanet J. Rare Dis. 2013, 8, 190. [Google Scholar] [CrossRef] [Green Version]
- Petrie, K.A.; Lee, W.H.; Bullock, A.N.; Pointon, J.J.; Smith, R.; Russell, R.G.G.; Brown, M.A.; Wordsworth, B.P.; Triffitt, J.T. Novel Mutations in ACVR1 Result in Atypical Features in Two Fibrodysplasia Ossificans Progressiva Patients. PLoS ONE 2009, 4, e5005. [Google Scholar] [CrossRef] [Green Version]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Wadhwa, S.; Tyagi, N.; Ahmad, I.; Kumar, S.; Sagar, S.; Zahra, S.; Kamai, A.; Shamim, U.; Kapoor, S.; et al. Generation of induced pluripotent stem cell line (IGIBi007-A) from a patient with a novel acromesomelic dysplasia, PRKG2 type (AMDP). Stem Cell Res. 2021, 53, 102340. [Google Scholar] [CrossRef]
- Chen, M.; Lin, S.M.; Li, N.; Li, Y.; Li, Y.; Zhang, L. An induced pluripotent stem cell line (GZHMCi003-A) derived from a fetus with exon 3 heterozygous deletion in RUNX2 gene causing cleidocranial dysplasia. Stem Cell Res. 2021, 51, 102166. [Google Scholar] [CrossRef] [PubMed]
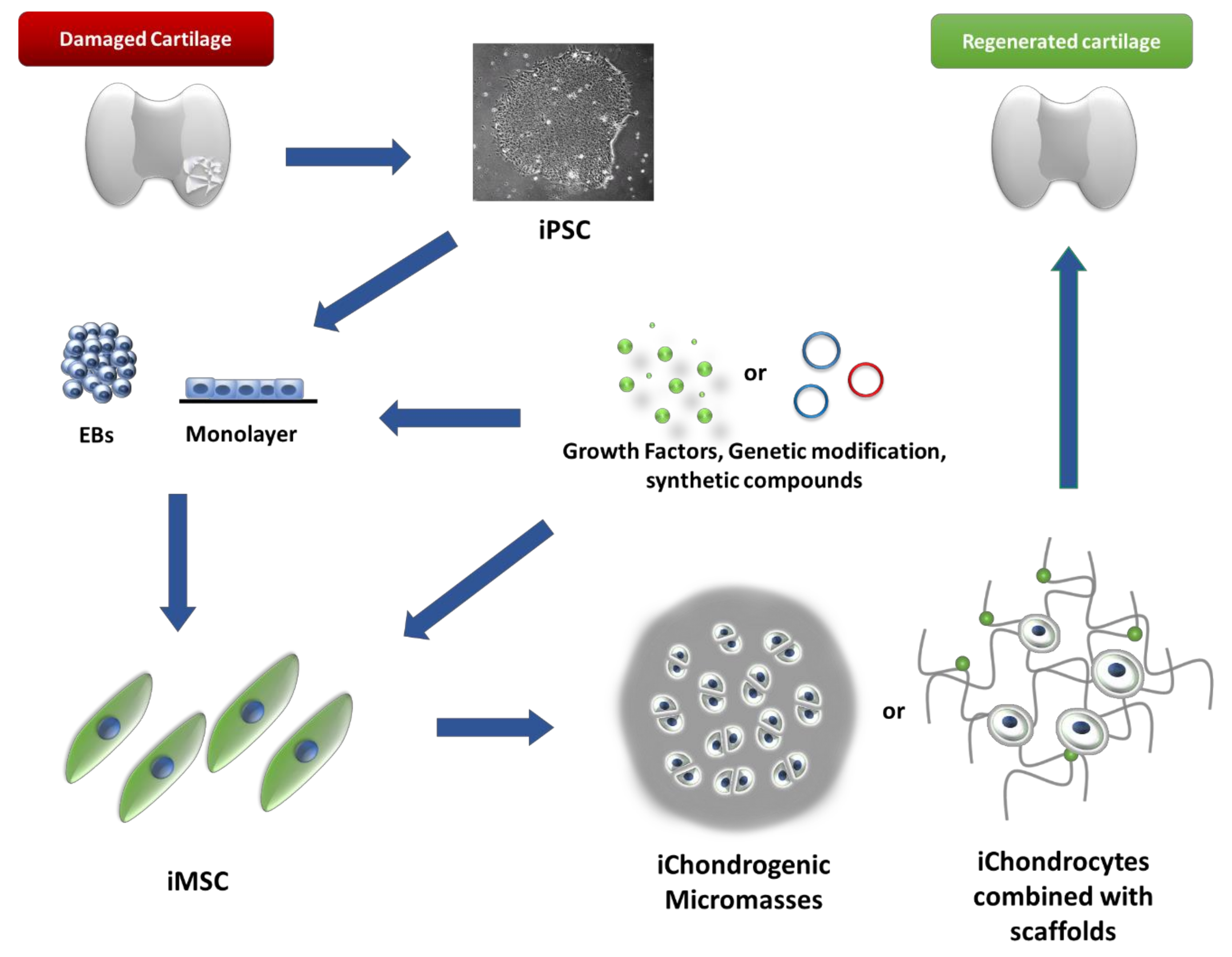

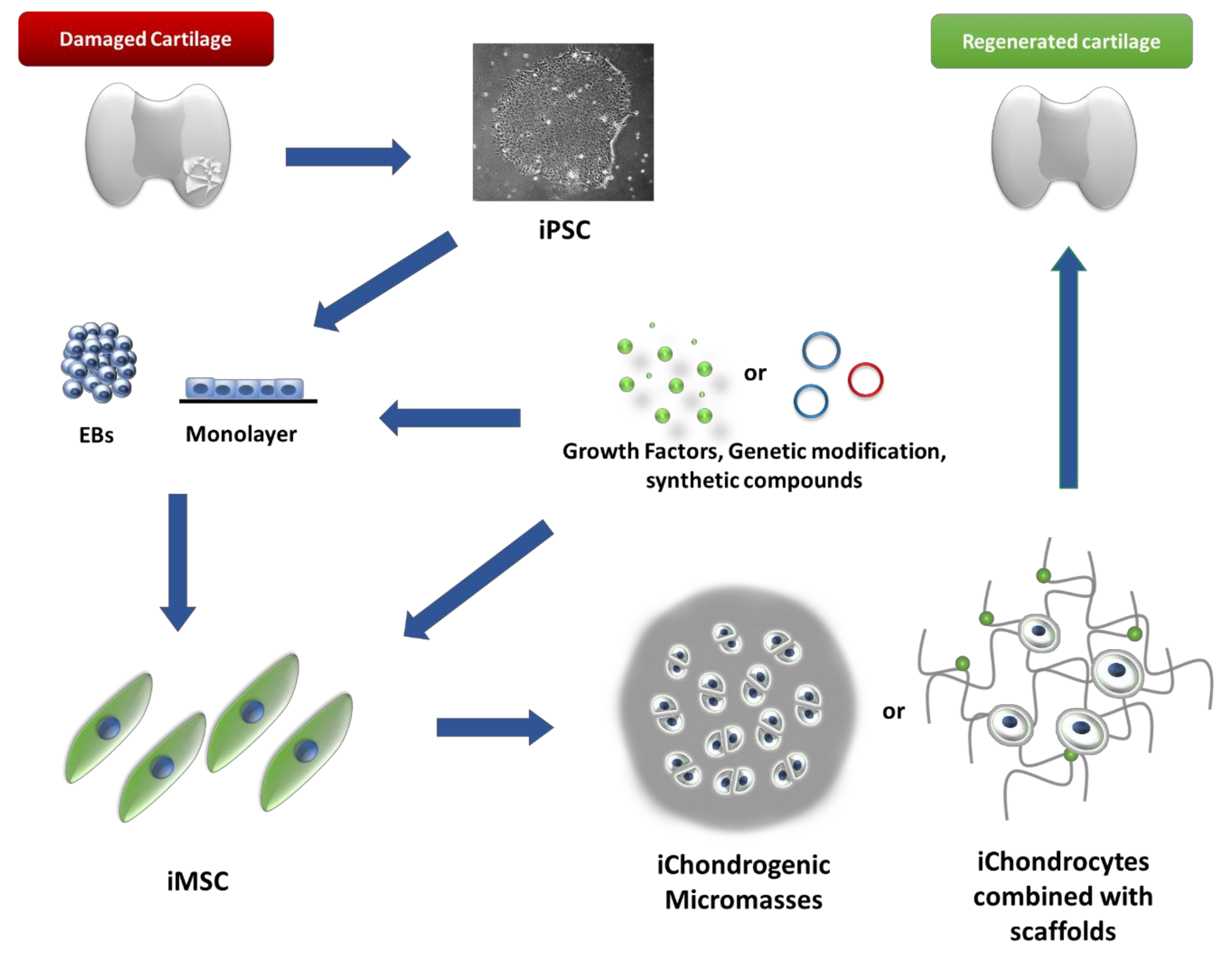{kind=link}
| Technique Repair | Advantages | Disadvantages | Ref |
|---|---|---|---|
| MACI |
|
| [23,24] |
| ACI |
|
| [22,25,26] |
| Microfracture |
|
| [4,27,28,29] |
| Mosaicplasty and OAT |
|
| [23,30,31,32,33,34] |
| Joint Arthroplasty |
|
| [35,36,37,38] |
| System of Culture | Differentiation Factor | Duration | In Vivo Confirmation | Serum-Free | Ref. |
|---|---|---|---|---|---|
| EB→3D PELLET | Stage 1: Mesenchymal differentiation through EB, ATRA (10−7 M) Stage 2: Chondrogenesis: alginate (2%), TGF-β3 (10 ng mL−1) | 33 days | Yes (osteochondral defect of the knee, rat) Outcome: 12 weeks after implantation, the macroscopic (ICRS) and histologic evaluation revealed better regenerative properties of iPSC-derived chondrocytes compared to the control group; Lack of teratoma formation was observed | No | Ko et al., 2014 [102] |
| EB→MONOLAYER | Stage 1: Mesenchymal induction through EB formation Stage 2: Chondrogenesis: TGF-β1 (10 ng mL−1) | 14 days | Yes (osteochondral defect of the knee, rat) Outcome: 15 weeks after implantation, the Micro-CT images and macroscopical evaluation has shown the repair of the damaged area on the surface, but the subchondral trabecular bone was not recovered; Histological analysis confirmed neocartilage formation and expression of COL2 and GAGs; Lack of teratoma development was notified | No | Zhu et al., 2016 [103] |
| EB→MONOLAYER → 3D PELLET | Stage 1: Collection of EB outgrowth Stage 2: Chondrogenic differentiation TGF-β1 (10 ng mL−1) | 48 days | Yes (kidney capsule, mice) Outcome: 6 weeks after implantation, the histological analysis revealed the GAGs deposition and presence of COL2 and COL10; Lack of teratoma was observed | No | Li et al., 2016 [104] |
| EB→MONOLAYER→3D PELLET | Stage 1: EB culture TGF-β1 (2 ng mL−1) Stage 2: Outgrowth of EB transfected with BMP-2 or TGF-β3 Stage 3: BMP2 or TGF-β3 transfected outgrowth EB mixed equally and cultured 3D | 47 days | Yes (osteochondral defect of the knee, rat) Outcome: 8 weeks after implantation of chondrogenic pellets, the damaged area was restored with the tissue containing a high level of GAG, COL2 and low amount of COL1 or COL10; Lack of teratoma formation was notified. | No | Rim et al., 2020 [105] |
| EB→MONOLAYER→3D PELLET | Stage 1: Collection of EB outgrowth Stage 2: Chondrogenic differentiation TGF-β3 (10 ng mL−1) | 42 days | No | No | Koyama et al., 2013 [106] |
| EB→MONOLAYER→3D PELLET | Stage 1: Activin A (2 ng mL−1); BMP-4 (3 ng mL−1); FGF2 (5 ng mL−1), CHIR99021 (1 µM) Stage 2: Dorsomorphin (4 µM); FGF2 (10 ng mL−1), SB431542 (5.4 µM) Stage 3: TGF-β3 (10 ng mL−1) | 25 days | Yes (subcutaneously, mice) Outcome: 12 weeks after implantation, the histological analysis of formed pellets has shown high expression of COL2, GAGs and low content of COL1; There was a lack of signs of mineralisation or teratoma formation | Yes | Craft et al., 2015 [107] |
| EB→MONOLAYER→3D PELLET | Stage 1: Mesoderm induction during EB formation: WNT3A (25 ng mL−1), Activin A (50, 25,10 ng mL−1), FGF2 (50 ng mL−1) and BMP4 (40 ng mL−1) Stage 2: FGF2 (50 ng mL−1), BMP4 (40, 20 ng mL−1), Follistatin (100 ng mL−1), NT4 (2 ng mL−1) and GDF5 (20, 40 ng mL−1) Stage 3: IGF (N.A.), FGF2 (N.A) | 42 days | Yes (subcutaneously, mice) Outcome: 4 weeks after implantation, the harvested pellet exhibited formation of cartilage containing a high amount of COL2 and ECM, without COL10 expression; Lack of development of teratoma | No | Lee et al., 2015 [108] |
| 3D PELLET→MONOLAYER→3D PELLET | Stage 1: Predifferentiation TGF-β1 (10 ng mL−1) Stage 2: Expansion (Human Serum) Stage 3: maturation TGF-β1 (10 ng mL−1) | 70 days | No | No | Boreström et al., 2014 [109] |
| MONOLAYER | Stage 1: WNT3A (25 ng mL−1); Activin A (50,25,10 ng mL−1); FGF2 (20 ng mL−1); BMP4 (40 ng mL−1); Follistatin (100 ng mL−1) Stage 2: BMP4 (40 ng mL−1); FGF2 (20 ng mL−1); GDF-5 (40 ng mL−1) | 14 days | No | Yes | Yang et al., 2012 [110] |
| MONOLAYER→3D DISK | Stage 1: WNT3A (25 ng mL−1), Activin A (50, 25, 10 ng mL−1), FGF2 (20 ng mL−1), BMP4 (40 ng mL−1) Stage 2: FGF2 (20 ng mL−1), BMP4 (40 ng mL−1), Follistatin (100 ng mL−1), NT4 (2 ng mL−1) Stage 3: NT4 (2 ng mL−1), FGF2 (20 ng mL−1), GDF-5 (20, 40 ng mL−1), BMP4 (20 ng mL−1) Stage 4: NT4 (2 ng mL−1), FGF2 (20 ng mL−1), GDF-5 (40 ng mL−1) | 21 days | Yes (osteochondral defect, mice) Outcome: 8 and 16 weeks after transplantation, the newly formed cartilage tissue was observed with dense ECM; one of the tested subjects has developed the teratoma | Yes | Saito et al., 2015 [111] |
| MONOLAYER→3D PELLET | Stage 1: Mesoendodermal induction: WNT3A (10 ng mL−1), Activin A (10 ng mL−1) Stage 2: Chondrogenesis: BMP2 (10 ng mL−1), TGF-β1 (10 ng mL−1), GDF5 (10 ng mL−1) | 42 days | Yes (subcutaneously, mice; Osteochondral defect, rat; Osteochondral defect, mini pig) Outcome: In mice, after 12 weeks, the implanted spheres exhibited microscopical morphology of hyaline cartilage with high ECM deposition and high COL2 expression; After 12 months, the implanted pellets underwent hypertrophy and maintained epiphyseal morphology; In rats, after 4 weeks, the osteochondral defect was restored with cartilage tissue expressing high COL2 and high GAG content with good integration with surrounding tissue; In mini-pigs, after 4 weeks the damaged cartilage was restored with visible integration with host tissue; Lack of teratoma formation in tested subjects | No | Yamashita et al., 2015 [112] |
| MONOLAYER | Stage 1: Mesoendoderm CHIR99021 (10 µM ) and TTNPB (100 nM) Stage 2: Chondrogenesis TTNPB (100 nM) | 9 days | Yes (subcutaneously, mice; osteochondral defect of the knee, mice) Outcome: After 8 weeks, histological analysis of subcutaneously implanted disc revealed high ECM and COL2 deposition without COL10 expression; After 6 months, histological analysis of the knee revealed the formation of hyaline cartilage enriched in COL2, ECM, and lack of COL10; Lack of teratoma was notified; The histological score was better than in the treated group. | Yes | Kawakata et al., 2019 [113] |
| MONOLAYER→3D PELLET | Stage 1: CHIR99021 (5 µM), FGF2 (4 ng mL−1) Stage 2: TGF- β1 (10 ng mL−1) | 56 days | No | No | Kreuser et al., 2020 [114] |
| MONOLAYER→3D PELLET | Stage 1: Mesodermal lineage induction: Activin A (30 ng mL−1), SB505124 (2 µM), CHIR99021 (4, 3 µM) , FGF2 (20 ng mL−1), C59 (1 µM), Dorsomorphin (4 µM) , PD173074 (500 nM), Purmorphamine (1 µM) Stage 2: Prechondrogenesis: BMP4 (20 ng mL−1) Stage 3: Chondrogenesis: TGF-β3 (10 ng mL−1) | 43 days | No | Yes | Adkar et al., 2019 [115] |
| MONOLAYER→3D PELLET | Stage 1: Mesenchymal induction through spontaneous differentiation, FGF2 (5 ng mL−1) Stage 2: Chondrogenic differentiation BMP2 (100 ng mL−1) | 50 days | No | No | Guzzo et al., 2013 [116] |
| MONOLAYER→3D PELLET | Stage 1: MSC induction spontaneous differentiation Stage 2: Chondrogenic differentiation TGF-β3 (10 ng mL−1) | 56 days | Yes (Osteochondral defect of the knee, rat) Outcome: After 6 weeks, the implanted spheres have shown good integration with the surrounding tissue, increased GAG and COL2 expression; The ICRS score was the highest in the iPS-derived chondrocytes | No | Nejadnik et al., 2015 [117] |
| MONOLAYER→3D PELLET | Stage 1: mesenchymal progenitor induction FGF (4 ng mL−1) Stage 2: Chondrogenesis TGF-β1 (10 ng mL−1) and BMP4 (100 ng mL−1) | 70 days | No | No | Diederichs et al., 2016 [118] |
| MONOLAYER→3D PELLET | Stage 1: mesenchymal differentiation: FGF (4 ng mL−1) Stage 2: Chondrogenesis: TGF-β1 (10 ng mL−1) | 42 days | No | No | Diederichs et al., 2019 [119] |
| MONOLAYER | Stage 1: Spontaneous differentiation into MSC in low glucose DMEM Stage 2: Chondrogenesis TGF-β1 (10 ng mL−1) | 49 days | Yes (osteochondral defect of the knee, rabbit) Outcome: After 12 weeks, histological analysis revealed better microscopical integration, increased ICRS score, higher aggrecan content and better regeneration than the control group; lack of teratoma formation | No | Chang et al., 2020 [120] |
| MONOLAYER→3D HYDROGELS | Stage 1: Mesenchymal induction through spontaneous differentiation, FGF2 (5 ng mL−1) Stage 2: Chondrogenic differentiation in the presence of TGF-β3 (10 ng mL−1) and mechanical loading | 50 days | No | No | Aisenbrey et al., 2019 [121] |
| 3D PELLET | Extracts from Li2C4S4 bioceramic (12.5, 6.25, 3.125 mg mL−1) in commercial MCDM medium | 14 days | No | Yes | Hu et al., 2020 [122] |
| MONOLAYER(HYALURONAN) | Co-culture with primary bovine chondrocytes, TGF-β3 (10 ng mL−1) | 21 days | No | No | Qu et al., 2013 [123] |
| MONOLAYER (SCAFFOLDS) | iPSC transduced with TGF-β1 and co-culture with chondrocytes | 14 days | Yes (Subcutaneously, mice) Outcome: After 6 weeks, histological analysis confirmed the highest expression of COL2 and formation of lacuna in TGF-β1/Alginate compared to other variants; lack of teratoma formation | No | Wei et al., 2012 [124] |
| Disease | Factor/Mutation | Results | Ref |
|---|---|---|---|
| Achondroplasia (ACH), Hypochondroplasia (HCH) and Thanatophoric dysplasia (TD) | ACH: FGFR3 c.1130G>A (p.G380R) HCH: FGFR3 c.1620C>G (p.N540K) TD1: FGFR3 c.742C>T (p.R248C) TD2: FGFR3 c.746C>G (p.S249C) | The FGFR inhibitor (NVP-BGJ398) has corrected the failure of growth plate formation during chondrogenic differentiation. | Kimura et al., 2018 [169] |
| Achondroplasia (ACH) and Thanatophoric dysplasia (TD) | ACH: FGFR3 c.1130G>A (p.G380R) TD1: FGFR3 c.742C>T (p.R248C) | The usage of statins during the chondrogenic differentiation of patient-derived iPSC has enabled the formation of cartilage micromasses and caused elongation of the bones in transgenic mice | Yamashita et al., 2014 [163] |
| Achondroplasia | ACH: FGFR3 c.1138G>A (p.G380R) | Genetically modified cell line using CRISPR/Cas9. Downregulation of IHH in mutated cells leads to impaired maturation of chondrogenic cells | Horie et al., 2017 [170] |
| Multiple epiphyseal dysplasia (MED) and metaphyseal chondrodysplasia type Schmid (MCDS) | MED: MATN3 c.359C>T (p.T120M) MATN3 c.659T>C (p.V220A) MATN3 c.626G>C (p.R209P) MCDS1: COL10A1 c.1841_1841delT (p.L614Rfs*8)) MCDS2: COL10A1 c.53G>A (p.G18E) COL10A1 c.1798T>C (p.S600P) | Intracellular retention of MATN3 and COL10 in mutated cells leads to UPR response, and its activation-dependent on the mutations. The in vitro model for drug testing and development was formed | Pretemer et al., 2021 [171] |
| Type II collagenopathy: -Achondrogenesis type II (ACGII) -Hypochondrogenesis (HCG) -Spondyloepiphyseal dysplasia (SPD) | ACGII-1: T>C, exon 41-intron 40, exon skipping ACGII-2: COL2A1 c.3545G>A (p.G1182A) HCG-1: COL2A1 c.1348G>C (p.G450R) SPD-1: COL2A1 c.4337dupG | Intracellular accumulation of COL2 decreased viability of obtained iChondrocytes. It increased ER-stress signalling due to a disturbed regulation of protein folding. The application of chemical chaperones could partially improve the secretion of COL2. | Okada et al., 2015 [172] |
| Familial Osteochondritis Dissecans (FOCD) | ACAN c.6907G>A (p.V2303M) | Increased accumulation of aggrecan in cells caused ER stress. Abnormal morphology of chondrocytes with a reduced ability to endure mechanical stress. The low amount of aggrecan protein in the ECM. | Xu et al., 2016 [173] |
| Neonatal-onset multisystem inflammatory disease (NOMID) | Patient 1: NLRP3 c.1709A>G (p.Y570C) Patient 2: NLRP3 c.919G>A (p.G307S) | The increased deposition of ECM and irregular endochondral ossification has been observed in mutant variants of chondrogenically differentiated iPSC. One of the potential causes was the upregulation of SOX9 in the cells mastered by the cAMP/PKA/CREB pathway. | Yokoyama et al., 2015 [174] |
| Metatropic dysplasia | TRPV4 c.1812C>G (p.I604M) | Reduced expression of genes responsible for cartilage growth markers in chondrogenic micromasses studied derived from TRPV4-iPSC. The increased expression of COL1 and irregular mineralisation patterns during chondrogenesis indicate abnormal differentiation of chondrocytes. | Saitta et al., 2014 [175] |
| Fibrodysplasia ossificans progressiva (FOP) | ACVR1 c.617G>A (p.R206H) | Enhanced expression of chondrogenesis related markers and production of ECM observed in FOP-iMSC. | Matsumoto et al., 2015 [176] |
| Hand Osteoarthritis | GDF5 SNP: rs143383 SMAD3 SNP: rs12901499 ALDH1A2 SNP: rs3204689 IL1R1 rs2287047 | Generated iPSC from patients with risk alleles correlated with the hOA exhibited decreased production of COL2 and proteoglycans in comparison with control | Castro-Viñuelas et al., 2020 [177] |
| Early-onset finger OA (efOA) | Genetic background not tested | The formation of vacuole-like structures was observed in chondrogenic masses formed in efOA with unknown aetiology. Compared to a healthy donor, the increased expression of hypertrophic markers and the secretion of cytokines and MMPS related with OA in chondrogenically differentiated efOA-iPSC. | Rim et al., 2021 [178] |
| Osteoarthritis | Artificial induced OA by addition IL-1β | The formation of mimicking osteochondral graft from iPSC cells enables us to observe changes during OA’s induction and study the biology of OA. This was shown by an increased expression of catabolic factors in constructed chips. | Lin et al., 2019 [179] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lach, M.S.; Rosochowicz, M.A.; Richter, M.; Jagiełło, I.; Suchorska, W.M.; Trzeciak, T. The Induced Pluripotent Stem Cells in Articular Cartilage Regeneration and Disease Modelling: Are We Ready for Their Clinical Use? Cells 2022, 11, 529. https://doi.org/10.3390/cells11030529
Lach MS, Rosochowicz MA, Richter M, Jagiełło I, Suchorska WM, Trzeciak T. The Induced Pluripotent Stem Cells in Articular Cartilage Regeneration and Disease Modelling: Are We Ready for Their Clinical Use? Cells. 2022; 11(3):529. https://doi.org/10.3390/cells11030529
Chicago/Turabian StyleLach, Michał S., Monika A. Rosochowicz, Magdalena Richter, Inga Jagiełło, Wiktoria M. Suchorska, and Tomasz Trzeciak. 2022. "The Induced Pluripotent Stem Cells in Articular Cartilage Regeneration and Disease Modelling: Are We Ready for Their Clinical Use?" Cells 11, no. 3: 529. https://doi.org/10.3390/cells11030529
APA StyleLach, M. S., Rosochowicz, M. A., Richter, M., Jagiełło, I., Suchorska, W. M., & Trzeciak, T. (2022). The Induced Pluripotent Stem Cells in Articular Cartilage Regeneration and Disease Modelling: Are We Ready for Their Clinical Use? Cells, 11(3), 529. https://doi.org/10.3390/cells11030529